

Abstract
Although liver fatty acid binding protein (L-FABP) binds fibrates and PPARα in vitro and enhances fibrate induction of PPARα in transformed cells, the functional significance of these findings is unclear, especially in normal hepatocytes. Studies with cultured primary mouse hepatocytes show that: 1) At physiological (6 mM) glucose, fibrates (bezafibrate, fenofibrate) only weakly activated PPARα transcription of genes in LCFA β-oxidation; 2) High (11–20 mM) glucose, but not maltose (osmotic control), significantly potentiated fibrate-induction of mRNA of these and other PPARα target genes to increase LCFA β-oxidation. These effects were associated with fibrate-mediated redistribution of L-FABP into nuclei—an effect prolonged by high glucose—but not with increased de novo fatty acid synthesis from glucose; 3) Potentiation of bezafibrate action by high glucose required an intact L-FABP/PPARα signaling pathway as shown with L-FABP null, PPARα null, PPARα inhibitor-treated WT, or PPARα-specific fenofibrate-treated WT hepatocytes. High glucose alone in the absence of fibrate was ineffective. Thus, high glucose potentiation of PPARα occurred through FABP/PPARα rather than indirectly through other PPARs or glucose induced signaling pathways. These data indicated L-FABP’s importance in fibrate-induction of hepatic PPARα LCFA β-oxidative genes, especially in the context of high glucose levels.
Keywords: fatty acid, β-oxidation, bezafibrate, L-FABP, PPARα, hepatocyte
INTRODUCTION
Resolving linkages between fatty acid and glucose signaling pathways is of great importance to our understanding of metabolic disorders such as diabetes mellitus, associated dyslipidemia (hypertriglyceridemia, decreased HDL cholesterol, increased small dense LDL), and increased cardiovascular disease (CVD) risk [1,2]. While statins lower LDL cholesterol and improve LDL subclass distribution in diabetics, they have limited effectiveness on hypertriglyceridemia and low HDL cholesterol levels—independent predictors of adverse cardiovascular events [2,3]. The liver has a key role in maintaining homeostasis of long chain fatty acid (LCFA) and glucose metabolism [4,5]. Hepatic peroxisome proliferator activated receptor-α (PPARα) is a major transcription factor controlling genes directing LCFA and lipoprotein metabolism [6–9]. Fibrates were first developed as less toxic analogues of branched-chain fatty acids—potent natural activators of PPARα [10–16]. A major beneficial effect of fibrate PPARα agonists is their effectiveness in treating hypertriglyceridemia [2,3,17–19]. Fibrates lower plasma triglyceride in large part by inducing hepatic PPARα transcription of key proteins in hepatic LCFA uptake (L-FABP, FATP) and LCFA β-oxidation (CPT1, CPT2, ACOX1) as well as proteins that increase lipolysis of plasma very low density lipoprotein (VLDL) triglyceride (LPL, apoAV, apoCIII, and others) [20–23]. Although not uniformly lowering triglyceride and CVD risk in all clinical studies, analysis of patient subgroups indicated that fibrate PPARα agonists lowered coronary heart disease events 2–6 fold more in diabetics with the most severe hypertriglyceridemia [2,3,17–19]. While PPARα agonists are also effective in reversing hepatic steatosis in various mouse models of NAFLD and NASH, their efficacy in human subjects has not yet been conclusively established due to limitations of existing studies such as small sample size, incomplete data and the use of agonists in combination with other strategies [24]. Understanding the processes accounting for the greater efficacy of fibrate triglyceride lowering therapies in the context of high glucose should significantly impact therapeutic approaches to treatment and prevention of accelerated CVD in diabetes.
Our and other labs have resolved a new pathway of PPARα regulation whereby liver fatty acid binding protein (L-FABP) facilitates not only rapid cellular LCFA uptake but also targeting into nuclei (rev. in [25–31]). Within nuclei L-FABP binds PPARα to deliver bound LCFA which in turn induce PPARα in transcription of enzymes in LCFA β-oxidation (rev. in [25–31]). Although L-FABP expression level correlates with fibrate induction of PPARα in HepG2 cells, a transformed liver-derived cell line cells [25,32], the physiological significance of these findings to normal liver hepatocytes is unclear. While important information has been gained with immortalized human and murine hepatocyte-derived cell lines, hepatoma lines differ significantly from normal hepatocytes in expressing no or less L-FABP [32–34] as well as being much less responsive to fibrates [35–38]. Further, hepatoma cells express only low Km glucose transporter GLUT1 and low Km hexokinase-1—opposite to normal hepatocytes, indicating potential differences in response to high glucose [39–41]. Critical studies demonstrated that PPARα activators such as bezafibrate are high affinity ligands of both PPARα [4,25,42] and L-FABP {Chuang, 2009 6264 /id;Chuang, 2008 6583 /id;Wolfrum, 2000 4496 /id;Di Pietro, 2000 4506 /id;Velkov, 2013 6944 /id} and that high glucose enhances L-FABP/PPARα interaction in vitro [29,30]. Glucose concentration in liver (~4mM) [48–50] is uniquely higher than in peripheral cells, and liver cytoplasmic glucose levels are more responsive to high extracellular glucose (20–30 mM) [48,49,51]. These data suggest that L-FABP-mediated fibrate signaling to PPARα in hepatocytes may be particularly sensitive to hyperglycemia [48,50].
Despite the importance of L-FABP and PPARα in hepatic LCFA and glucose metabolism, the extent to which L-FABP impacts fibrate-mediated PPARα transcriptional activity in liver hepatocytes, especially in the context of high glucose, is unknown (rev. in [25,29]. The present study used cultured primary hepatocytes from wild-type, L-FABP null, and PPARα null mice as well as wild-type hepatocytes treated with the PPARα inhibitor MK886 to determine: i) L-FABP’s role in fibrate-mediated activation of PPARα at physiologically normal glucose; ii) impact of high glucose on L-FABP mediated fibrate activation of PPARα; iii) extent to which fibrate and high glucose induced L-FABP targeting to the nucleus.
EXPERIMENTAL
Materials
Albumin fraction V-fatty acid-free (10% solution for tissue culture), stearic acid (C18:0), insulin from bovine pancreas, dexamethasone, sodium DL-lactate, D(+) glucose, D(+) maltose monohydrate, and collagen type I from rat tail were from Sigma-Aldrich (St. Louis, MO). Bezafibrate and fenofibrate were from Santa Cruz Biotechnology (Santa Cruz, CA). Collagenase B was purchased from Roche (Life technologies, Carlsbad, CA). MK886 was from Cayman Chemical (Ann Arbor, MI). Dulbecco’s modified Eagle medium DMEM/F12 (1:1), glucose-free DMEM, standard Williams medium E, Hank’s balanced salt solution free of calcium and magnesium (HBSS), fetal bovine serum, and gentamycin were from Gibco/Invitrogen (by Life Technologies, Carlsbad, CA). Glucose-free Williams medium E was custom made by Gibco, Invitrogen Corporation (Carlsbad, CA). RNA-easy kit and RNA-ase free DNA-ase set were from Qiagen Sciences (Maryland, USA) and Qiagen GmbH (Hilden, Germany), respectively. TaqMan, One-Step RT-PCR Master Mix reagents, TaqMan Gene Expression Assays for carnitine-palmitoyl-transferase 1 A (CPT1A), carnitine-palmitoyl-transferase 2 (CPT2), acyl-coenzyme A oxidase 1 (ACOX1), liver fatty acid binding protein (L-FABP), acyl CoA binding protein (ACBP), peroxisome proliferator activated receptor-α (PPAR-α), hepatocyte nuclear factor 4α (HNF4α), and hepatocyte nuclear factor 1α (HNF1-α) were purchased from Applied Biosystems (by Life Technologies, Carlsbad, CA). [1-3H]-glucose (8.00 Ci/mmol) was from Amersham/GE Healthcare (Piscataway, NJ). [9,10-3H]-stearic acid (1m Ci/ml in EtOH) was from Moravek Biochemicals (Brea, CA, USA). Rabbit polyclonal antibody against rat L-FABP was produced in our laboratory as described [52]. TO-PRO-3 monomeric cyanine nucleic acid stain and SlowFade reagent were from Invitrogen, Molecular Probes (Eugene, OR). Goat anti-rabbit IgG conjugated to FITC was from Sigma-Aldrich (St. Louis, MO). For immunogold EM localization experiments, LR White resin, donkey anti-rabbit IgG conjugated to 15 nm gold were from Electron Microscopy Sciences (Hatfield, PA).
Animals
All mice were on the C57BL/6N background. Wild-type C57BL/6N mice from Charles River Laboratories (Wilmington, MA) were obtained through the National Cancer Institute (Frederick Cancer Research and Development Center, Frederick, MD). Congenic L-FABP null mice were bred in-house to greater than N10 (99.9% homogeneity) backcross from N2 generation L-FABP gene-ablated (−/−, null) mice [53]. PPARα null mice >N10 backcross generation on the C57BL/6N background were generously provided by Dr. Frank Gonzalez (National Institutes of Health, Bethesda, MD). Livers were collected from male mice aged 3–6 mo. for hepatocyte isolation. All animal protocols were approved by the Institutional Animal Care and Use Committee (IACUC) at Texas A&M University. Animals were kept under constant 12:12 light-dark cycles and had access to food and water ad libitum.
Methods
Hepatocyte isolation, culture and treatments
Primary hepatocytes, isolated from livers of wild type (WT), L-FABP null, and PPARα null mice, were cultured and viability determined as described previously by our lab [52,54,55]. Since expression of some proteins, enzymes, and receptors may be lost with increasing time in culture, it was important to assure that the expression of those key in glucose and LCFA uptake/metabolism was maintained during the time period of the experiments. Western blotting established that expression of GLUT2, GLUT1, glucokinase, and insulin receptor in these primary hepatocytes was similar to that in liver and was constant for ≥ 3 days in culture [56]. The levels of L-FABP, SCP-2, and membrane LCFA transporters (FATP5, GOT, FATP2, FATP4) were also similar to those in liver and remained constant for 2–3 days in culture [54–56]. Likewise, expression of proteins involved in hepatic cholesterol uptake and efflux (SRB1, ABCA1, ABCG1, ABCG5, ABCG8, and NPCL1) were constant for 2–3 days in culture [56]. Finally, expression of nuclear receptors (PPARα, PPARβ, LXR, ChREBP, and SREBP) involved in expression of these proteins was also similar to that in liver and constant for 2–3 days in culture (not shown)—consistent with findings of others [57]. Therefore, all experiments were performed with mouse primary hepatocytes maintained in culture ≤ 2 days.
Hepatocyte culture with PPARα ligands at physiological and high glucose
Hepatocytes were first plated on collagen type I coated dishes (6 well- or 100 mm tissue culture dishes for RNA analysis, or 2-well chamber glasses for immunostaining and confocal imaging [54], or slides for immunostaining and electron microscopy [29,30]. Cells were then incubated in DMEM/F12 (1:1) supplemented with 10 mM HEPES pH 7.4, 0.1 mg/ml gentamycin sulfate and 5% fetal bovine serum, overnight at 37°C, 5% CO2. The medium was changed the next morning to glucose-free Williams’ medium E supplemented as described previously [58]. After further incubation in this medium for 1hr, glucose (6, 11, 20 or 30 mM) and either 40 μM albumin alone or in complex with bezafibrate (200 μM), fenofibrate (40 μM), or stearic acid (C18:0, 200 μM) were added to the culture medium, and incubated for 5 h. At these levels bezafibrate and fenofibrate exhibit little toxicity to hepatocytes [59]. Hepatocytes were collected for RNA isolation as described in below. Fibrate and stearic acid complexes with albumin were prepared as described earlier [60]. The range of glucose concentrations chosen was based on previously established levels in mouse serum under the following conditions: 5–6 mM, similar to the effect of overnight fasting (normal), 9–11 mM postprandial (transient normal; diabetic), 14–25 mM overnight fasting (high fat diet induced obese; ob/ob; diabetic) or uncontrolled diabetes (Jackson Labs mouse genome database, http://phenome.jax.org/). The lipid ligand concentration of 200 μM was based on previously established concentrations for maximal LCFA activation of PPARα in hepatocytes [58]. The 5 h incubation time was chosen based on prior studies showing maximal LCFA induction of PPARα regulated gene transcription in cultured primary hepatocytes [58].
RNA isolation and gene expression analysis by quantitative real time PCR
Hepatocytes were cultured for 5 hr as above in glucose-free Williams’ medium E supplemented as described previously [58] to which glucose (6, 11, or 20 mM) and either 40 μM albumin alone or albumin complexed with fenofibrate, bezafibrate, or stearic acid were added. Hepatocytes were then collected for total RNA isolation using the RNA-easy mini kit from Qiagen Sciences (MD, USA) according to the manufacturer’s instructions. RNA concentrations were determined spectrophotometrically. Total RNA isolated from either wild-type (WT), L-FABP null, or PPARα null hepatocytes as well as from wild-type hepatocytes treated with MK886, a PPARα inhibitor [61], was used to measure the relative level of mRNA expression for CPT1A, CPT2, ACOX1, L-FABP, ACBP, PPARα, HNF4α, and HNF1α genes by quantitative real time PCR using an ABI PRISM 7000 Sequence Detection System (SDS) from Applied Biosystems (Foster City, CA) with the following thermal protocol: 48°C for 30 min, 95°C for 10 min before the first cycle, 95°C for 15 sec, and 60°C for 60 sec, repeated 40 times. The reverse transcription and PCR reagents [TaqMan One-Step Master Mix and Gene Expression Assays for mouse CPT1A (Mm 00550438_m1), CPT2 (Mm00487202_m1), ACOX1 (Mm00443579_m1)], L-FABP (Mm00444340_m1), ACBP (Mm03048192_g1), PPARα (Mm00440939_m1), HNF4α (Mm00433959_m1), and HNF1α (Mm00493434_m1) were from Applied Biosystems (Life Technologies, Carlsbad, CA). Experiments were performed in triplicate and analyzed with ABI PRISM 7000 SDS software (Applied Biosystems) to determine ΔCt for each well of a 96 well plate, relative to 18S as a housekeeping gene used as positive control. The relative abundance of CPT1A, CPT2, ACOX1, L-FABP, ACBP, PPARα, HNF4α, and HNF1α mRNA was calculated for each glucose and albumin/fibrate or albumin/stearic acid treatment of hepatocytes, as compared to the mRNA levels in hepatocytes treated with 6 mM glucose and albumin only. The comparative 2−ΔΔCt calculation method was used as described in the manufacturer’s User Bulletin 2, ABI Prism 7000 SDS (Applied Biosystems) and earlier [62].
Intracellular glucose concentration measurements
Hepatocytes were isolated from livers of wild-type C57BL/6N mice and plated at 2×105 hepatocytes per well in the 12-well tissue culture plates (Becton Dickinson, Franklin Lakes, NJ) as described above. The plates were coated with collagen the night before. Hepatocytes were incubated with DMEM/F-12 media supplemented with 5% fetal bovine serum at 37°C, 5% CO2 overnight. Media were removed, hepatocytes were washed with PBS three times, and cells further incubated at 5% CO2 for 1 hour at 37°C with glucose-free DMEM (instead of Williams medium E) supplemented as indicated previously [58] plus 6, 20, or 30 mM glucose. Hepatocytes were washed quickly with ice old solution of MgCl2 (100 mM) with 0.1 mM phloretin, a glucose transport inhibitor [63]. Hepatocytes were then quickly scraped from the dishes with PBS and protease inhibitor at 4°C followed by disruption of hepatocytes at 4°C with a probe sonicator (Sonic Dismembrator 550, Fisher Scientific, Waltham, MA). Samples were centrifuged at 10,000 g, 4°C, for 20 min and supernatant used for glucose analysis as in [63]. Glucose quantitation was done with Amplite™ Glucose Quantitation Kit (AAT Bioquest, Inc. Sunnyvale, CA) according to the manufacturer’s instructions. Glucose concentration was calculated based on comparing hepatocyte protein to a standard curve of a known number of hepatocytes and known hepatocyte cell volume (i.e. 7.4×10−12 liter/cell) [64].
[1-3H]-Glucose-derived radioactivity incorporation into de novo synthesized fatty acids, triglycerides and total lipid mass
Mouse hepatocytes were cultured as above with glucose-free DMEM/F12 supplemented with fatty acid-free BSA (40 μM) and 6 or 20 mM glucose to which tracer amounts of [1-3H]-glucose were added at the same specific activity. At 0, 1, 2, and 6 hours incubation, medium and cells were collected to determine cytosolic glucose as described above and earlier [63], 3H incorporation into triacylglycerols (TG) and unesterified LCFAs as shown previously [52], and mass of TG and unesterified LCFAs as described [52].
Bezafibrate- and glucose-induced alterations in uptake, oxidation, and esterification of stearic acid by wild type and L-FABP null hepatocytes
Isolated primary wild type (WT) or L-FABP null (LKO) hepatocytes were plated on collagen-coated 6 well dishes at 1×106 cells/well and incubated overnight in complete D-MEM:F12 media. The medium was then removed, the cells washed twice with PBS, and incubated for 24 hours in PBS supplemented with 40 μM BSA or 200 μM bezafibrate/40 μM BSA with 6 mM or 20 mM glucose. The media was removed, the cells washed twice with PBS, and incubated for additional 24 hours in PBS supplemented with 200 μM cold stearate/40 μM BSA and 0.01 nmol/well 3H-stearate (1nmol/63.5 μCi stearic acid [9,10-3H], Moravek Biochemicals, Brea, CA) with 6 mM or 20 mM glucose. Medium was removed and saved, the cells were washed twice with PBS, and the washes pooled with the appropriated medium samples. The cells were snap frozen with liquid nitrogen, scraped in a hexane: isopropanol (3:2, v/v) mixture, centrifuged at 1500 rpm for 15 minutes, and the lipid extracts transferred to acid-washed glass tubes. The precipitated protein samples were dried overnight at room-temp, dissolved overnight in 1 ml/sample of 0.2 M KOH, and quantified by Bradford colorimetric assay for protein concentration. Cellular lipids were dried under nitrogen gas, resolved by TLC using silica gel G plates (Analtech, Inc., Newark, DE) and a petroleum ether: diethyl ether: methanol: acetic acid (180:14:4:1, v/v/v/v) solvent system, and individual lipid bands were identified with lipid standards (TLC 18.5A, Nu-Chek Prep, Inc., Elysian, MN) then removed from the plate for DPM quantification by scintillation counting. An extraction using a chloroform: methanol (2:1, v/v) solution and a 30 min, 1500 rpm centrifugation was used to separate radiolabeled aqueous (oxidation products) and organic (excreted lipids and excess labeled probe) phases from the medium/wash samples. The organic phase samples were dried under nitrogen gas, resolved by TLC, and individual lipid bands as well as the aqueous oxidation product samples were quantified for DPM as above and as described earlier for other radiolabeled LCFAs [52]. Samples extracted from cell-free wells and unlabeled feedings were used to control for loss/precipitation of the labeled probe and label specificity/background respectively.
Nuclear distribution of L-FABP: confocal microscopy and image analysis
Freshly isolated mouse hepatocytes were plated in LabTek chamber slides (Nalgene Nunc International, purchased from VWR) as described above. After incubation with glucose and BSA/lipidic ligand complexes, the cells were quickly washed with Hanks’s balanced salt solution (HBSS) 3 times, and fixated with 3.3% paraformaldehyde in HBSS (Electron Microscopy Sciences, Hatfield, PA) for 1 hr at room temperature (RT); the excess paraformaldehyde was washed with HBSS and neutralized with ammonium chloride (50 mM NH4Cl for 15 min at RT). The cells were then blocked against nonspecific protein binding with 5% fetal bovine serum in HBSS for 30 min at RT, and immunolabeled by incubation with L-FABP rabbit polyclonal serum for 1 hr, followed by goat anti-rabbit IgG conjugated to FITC for 1 hr. Nuclei were counterstained by further incubation of fixed cells with 1 μM of TO-PRO-3 monomeric cyanine nucleic acid stain from Invitrogen, Molecular Probes (Eugene, OR) for 30 min. Cells were extensively washed with HBSS, air-dried and treated with SlowFade reagent from Molecular Probes (Eugene, OR). Immunofluorescence labeled L-FABP and the nuclear stain TO-PRO-3 localization in hepatocytes was observed simultaneously through two separate photomultipliers with an MRC-1024 Bio-Rad laser scanning confocal microscopy (LSCM) system (Carl Zeiss Micro Imaging, LLC, One Zeiss Drive, Thornwood, NY 10594 USA). Excitation of FITC at 488 nm, and TO-PRO-3 at 647 nm with a krypton/argon ion laser resulted in fluorescence images that were detected in separate PMTs equipped with 540/30 (for FITC) and 680/32 (for TO-PRO-3) emission filters. The confocal images were analyzed with Image J program (http://rsbweb.nih.gov/ij/). Fluorescence intensities of nuclei and cytoplasmic areas of hepatocytes were obtained as average of fluorescence intensity per surface unit, and nuclear versus cytoplasmic ratios of these were plotted for various treatments of cells with glucose, long chain fatty acids and PPAR ligands.
Nuclear distribution of L-FABP: immunogold electron microscopy
Cultured primary hepatocytes freshly isolated mouse hepatocytes were plated on slides for electron microscopy as we described earlier [29,30]. After incubation with glucose and BSA/lipidic ligand complexes as above, the cells were quickly washed with Hanks’s balanced salt solution (HBSS) 3 times, and fixated by immersion in 4% formaldehyde, 0.1% glutaraldehyde in 0.1M sodium phosphate buffer (pH 7.4) for 20 h at 4 °C. The fixed hepatocytes were then washed with 0.1M sodium phosphate, dehydrated in an ethanol series then embedded in LR White resin at 48 °C for 2 days. Ultrathin sections (60 to 80 nm) were placed on Formvar-coated nickel grids and immunogold labeled with donkey anti-rabbit IgG conjugated to 15 nm gold [29]. L-FABP null hepatocytes incubated with anti-L-FABP hepatocytes and hepatocytes without primary antibodies were used as controls. Sections were briefly post stained with aqueous 2% uranyl acetate and Reynold’s lead citrate followed by imaging with a Zeiss 10c TEM (Carl Zeiss Micro Imaging Inc., Thornwood, New York). Anti-L-FABP labeling particle density in whole cell, nucleoplasm and cytoplasm were determined [29] and statistically analyzed [65]. Identification and manually counting of the gold particles in the cytoplasm and nucleoplasm was done using Meta Morph Image Analysis Software (Molecular Devices, Sunnyvale, CA).
Statistical Analysis
The data values were expressed as mean ± standard error with sampling size for each experiment as indicated. Statistical variance between mean values within groupings of three or more were performed using one-way ANOVA and a Newman-Keuls post-hoc test or otherwise unpaired t tests with a level of significance of P ≤ 0.05. GraphPad Prism (GraphPad Software Inc., La Jolla, CA) was used for all statistical analysis.
RESULTS
High glucose potentiated the ability of fibrates to induce PPARα transcription of LCFA β-oxidative genes in wild-type (L-FABP+/+) but not L-FABP null (L-FABP−/−) mouse hepatocytes
Since most prior studies of ligand-induced transcription of PPARα-regulated genes LCFA β-oxidation in cultured primary hepatocytes were performed with culture media containing high (11–28 mM) glucose, it was important to first resolve the extent to which glucose level itself influences fibrate induced PPARα transcriptional activity in hepatocytes cultured [28,58,66]. Therefore, the impact of glucose level on the ability of bezafibrate (pan-PPAR agonist), fenofibrate (strong PPARα agonist), and C18:0 (not agonist) to enhance transcription of three key LCFA β-oxidative genes regulated by PPARα, but not by SREBP1 or ChREBP (rev. in [67], was examined: carnitine palmitoyl transferase 1A (CPT1A, rate limiting in mitochondrial LCFA β-oxidation), mitochondrial carnitine palmitoyl transferase 2 (CPT2), and peroxisomal acyl CoA oxidase 1 (ACOX1, rate limiting in peroxisomal LCFA β-oxidation).
To determine the extent to which L-FABP contributes to basal PPARα transcription of LCFA β-oxidative enzymes, basal levels of CPT1A, CPT2, and ACOX1 mRNAs were determined at 6 mM glucose in hepatocytes from L-FABP(+/+) WT and L-FABP(−/−) null mice. As compared to wild-type hepatocytes, L-FABP null hepatocytes exhibited small (15–30%) decreases in basal PPARα transcription of the rate-limiting enzymes in mitochondrial (CPT1A) and peroxisomal (ACOX1) LCFA β-oxidation, but not that of CPT2 (Table 1). Thus, PPARα transcriptional activity of rate limiting enzymes in LCFA β-oxidative enzymes was dependent at least in part on L-FABP expression.
Table 1. Impact of L-FABP gene ablation on bezafibrate-mediated PPARα target gene expression in cultured mouse primary hepatocytes.
Mouse primary hepatocytes were cultured in 6 mM glucose media as described in legend to Fig. 1, total RNA was extracted from the hepatocytes and mRNA levels of CPT1A, CPT2, and ACOX1 were determined relative to an internal house-keeping gene as described in Methods. The relative impact of L-FABP gene ablation on the respective CPT1A, CPT2, and ACOX1 mRNA levels in response to each treatment was then calculated as the ratio of [mRNA in L-FABP (−/−)/mRNA in L-FABP (+/+)] for CPT1A, CPT2, and ACOX1, respectively.
| Treatment | Ratio of [mRNA for L-FABP (−/−)]/[mRNA for L-FABP (+/+)] | ||
|---|---|---|---|
| CPT1A | CPT2 | ACOX1 | |
| Albumin | 0.85 ± 0.09* | 1.25 ± 0.15 | 0.79 ± 0.08* |
| Bezafibrate (BZ) | 1.05 ± 0.14 | 1.14 ± 0.13 | 0.71 ± 0.08* |
| Stearic Acid (C18:0) | 0.71 ± 0.08* | 1.04 ± 0.15 | 0.57 ± 0.07* |
Mean ± standard error (SE), n= 3–4.
p<0.05.
To determine the impact of L-FABP expression on fibrate-induced PPARα transcription of LCFA β-oxidative enzymes at normal physiological (6 mM) glucose, the above experiments were repeated with bezafibrate (pan-PPAR agonist) and fenofibrate (PPARα specific agonist). Bezafibrate (Fig. 1A–C; black bars) and fenofibrate (Fig. 1D–F, black bars) only weakly enhanced CPT1A, CPT2, and ACOX1 mRNAs by 1.5–2.3 fold while stearic acid (C18:0) did not affect transcription of these genes in L-FABP(+/+) hepatocytes (Fig. 1A–C; black bars). Confirmation that the modest bezafibrate-mediated activation of PPARα at normal physiological (6 mM) glucose was dependent at least in part on L-FABP was performed using studies with L-FABP (−/−) hepatocytes. L-FABP gene ablation decreased bezafibrate-mediated PPARα transcription of the rate-limiting enzyme in peroxisomal LCFA β-oxidation (ACOX1), but not that of CPT1A or CPT2 (Table 1). L-FABP null hepatocytes cultured with stearic acid (C18:0) also exhibited decreased PPARα transcription of CPT1A and ACOX1, but not CPT2 (Table 1).
Figure 1. High glucose enhances fenofibrate- and bezafibrate-induced expression of fatty acid oxidation genes at the transcriptional level in wild-type L-FABP (+/+) but not L-FABP (−/−) mouse hepatocytes.

Fold-change in mRNA levels for CPT1A (A, D, G), CPT2 (B, E, H), and ACOX1 (C, F, I) in WT L-FABP (+/+) hepatocytes (A, B, C and D, E, F) and L-FABP (−/−) hepatocytes (G, H, I) were determined by qPCR in hepatocytes incubated with serum-free medium containing 6, 11 or 20 mM glucose and fatty acid-free albumin (Alb, 40 μM) or Alb complexed with bezafibrate (BZ, 200 μM), fenofibrate (FF, 40 μM), or stearic acid (C18:0, 200 μM) as indicated. At these levels bezafibrate and fenofibrate exhibit little toxicity to hepatocytes [59]. Mean ± standard error (SE), n= 3–4. * = p<0.05 for lipid ligand treatment vs. albumin at each glucose concentration; # = p<0.05 for 11, 20 mM glucose treatments vs. 6 mM glucose concentration within each group of lipid.
In contrast, when L-FABP(+/+) hepatocytes were cultured similarly except at high (11–20 mM) glucose, an interesting synergistic effect of glucose with fibrate on PPARα target gene expression was found. At 11–20 mM glucose, bezafibrate (Fig. 1A, white and cross-hatched bars) and fenofibrate (Fig. 1D; cross-hatched bar) enhanced PPARα transcription of CPT1A mRNA more strongly as compared to effect of bezafibrate (Fig. 1A; black bar) and fenofibrate (Fig. 1D; black bar) at 6 mM glucose. Likewise, at 20 mM glucose bezafibrate (Fig. 1B; cross-hatched bar) and fenofibrate (Fig. 1E; cross-hatched bar) enhanced PPARα transcription of CPT2 mRNA more strongly as compared to their effects at 6 mM glucose (Fig. 1B and E; black bars). Finally, 20 mM glucose also potentiated the effects of bezafibrate (Fig. 1C; cross-hatched bar) and fenofibrate (Fig. 1F; cross-hatched bar) on PPARα transcription of ACOX1 mRNA. Since the ΔCt values from qPCR measurements indicated that ACOX1 mRNA was much more abundant than those of CPT1A and CPT2 in hepatocytes, this fact may explain the much smaller impact on message upregulation of this particular gene in L-FABP(+/+) WT hepatocytes. Importantly, as shown with L-FABP (−/−) hepatocytes, the ability of high glucose to potentiate bezafibrate-induction of PPARα was highly dependent on L-FABP. L-FABP gene ablation completely abolished the ability of high glucose to potentiate bezafibrate-mediated transcription of CPT1A (Fig. 1G vs 1A), CPT2 (Fig. 1H vs 1B), and ACOX1 (Fig. 1I vs 1C). In contrast, stearic acid (C18:0) was ineffective in inducing transcription of CPT1A (Fig. 1G vs 1A), CPT2 (Fig. 1H vs 1B), and ACOX1 (Fig. 1I vs 1C) in either L-FABP(+/+) WT (Fig. 1A,B,C) or L-FABP(−/−) (Fig. 1G,H,I) hepatocytes, regardless of glucose level. Thus, high glucose did not confer on stearic acid (C18:0) the ability to induce transcription of PPARα-regulated LCFA β-oxidative enzymes (Fig. 1A–C; C18:0, open or cross-hatched bars vs black bars).
In summary, the modest impact of fibrates on PPARα transcription of CPT1A, CPT2 and ACOX1 in L-FABP (+/+) hepatocytes cultured with physiological 6 mM glucose was significantly potentiated in the presence of high glucose. In contrast, the negative control saturated fatty acid stearic acid (C18:0) is weakly bound by PPARα [42,68] and did not stimulate PPARα transcription of CPT1A, CPT2, or ACOX1 in hepatocytes regardless of the glucose level in the culture medium. It is important to note, that the high glucose potentiation of fibrate ligand-induced PPARα transcription of LCFA β-oxidative enzymes required the presence of L-FABP and correlated with the ability of high glucose to increase L-FABP/PPARα binding affinity [29,30]. Finally, high glucose potentiation of fibrate-mediated PPARα transcription was not due to high glucose itself, since incubating hepatocytes with high glucose (11 to 20 mM) in the absence of lipidic ligand did not induce or potentiate endogenous fatty acids to stimulate PPARα transcription of CPT1A, CPT2, or ACOX1 at any glucose level examined (Fig. 1A–F; Alb).
High glucose potentiated bezafibrate-mediated PPARα transcription of genes in LCFA/LCFA-CoA intracellular transport (L-FABP, ACBP) in L-FABP(+/+) but not L-FABP(−/−) mouse hepatocytes
L-FABP and ACBP are PPARα-regulated cytosolic proteins with important functions in hepatic LCFA and/or LCFA-CoA uptake and transport to LCFA β-oxidative organelles [52,69–72]. Therefore, the impact of glucose level in the culture medium on the ability of bezafibrate and stearic acid to impact PPARα transcription of L-FABP and ACBP was examined.
Increasing glucose level in the culture medium differentially altered bezafibrate-mediated PPARα transcription of L-FABP and ACBP in L-FABP(+/+) hepatocytes. In L-FABP(+/+) hepatocytes cultured with normal physiological glucose (6 mM), bezafibrate weakly stimulated transcription of L-FABP (Fig. 2A), but not ACBP (Fig. 2B). In contrast, high glucose (20 mM) potentiated the ability of bezafibrate to stimulate transcription of L-FABP (Fig. 2A) and conferred near equal enhancement of transcription of ACBP (Fig. 2B). In contrast, glucose did not impact the ability of stearic acid, a very weak/poor PPARα ligand [42,68], to induce PPARα transcription of L-FABP and ACBP in L-FABP(+/+) hepatocytes. In L-FABP(+/+) hepatocytes cultured with normal physiological glucose (6 mM), stearic acid induced transcription of ACBP (Fig. 2B), but not L-FABP (Fig. 2A). However, high glucose (20 mM) did not further increase the ability of stearic acid to stimulate transcription of ACBP (Fig. 2B) or alter transcription of L-FABP (Fig. 2A).
Figure 2. Impact of high glucose on bezafibrate-mediated transcription of LCFA/LCFA-CoA binding proteins (L-FABP, ACBP) and nuclear receptors (PPARα, HNF4α, HNF1α).

WT (A, B, D, F, G) and L-FABP (−/−) (C, E) mouse hepatocytes treated with albumin only (Alb, 40 μM), bezafibrate (BZ, 200 μM), or stearic acid (C18:0, 200 μM) and 6 or 20 mM glucose for 6 hrs as described in Methods. Total RNA was extracted and analyzed by qPCR as described in Methods to determine transcription of L-FABP (A), ACBP (B, C), PPARα (D, E), HNF4α (F), or HNF1α (G) mRNAs. L-FABP mRNA was not detected in L-FABP (−/−) hepatocytes as expected (not shown). Mean ± SE, n=4, p < 0.005. (*), significant difference between ligand/albumin and albumin at constant concentration of glucose; (#), significant difference between 6 and 20 mM glucose for the same ligand.
These effects of glucose and bezafibrate on PPARα transcription of ACBP were highly dependent on the presence of L-FABP. Ablation of L-FABP completely abolished the ability of bezafibrate to induce transcription of ACBP, regardless of glucose level in the culture medium (Fig. 2C).
Taken together, these data show that high glucose not only potentiated bezafibrate-mediated transcription of L-FABP but also ACBP, a protein not induced at normal physiological glucose. In contrast, high glucose did not potentiate or confer on stearic acid the ability to induce either L-FABP or ACBP. Interestingly, the synergistic effect of high glucose/bezafibrate on the expression of ACBP mRNA was abolished in L-FABP(−/−) mouse hepatocytes, underscoring the critical role of L-FABP in glucose/fibrate-initiated signal transduction and transcription regulation of another important PPARα controlled gene such as ACBP.
High glucose potentiated the ability of bezafibrate to induce PPARα mediated transcription directly through PPARα rather than other key nuclear receptors (HNF4α and HNF1α) known to induce PPARα and L-FABP in wild-type L-FABP(+/+) but not L-FABP(−/−) null mouse hepatocytes
Consistent with the presence of a DR1 response element in the PPARα promoter, transcription of PPARα itself is regulated by PPARα ligands and by HNF4α [73]. Further, PPARα regulated genes such as L-FABP are also induced by HNF1α, albeit through a different response element [74–76]. Therefore, the possibility that high glucose potentiated transcription of PPARα directly and/or indirectly through HNF4α and/or HNF1α was examined.
Bezafibrate differentially impacted transcription of PPARα as compared to HNF4α and HNF1α. Bezafibrate induced transcription of PPARα at low glucose (Fig. 2D; black) but not in the absence of L-FABP (Fig. 2E, black). In contrast, bezafibrate decreased transcription of HNF4α (Fig. 2F; black) and HNF1α (Fig. 2G; black). High glucose potentiated bezafibrate-mediated transcription of PPARα itself (Fig. 2D; white) but not in the absence of L-FABP (Fig. 2E; white). In contrast, high glucose did not enhance transcription of HNF4α (Fig. 2F; white) and only weakly increased that of HNF1α—but not to basal level in the absence of bezafibrate (Fig. 2G; white).
Taken together these findings showed that high glucose synergized with bezafibrate to induce transcription of PPARα as long as L-FABP was present. However, this was not due to indirect upregulation of HNF4α and/or HNF1α.
Potentiation of bezafibrate-mediated PPARα transcription of LCFA β-oxidative genes in cultured primary mouse hepatocytes was not due to an osmotic effect
To determine if high glucose enhancement of fibrate-mediated PPARα transcription was due to higher osmolality, glucose was replaced with maltose—a disaccharide sugar not taken up [77]. Hepatocytes were incubated with medium containing either albumin or albumin-bezafibrate complex to which was added 6 mM glucose or [6 mM glucose + 14 mM maltose] as described in Methods. Incubation with [6 mM glucose + 14 mM maltose] did not potentiate transcription of CPT1A, CPT2, or ACOX1 (Suppl. Fig. 1A–C). Finally, incubation with medium containing either alb or albumin-C18:1 complex to which was added 6 mM glucose or [6 mM glucose + 14 mM maltose] also did not potentiate transcription of CPT1A, CPT2, or ACOX1 (Suppl. Fig. 1A–C). Taken together, these findings suggested that high glucose did not potentiate bezafibrate-mediated PPARα transcription of LCFA β-oxidative enzymes due to increased osmolality at high glucose.
High glucose potentiation of bezafibrate-mediated transcriptional of LCFA β-oxidative genes was primarily regulated through PPARα isoform in liver hepatocytes
In liver hepatocytes CPT2A is regulated by PPARα while CPT1A and ACOX1 are regulated by both PPARα and PPARβ [78]. Thus, it is essential to examine if the ability of high glucose to potentiate L-FABP mediated fibrate induction of CPT1A, CPT2, and ACOX1 transcription is facilitated primarily by PPARα. This issue was addressed in a four-fold approach.
First, in the absence of bezafibrate the presence of high (20 mM) glucose in the culture medium had no effect on basal PPARα transcription of CPT1A, CPT2, and ACOX1 in either wild-type L-FABP (+/+) (Fig. 1A, B, C; alb), wild-type PPARα (+/+) (Fig. 3A, C, E; Alb), or PPARα (−/−) (Fig. 3B, D, F; Alb) hepatocytes.
Figure 3. PPARα inhibitor MK886 treatment of L-FABP (+/+) hepatocytes and PPARα gene ablation reduced or abolished the ability of bezafibrate to induce fatty acid oxidation gene expression in PPARα (−/−) mouse hepatocytes.

CPT1A (A,B), CPT2 (C,D), and ACOX1 (E,F) mRNA levels were measured in PPAR (+/+) hepatocytes without or with MK886 treatment and in PPARα (−/−) mouse hepatocytes treated with 6 or 20 mM glucose and fatty acid-free albumin (Alb, 40 μM) or Alb complexed with bezafibrate (BZ, 200 μM) or stearic acid (C18:0, 200 μM). Mean ± SE, n= 3–4. * = p<0.05 for lipidic ligand treatment vs. albumin at each glucose concentration; # = p<0.05 for 20 mM vs. 6 mM glucose concentration within each group.
Second, high (20 mM) glucose in the culture medium potentiated the ability of the PPARα-specific agonist fenofibrate to induce PPARα transcription of CPT1A, CPT2, and ACOX1 in wild-type L-FABP (+/+) hepatocytes (Fig. 1D, E, F; FF).
Third, the impact of ablating or inhibiting PPARα on the ability of high glucose to potentiate bezafibrate-mediated transcription of LCFA β-oxidative genes was examined in cultured primary hepatocytes from PPARα(+/+) and PPARα(−/−) mice. Again, to facilitate comparisons, the basal (albumin only) levels of CPT1A, CPT2, and ACOX1 mRNA were set as 1 in both wild-type (Fig. 3A, C, E) and PPARα null (Fig. 3B, D, F) hepatocytes. PPARα gene ablation abolished or diminished basal bezafibrate induction of PPARα transcription of CPT1A, CPT2, and ACOX1 at both 6 mM glucose (Fig. 3B, D, F vs 3A, C, E; BZ, black bars) and even more so at high (20 mM) glucose (Fig. 3B, D, F; BZ, white bars). Again, stearic acid (C18:0), a weak PPARα ligand, did not induce transcription of these enzymes in PPARα (−/−) hepatocytes regardless of glucose concentration (Fig. 3B, D, F; C18:0).
Fourth, treatment of wild-type PPARα (+/+) mouse hepatocytes with MK886, an inhibitor of PPARα [61], prevented bezafibrate-induced transcription of CPT1A, CPT2, and ACOX1 at physiological (6 mM), and even more so at high (20 mM) glucose (Fig. 3A, B, C; BZ+MK).
Taken together, these data indicate that the ability of high glucose to potentiate transcription of LCFA β-oxidative enzymes such as CPT1A, CPT2 and ACOX1 was primarily mediated through PPARα.
Impact of high glucose and bezafibrate on uptake of 3H-stearic acid in cultured primary hepatocytes from wild-type and L-FABP null mice
While overexpression of L-FABP, a PPARα-regulated cytosolic protein, enhances LCFA uptake in transformed cells, L-FABP antisense treatment or L-FABP gene ablation inhibits LCFA uptake in cultured hepatocytes and in vivo [12,31,32,52,53,79]. As shown in Fig. 2A, bezafibrate enhanced PPARα transcription of L-FABP in cultured primary mouse hepatocytes from wild-type L-FABP(+/+) mice–especially in the context of high glucose. Therefore, the effect of bezafibrate (BZ) and high glucose on uptake of exogenous 3H-stearic acid was determined in cultured primary hepatocytes preincubated 24 h with [6 or 20 mM glucose + BSA/BZ] followed by 24 h incubation with [6 or 20 mM glucose + 40 μM BSA/200 μM C18:0 containing tracer amount of 3H-stearic acid] as described in Methods.
At normal physiological (6 mM) glucose, bezafibrate significantly increased the uptake of 3H-stearic acid in an L-FABP dependent manner in cultured primary mouse hepatocytes. Bezafibrate increased the uptake of 3H-stearic acid 2-fold in cultured primary hepatocytes from wild-type L-FABP (+/+) mice (Fig. 4A; BZ, open vs black bars). L-FABP gene ablation abolished this increase in 3H-stearic acid uptake as shown in hepatocytes from L-FABP(−/−) mice (Fig. 4A; BZ, open vs black bars).
Figure 4. Total uptake, oxidation, and esterification of stearate by mouse hepatocytes.

Total uptake (A, B), oxidation (C, D), and esterification (E, F) of stearate by L-FABP (+/+) and L-FABP (−/−) hepatocytes after pretreatment with BSA (black bars) or bezafibrate/BSA (open bars) and 6 mM (A, C, and E) or 20 mM (B, D, and F) glucose as described in Methods. Values shown as mean nmol stearate per mg protein (x104) ± standard error (n = 4 to 6). * = P<0.05 BSA vs. Bezafibrate/BSA; $ = P<0.05 L-FABP (+/+) vs. L-FABP (−/−); @ = P<0.05 6 mM glucose vs. 20 mM glucose.
High glucose (20 mM) potentiated the ability of bezafibrate to enhance 3H-stearic acid uptake, again in an L-FABP dependent manner. In wild-type L-FABP(+/+) hepatocytes cultured with high glucose (20 mM), bezafibrate stimulated the uptake of 3H-stearic acid by 4.3-fold (Fig. 4B; BZ, open bar) as compared to culturing with normal physiological (6mM) glucose (Fig. 4A; BZ, black bar). L-FABP gene ablation markedly reduced these effects as shown in hepatocytes from L-FABP(−/−) mice.
Since stearic acid in the presence of high glucose (20mM) also trended to increase transcription of L-FABP slightly (not statistically significant), it was expected that in the presence of high glucose (20 mM) the 200 μM stearic acid/BSA in the incubation medium could perhaps increase 3H-stearic acid uptake. Indeed, high glucose (20 mM) together with 40 μM BSA/200 μM stearic acid complex also increased uptake somewhat (Fig. 4B; BSA, black bar) as compared to culturing at normal physiological (6 mM) glucose (Fig. 4A; BSA, black bar).
Thus, bezafibrate significantly enhanced the uptake of 3H-stearic acid which in turn was potentiated by high glucose. These effects were both highly dependent on L-FABP expression.
Impact of high glucose and bezafibrate on β-oxidation of 3H-stearic acid in cultured primary hepatocytes from wild-type mice
L-FABP overexpression in transformed cells enhances while L-FABP gene ablation inhibits LCFA β-oxidation in cultured primary hepatocytes and in vivo [12,31,52]. L-FABP directly transports bound LCFA-CoA to CPT1A (rate limiting enzyme in mitochondrial β-oxidation) [71] as well as enters nuclei to facilitate ligand-mediated PPARα transcription of CPT1A [25,27,28,31]. Therefore, the effect of bezafibrate (BZ) and high glucose on LCFA β-oxidation was determined in cultured primary hepatocytes preincubated 24 h with [6 or 20 mM glucose + BSA/BZ] followed by 24 h with [6 or 20 mM glucose + 40 μM BSA/200 μM C18:0 containing tracer amount of 3H-stearic acid] as detailed in Methods.
When cultured primary hepatocytes were incubated with glucose at normal physiological concentrations (6 mM), bezafibrate increased the β-oxidation of 3H-stearic acid by 53 ± 1 % in hepatocytes from wild-type L-FABP(+/+) but not L-FABP(−/−) mice (Fig. 4C; BZ). These data correlated with bezafibrate induction of LCFA β-oxidative enzymes in wild-type L-FABP(+/+) hepatocytes at normal physiological 6 mM glucose level (Fig. 1).
In L-FABP(+/+) but not L-FABP(−/−) hepatocytes cultured with both high (20 mM) glucose and bezafibrate, LCFA β-oxidation was induced to the highest extent. 3H-stearic acid β-oxidation was increased 3.1 ± 0.2 fold vs BSA at 6 mM glucose (Fig. 4D vs 4C) and to even higher level at 20 mM glucose (Fig. 4D). Again, these data correlated with bezafibrate induction of LCFA β-oxidative enzymes in wild-type L-FABP(+/+) hepatocytes at high (20 mM) glucose level (Fig. 1).
As expected, at high (20 mM) glucose together with 200 μM stearic acid/BSA complex also increased β-oxidation of stearic acid oxidation by 65 ± 2 % (Fig. 4D; BSA vs 4C; BSA). This effect was reduced by L-FABP gene ablation. These findings were consistent with 200 μM stearic acid/BSA complex directly being transported to mitochondria for β-oxidation by L-FABP at high glucose in wild-type L-FABP (+/+) hepatocytes.
Taken together, these data support the qPCR data indicating that high glucose potentiated LCFA β-oxidation in cultured primary hepatocytes. In contrast, these effects were not observed or markedly reduced with similarly treated cultured primary hepatocytes from L-FABP gene ablated mice. The latter finding was consistent with fibrates not inducing the levels of mRNAs (Fig. 1) encoding LCFA β-oxidative enzymes in L-FABP(−/−) null hepatocytes.
Effect of bezafibrate on esterification of 3H-stearic acid in primary hepatocytes cultured in the presence of normal or high glucose
L-FABP directly transports/targets LCFA-CoA to microsomal esterification enzymes: i) glycerol-3-phosphate acyltransferase (GPAT)—the rate limiting hepatic enzyme in incorporation of LCFA into glycerides [80–82], and ii) acyl CoA cholesterol acyltransferase (ACAT) [83,84]. Since bezafibrate, especially in the context of high glucose, increased PPARα regulated transcription of L-FABP (Fig. 2A), their impact on 3H-stearic acid esterification into glycerides and cholesteryl ester was determined in cultured primary hepatocytes preincubated 24 h with [6 or 20 mM glucose + BSA/BZ] followed by 24 h incubation with [6 or 20 mM glucose + 40 μM BSA/200 μM C18:0 containing tracer amount of 3H-stearic acid] as described in Methods.
In terms of 3H-stearic acid incorporation into total esterified lipids (cellular + secreted), primary hepatocytes cultured with normal physiological (6 mM), bezafibrate increased 3H-stearic acid esterification 2.4-fold in hepatocytes from wild-type L-FABP(+/+) but much less so in hepatocytes from L-FABP(−/−) mice (Fig. 4E; BZ). High glucose (20 mM) and bezafibrate potentiated 3H-stearic acid esterification by 5.2-fold in hepatocytes from wild-type L-FABP(+/+) but very little in hepatocytes from L-FABP(−/−) mice (Fig. 4F; BZ). As expected, high (20 mM) glucose together with 200 μM stearic acid/BSA complex also increased total esterification of 3H-stearic acid by 1.7-fold (Fig. 4F; BSA vs 6E; BSA). This effect was abolished by L-FABP gene ablation (Fig. 4E, F). Taken together, these data showed that at both physiological 6 mM glucose and at high glucose the basal (no bezafibrate) proportion of 3H-stearic acid appearing oxidized and esterified was similar. In contrast, although bezafibrate increased 3H-stearic acid, it increased 3H-stearic acid esterification even more. Thus, in the presence of bezafibrate the proportion of 3H-stearic acid appearing esterified was about 2-fold higher than that oxidized regardless of glucose level in the culture medium. These effects were highly dependent on L-FABP expression.
Figure 6. Esterification and secretion of stearate after uptake by mouse hepatocytes.

Incorporation and secretion of stearate in phospholipids (A, B), triacylglycerides (C, D), and cholesteryl ester (E, F) by L-FABP (+/+) and L-FABP (−/−) hepatocytes after pretreatment with BSA (black bars) or bezafibrate/BSA (open bars) and 6 mM (A, C, E) or 20 mM (B, D, F) glucose as described in Methods. Values shown as mean nmol stearate per mg protein ± standard error (n = 4 to 6). * = p<0.05 BSA vs. Bezafibrate/BSA; $ = p<0.05 L-FABP (+/+) vs. L-FABP (−/−); @ = p<0.05 6 mM glucose vs. 20 mM glucose.
To determine the relative contribution of cellular lipids to esterified 3H-stearic acid in response to high glucose and bezafibrate, lipids were extracted separately from the primary hepatocytes cultured with normal physiological (6 mM) or high (20 mM) glucose in the presence of bezafibrate as above. In the lipids of L-FABP(+/+) primary hepatocyte cultured with normal physiological (6 mM) glucose, 3H-stearic acid was esterified primarily into triglycerides (Fig. 5C; TG) > phospholipids (Fig. 5A; PL) > cholesteryl-esters (Fig. 5E; CE). At physiological glucose (6 mM), bezafibrate stimulated esterification of 3H-stearic acid modestly about 2-fold into each lipid class. High (20 mM) glucose potentiated bezafibrate stimulated esterification of 3H-stearic acid into each lipid class about 5–6 fold into each lipid class. As expected, high (20 mM) glucose together with 200 μM stearic acid/BSA complex also increased esterification of 3H-stearic acid into these lipid classes (Fig. 5B, D, F; BSA vs 7A, C, E; BSA), but much less so than with bezafibrate or bezafibrate/high glucose. This effect was also markedly diminished by L-FABP gene ablation.
Figure 5. Esterification and retention of stearate after uptake by mouse hepatocytes.
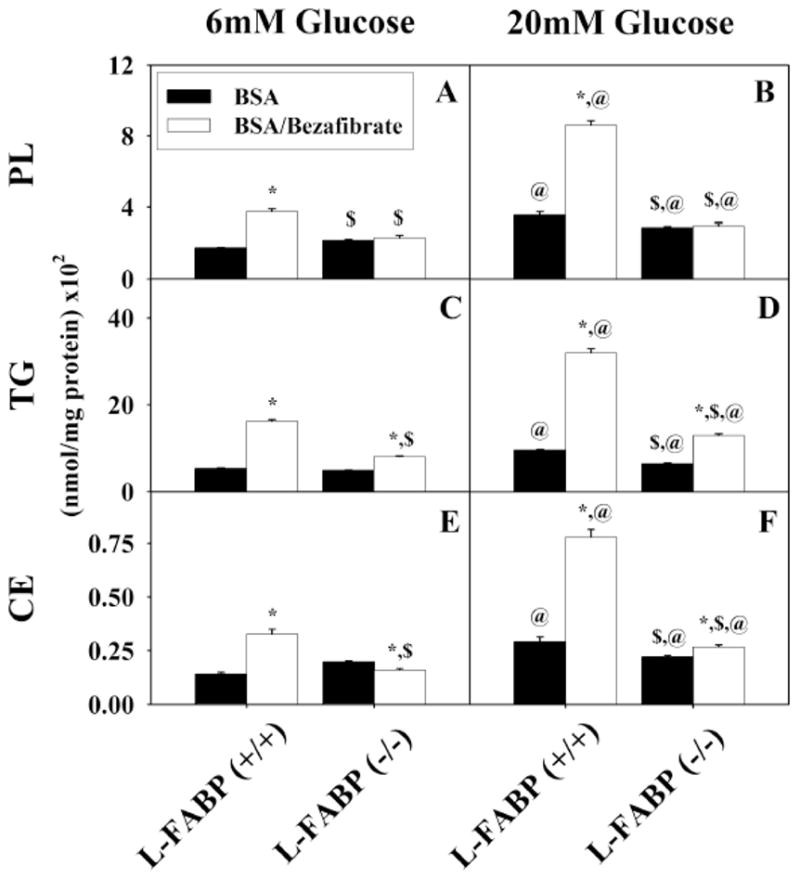
Incorporation of stearate into cellular phospholipids (A, B), triacylglycerides (C, D), and cholesteryl ester (E, F) by L-FABP (+/+) and L-FABP (−/−) hepatocytes after pretreatment with BSA (black bars) or bezafibrate/BSA (open bars) and 6mM (panels A, C, and E) or 20 mM (panels B, D, and F) glucose as described in Methods. Values shown as mean nmol stearate per mg protein (x102) ± standard error (n = 4–6). * = p<0.05 BSA vs. Bezafibrate/BSA; $ = p<0.05 L-FABP (+/+) vs. L-FABP (−/−); @ = p<0.05 6 mM glucose vs. 20 mM glucose.
Figure 7. Quantitative analysis of shows that bezafibrate significantly increases nuclear distribution of L-FABP in cultured mouse hepatocytes.

Panels A and B: WT mouse hepatocytes were cultured overnight, washed, and further incubated for 0.5–24 h with serum-free, glucose-free Williams’ medium E plus 6 mM (A) or 20 mM (B) glucose, insulin, dexamethasone, and LCFA-free BSA (Alb) or BSA/bezafibrate (200 μM) as described in Methods. Cells were fixed, labeled with FITC-anti L-FABP and TO-PRO DNA dye, and multiple cells analyzed by confocal microscopy to determine nuclear and cytoplasmic L-FABP labeling intensity in order to calculate the nuclear/cytoplasm L-FABP ratio (Nucl/Cyto FI L-FABP) described in Methods [29,30,117]. Means ± SEM, n = 40. *p < 0.05 vs albumin; # p < 0.05 as compared at 0.5 h within each group. C: WT hepatocytes were cultured as above and incubated with 6mM glucose and either LCFA-free BSA or BSA/bezafibrate (200 μM) for 24 hrs, fixed, anti-L-FABP immunogold labeled, and antibody-L-FABP labeling particle density in the whole cell, nucleoplasm and cytoplasm determined as we described [29]. Statistics was performed using a one-way ANOVA and a Newman-Keuls post-test. Mean ± SEM, n = 20 @ p<0.05 vs Alb whole cell; # p<0.05 vs Alb nucleus; $ p<0.05 vs Alb cyto; ^ p<0.05 vs BZ whole cell; & p<0.05 vs BZ nucleus.
A small amount of 3H-stearic acid esterified by hepatocytes appeared secreted into the culture medium, but in very different proportion than 3H-stearic acid appearing in the cellular esterified lipids. In the lipids secreted by L-FABP(+/+) primary hepatocyte cultured with normal physiological (6 mM) glucose, 3H-stearic acid appeared primarily into cholesteryl-esters (Fig. 6E; CE) ≫ triglycerides (Fig. 6C; TG) > phospholipids (Fig. 6A; PL ), essentially opposite to the order in the cellular lipids. Bezafibrate did not increase esterification of 3H-stearic acid into any of these secreted lipid classes at physiological (6 mM) glucose (Fig. 6A, C, E; BZ) nor did high (20 mM) glucose potentiate bezafibrate action (Fig. 6B, D, F; BZ). However, high (20 mM) glucose together with 200 μM stearic acid/BSA complex increased esterification of 3H-stearic acid into secreted PL and TG, but not CE (Fig. 6B, D, F; BSA vs 7A, C, E; BSA). L-FABP gene ablation reduced this effect.
Since neither GPAT nor ACAT are regulated by PPARα [85,86], the bezafibrate-mediated increase in LCFA esterification (especially in the context of high glucose) was associated at least in part with: i) bezafibrate-mediated PPARα transcription of L-FABP (Fig. 2A)—which in turn enhanced delivery of LCFA/LCFA-CoA to the endoplasmic reticulum for esterification; ii) high glucose itself inducing lipogenic enzymes including GPAT and ACAT [85,86].
Lipidic ligands (bezafibrate, stearic acid), but not glucose, increased the nuclear distribution of L-FABP in cultured primary hepatocytes: immunofluorescence confocal imaging and immunogold electron microscopy (EM)
Although L-FABP enhances the distribution of bound lipidic ligands to the nucleus, it is not known if the converse is true (rev. in [28,31,87]. Therefore, the impact of bezafibrate and stearic on L-FABP distribution to the nucleus was examined by immunolabeling confocal microscopy and immunogold EM of cultured primary mouse hepatocytes cultured with physiologically normal (6 mM) and high (20 mM) glucose medium with BSA, bezafibrate/BSA, or stearic acid/BSA.
Confocal microscopy revealed that L-FABP was highly prevalent not only in cytosol, but also in nuclei, of mouse primary hepatocytes. Representative confocal fluorescence images of L-FABP in hepatocytes (Suppl. Fig. 2A–D; 1st columns), nuclei (Suppl. Fig. 2A–D; 2nd columns), and colocalized pixels of both L-FABP and nuclei (Suppl. Fig. 2A–D; 3rd columns) are shown for hepatocytes incubated with albumin without exogenous lipidic ligands for 1hr (Suppl. Fig. 2A, C) and 24 hr (Suppl. Fig. 2B, D). Qualitative analysis of confocal images showed that hepatocytes cultured with 6 mM glucose and albumin (as negative control) in the absence of BZ or C18:0 for 1 h (Suppl. Fig. 2A, B; upper row of pictures) and 24 h (Suppl. Fig. 2C–D; upper row of pictures) had the lowest degree of L-FABP colocalization with nuclear stain, primarily in the perinuclear region. Quantitative analysis of multiple confocal images of hepatocytes showed that the nuclear/cytoplasmic ratio of L-FABP distribution (i.e. L-FABP localizing with the nuclear dye/L-FABP not localizing with the nuclear dye) was near 0.72 when cells were treated with 6 mM glucose and lipid-free albumin as negative control (Fig. 7A; Alb).
Due to the much lower limit of resolution of confocal microscopy near 2000 nm potentially resulting in overestimation of the nuclear/cytoplasmic ratio of L-FABP, the distribution of L-FABP in nuclei was also examined by immunogold EM, a technique with higher limit of resolution < 0.05 nm. The anti-L-FABP antibody used was relatively specific for L-FABP since labeling was observed only in WT hepatocytes, but very little in hepatocytes from L-FABP null mice [29]. Immunogold EM detected L-FABP in both the cytoplasm and nucleus—again at slightly lower level (Fig. 7C; Alb). The L-FABP nuclear/cytoplasmic ratio calculated from the immunogold EM data was near 0.8 (Fig. 7C; Alb)—close to that obtained from confocal immunofluorescence imaging near 0.72 (Fig. 7A; Alb). Thus, the nuclear/cytoplasmic distribution of L-FABP determined by confocal imaging of fluorescent-labeled L-FABP colocalizing with the nuclear dye/L-FABP was basically confirmed by higher resolution immunogold EM—consistent with an earlier study from our lab [29].
Lipidic ligands (bezafibrate, stearic acid) elicited a higher level of colocalization of L-FABP with nuclear stain in hepatocytes cultured with normal physiological (6mM glucose) for 1 h, (Suppl. Fig. 2A; middle row). Interestingly, this effect of BZ on L-FABP nuclear translocation appeared qualitatively reversed by 24 h (Suppl. Fig. 2B; middle row). Quantitative analysis of multiple hepatocytes showed that bezafibrate and stearic acid rapidly (within 0.5 h) increased the nuclear/cytoplasmic ratio of L-FABP distribution (i.e. L-FABP localizing with the nuclear dye/L-FABP not localizing with the nuclear dye) from near 0.72 to 0.83 and 0.87, respectively (Fig. 7A; Alb). However, by 24 h this ratio returned to baseline value of 0.71 in bezafibrate treated hepatocytes while that of stearic acid treated cells was even further reduced to 0.62 (Fig. 7A). Immunogold EM established that bezafibrate increased the amount of immunodetectable cellular L-FABP (Fig. 7C)—consistent with L-FABP being induced by PPARα ligands such as bezafibrate [4,9,88]. Further, immunogold EM showed that bezafibrate preferentially increased the level of immunodetectable L-FABP in hepatocyte nuclei more so than in cytosol (Fig. 7C). Thus, both immunofluorescence confocal microscopy and immunogold EM showed that bezafibrate increased the L-FABP nuclear distribution.
High (20 mM) glucose itself (i.e. in absence of lipidic ligand) much more weakly and transiently increased L-FABP nuclear distribution. When mouse hepatocytes were treated with high (20 mM) glucose and albumin with no lipidic ligands present in the medium for 1 h (Suppl. Fig. 2C; upper row) and 24 h (Suppl. Fig. 2D; upper row), L-FABP was again more cytoplasmic with lower colocalization of L-FABP with nuclear stain. Quantitative analysis of multiple hepatocytes incubated without lipidic ligand at 20 mM glucose (Fig. 7B) showed that the nuclear/cytoplasmic ratio of L-FABP distribution (i.e. L-FABP localizing with the nuclear dye/L-FABP not localizing with the nuclear dye) was slightly increased versus incubation at 6 mM glucose (Fig. 7A). Again, this increase was also transient with increasing incubation time.
High glucose did not further exacerbate, but extended for longer time the lipidic ligand (bezafibrate, stearic acid) induced redistribution of L-FABP into hepatocyte nuclei. Upon addition of bezafibrate/albumin to 20 mM glucose in the cell culture medium, L-FABP was largely detected inside nuclei at 1h (Suppl. Fig. 2C; middle row) and even after 24 h incubation (Suppl. Fig. 2D; middle row). Quantitative analysis of multiple hepatocytes incubated with 20 mM glucose showed that the nuclear/cytoplasmic ratio of L-FABP distribution (i.e. L-FABP localizing with the nuclear dye/L-FABP not localizing with the nuclear dye) increased from near 0.67 in the absence of lipidic ligand (Fig. 7B; Alb) to maximum near 0.86 and 0.82 for longer time (e.g. 24h) in the presence of bezafibrate (Fig. 7B; BZ) and prevented inhibition by stearic acid at longer time of 24h (Fig. 7B; C18:0), respectively.
Taken together, these findings indicated that at normal physiological (6 mM) glucose the lipidic ligands bezafibrate and stearic acid induced rapid L-FABP nuclear translocation for a relatively short amount of time, up to 4 h, after which a homeostasis process brings the L-FABP from nuclei back into cytoplasm to return the nuclear/cytoplasmic L-FABP ratio back to near. In contrast, for cells incubated with high (20 mM) glucose and lipidic ligand (bezafibrate, stearic acid), the nuclear/cytoplasmic L-FABP ratio rapidly increased to a higher maximum of 0.83 at 1 h, and stayed above its negative control (20 mM glucose and lipid-free albumin) level near 0.7 for all time points up to 24 h. These data suggest that high glucose may affect the interaction of L-FABP with nuclear import and export proteins—analogous to high glucose impacting L-FABP/PPARα binding [29,30]. Since a slower rate of increase in the nuclear/cytoplasmic L-FABP ratio was measured for 20 mM glucose as compared to 6 mM, it is more plausible to hypothesize that higher glucose concentration impaired the export of L-FABP from nuclei into the cytoplasm. However, a further investigation of this molecular mechanism was beyond the scope of the current study.
Cytosolic glucose levels increased with increasing extracellular glucose
Although glucose levels in liver and liver derived hepatoma cells are about 80% of extracellular, near 4 mM, and responsive to extracellular glucose [48–51], it is not known if these properties are maintained in cultured primary hepatocytes. To address this issue, the uptake of 3H-glucose uptake and intracellular glucose levels were determined in cultured primary mouse hepatocytes as described in Methods.
Uptake of 3H-glucose by mouse hepatocytes was rapid, half-maximal within minutes, reaching an intracellular level of 2.4 mM and 10.5 mM when extracellular glucose was 6 and 20 mM, respectively (Fig. 8A). This pattern was very similar to that determined by non-invasive imaging of hepatic cells with glucose nanosensors [51]. Results obtained with 3H-glucose were confirmed by chemical analysis of intracellular glucose mass. Incubating mouse primary hepatocytes with high 20 mM glucose increased intracellular glucose from 3.0 mM to 12 mM (Fig. 8B). Thus hepatocyte cytosol was 40–50% that of extracellular glucose and highly responsive to extracellular glucose. Interestingly, glucose nanosensors also showed that nucleoplasmic and cytoplasmic glucose levels were very similar [30,89,90].
Figure 8. Intracellular glucose level in cultured mouse primary hepatocytes is proportional to extracellular concentration.

(A), Hepatocytes from wild type L-FABP (+/+) mice were treated with fatty acid-free Alb (40 μM), 6 (open symbols) and 20 mM (solid symbols) glucose, and a tracer amount of [1-3H]-glucose at the same specific activity for up to 6 hr. A: at indicated time points, medium and cells were collected to determine cytosolic glucose concentration as described in the Methods. B: L-FABP (+/+) hepatocytes were treated with 6, 20, and 30 mM glucose for 1 hr, and intracellular glucose concentrations were determined as described in Methods.
De novo LCFA synthesis from high glucose did not significantly contribute to PPARα activation
Our finding that high glucose without exogenous lipidic ligand did not induce PPARα transcription of fatty acid β-oxidative enzymes (Fig. 1A–C; albumin only) suggested that de novo synthesis of LCFAs from high glucose did not likely account for glucose potentiation of bezafibrate-induction of transcription. To further establish this possibility hepatocytes were cultured with 6 or 20 mM glucose containing trace amounts of 3H-glucose. Incorporation of 3H-glucose-derived 3H into hepatocyte lipids was then determined with increasing incubation time as described in Methods.
Incubating hepatocytes with 6 mM 3H-glucose resulted in very little 3H-glucose-derived radioactivity appearing in lipids and that within lipids was primarily incorporated into triglycerides (Fig. 9A; inverted open triangles) rather than in LCFAs (Fig. 9A; open diamonds). Less than 3% of LCFA and TG mass was derived from 3H-glucose after 6 h (Fig. 9A–B). While 6 h incubation with high glucose (20 mM) increased 3H-glucose incorporation into LCFAs 2-fold (Fig. 9A; solid squares vs open diamonds), incorporation into triglycerides was not increased (Fig. 9A; solid circles vs inverted open triangles). Most importantly, incubation for 6 h with high (20 mM) glucose did not increase the total mass of LCFAs or triglycerides (Fig. 9B). Almost no 3H-glucose-derived lipid was detected in the medium. These data indicated short-term incubation (6 h) did not increase de novo LCFA synthesis from glucose.
Figure 9. Effect of high glucose on de novo synthesis (A) and mass (B) of LCFAs and triglycerides (TG) in cultured mouse primary hepatocytes.
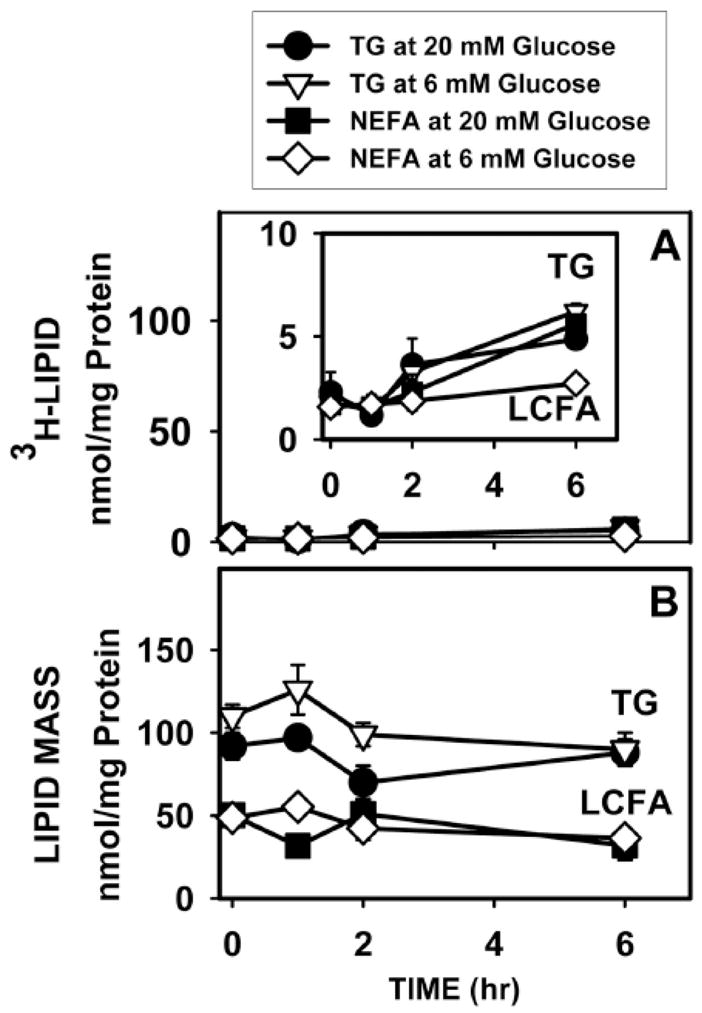
Wild-type L-FABP (+/+) hepatocytes were incubated with fatty acid-free Alb (40 μM) and 6 mM (open symbols) or 20 mM (solid symbols) glucose to which tracer amounts of [1-3H]-glucose were added at the same specific activity. At indicated times, medium and cells were collected for quantitation as described in Methods: A, 3H incorporation into TG (open triangles, closed circles) and LCFAs (open squares, closed diamonds) [52]; B, mass of TG (open triangles, closed circles) and LCFAs (open squares, closed diamonds). Values represent the mean ± SE (n = 3).
DISCUSSION
Since nearly 30% of diabetics are poorly compliant with glucose lowering therapy, serum glucose levels remain elevated in these individuals [91–93]. Paradoxically, the fibrates [2,3,17–19] and other lipidic [94–99] PPARα agonists may be more effective in lowering serum triglycerides in diabetics with the most severe hypertriglyceridemia [91–93]. However, the role of high glucose in impacting the effects of fibrate PPARα agonists under normal physiologic conditions, much less diabetic states, is not clear. Studies with cultured primary hepatocytes from normal mice have also not addressed the role of high glucose alone since the culture media used contained high (11–28 mM) glucose without comparisons to normal physiological glucose [28,58,66]. Thus, little is known regarding the molecular mechanism(s) whereby high glucose may contribute the effectiveness of fibrates even under normal physiological conditions. The results presented herein with bezafibrate and fenofibrate treated primary mouse hepatocytes cultured with normal physiological (6 mM) or high (11–20 mM) glucose tested the hypothesis that: i) fibrate PPARα agonists are more effective in inducing transcription of LCFA β-oxidative enzymes in the context of high glucose than normal physiological glucose, ii) this effect requires an intact L-FABP/PPARα signaling pathway. The following new insights were obtained:
First, fibrates such as bezafibrate (pan-PPAR agonist) and fenofibrate (PPARα-specific agonist) only weakly/modestly stimulated PPARα transcription of mitochondrial (CPT1A, CPT2) and peroxisomal (ACOX1) LCFA β-oxidative enzymes when primary mouse hepatocytes were cultured with normal physiological (6 mM) glucose. This stimulation required an intact L-FABP/PPARα signaling pathway—consistent with in vivo studies of control-chow fed L-FABP(−/−) mice [15,31,52,53,100,101] and PPARα (−/−) mice [102].
Second, the ability of bezafibrate to induce transcription of PPARα-regulated LCFA β-oxidative enzymes (CPT1A, CPT2, ACOX1) and other PPARα-regulated proteins (L-FABP, ACBP, PPARα) was dramatically enhanced in the context of high glucose. High glucose potentiation of fibrate-mediated PPARα transcription of these enzymes and proteins was mediated primarily through L-FABP and PPARα as it was not observed with L-FABP(−/−) or PPARα(−/−) hepatocytes and not with L-FABP(+/+) hepatocytes treated with the PPARα inhibitor MK886. Although other nuclear receptors such as HNF4α (regulates transcription of PPARα itself) and HNF1α (induces PPARα regulated genes through a different response element) also influence expression of PPARα regulated genes [73–76], bezafibrate inhibited transcription of HNF1α and HNF4α which were only partially or not restored, respectively, by high glucose. These findings may be explained by the impact of high glucose on the L-FABP/PPARα signaling pathway where L-FABP binds fibrates [43–45,103–106], directly interacts with PPARα to transfer bound ligand {Hostetler, 2009 5158 /id;Hostetler, 2010 6409 /id;Velkov, 2013 6944 /id}, and high glucose enhances this L-FABP/PPARα interaction [30]. The data presented herein show for the first time that L-FABP is important for fibrate signaling to and activation of PPARα in primary hepatocytes—especially in the context of high glucose.
Third, high glucose potentiated LCFA β-oxidation. By enhancing bezafibrate mediated PPARα transcription of L-FABP as well as LCFA β-oxidative enzymes, LCFA β-oxidation was increased. These findings were consistent with the literature showing that mRNA and protein levels of PPARα regulated genes (CPT1A, CPT2, ACOX1, L-FABP, PPARα) directly correlate with LCFA uptake, targeting to mitochondria for oxidation, and targeting to the nucleus for increasing transcription of LCFA β-oxidative enzymes [31,69,70,107]. Further, high glucose alone (without bezafibrate) did not induce PPARα transcriptional activity—indicating that potentiation of bezafibrate action by high glucose was not indirectly due to high glucose activating other signaling pathways.
Fourth, high glucose also potentiated esterification of exogenous LCFA. This was despite the fact that neither glycerol-3-phosphate acyltransferase (GPAT) nor acyl CoA cholesteryl acyltransferase (ACAT) are directly regulated by PPARα [85,86]. However, the activities of these enzymes may be stimulated indirectly through increased expression of L-FABP in response to bezafibrate induced PPARα transcription of L-FABP, especially at high glucose. Consistent with this possibility, in vitro studies show that L-FABP directly stimulates LCFA-CoA utilization by glycerol-3-phosphate acyltransferase (GPAT, rate limiting in hepatic glyceride formation) [80–82]. While fibrates are thought to lower serum triglycerides at least in part by inducing hepatic PPARα transcription of LCFA β-oxidative enzymes, effects on liver triglyceride level are dose dependent since fibrates also induce transcription of enzymes in de novo LCFA synthesis, desaturation, elongation, and triglyceride synthesis [20–23,108]. Partitioning of LCFA- or glucose-derived acetyl-CoAs toward oxidative vs synthetic paths determines net effect on hepatic triglyceride level and treatment outcome [20,109].
Fifth, bezafibrate induced L-FABP redistribution to the nucleus. Our and other labs have shown that L-FABP binds fibrates [43,44], binds PPARα in vitro and in vivo [29,30], and stimulates bezafibrate-mediated PPARα transcription in HepG2 cells directly proportional to L-FABP protein expression level [26]. Our confocal anti-L-FABP immunofluorescence colocalization with DNA dye and immunogold EM imaging showed that in cultured primary mouse hepatocytes the ratio of L-FABP protein in the nucleus/cytoplasm was near 0.8—confirming our earlier immunogold electron microscopic findings of L-FABP distribution in mouse liver hepatocytes [29]. As shown herein, fibrate induced L-FABP translocation to nuclei and maintained increased L-FABP distribution to the nuclei for hours—especially in the context of high glucose. Since the concentration of L-FABP in hepatocytes is quite high, i.e. in the 200–400 μM range [110] and at high glucose bezafibrate maximally shifted the nuclear/cytoplasmic ratio by as much as 25%, significant amounts of L-FABP protein were redistributed to nuclei. The bezafibrate-induced redistribution of L-FABP to nuclei did not require high affinity ligand binding by PPARα since both high (bezafibrate) and very weak [stearic acid (C18:0)] affinity ligands of PPARα equally well induced L-FABP redistribution to cultured primary hepatocyte nuclei. Since stearic acid is very weakly bound by PPARα, stearic acid did not significantly increase the transcriptional activity of PPARα. These findings suggest that fibrates may ‘piggy-back’ on the recently-discovered L-FABP-mediated LCFA signaling pathway for uptake, cytosolic transport, and nuclear targeting to PPARα (rev. in {Schroeder, 2008 5775 /id;Wolfrum, 2001 4357 /id;Velkov, 2013 6944 /id}).
Sixth, high glucose in the culture medium increased intracellular glucose level in cultured primary hepatocytes. Cytosolic glucose concentration is determined by type and number of glucose transporters and hexokinases, insulin responsiveness, and metabolic activity [48,50,90,111,112]. In most peripheral cells, cytosolic glucose is >100-fold lower than extracellular [63,89,111,113]. Due to higher Kms of GLUT2 and glucokinase as well as different insulin sensitivity and metabolic activity, liver cytosolic glucose is in the mM range (~4 mM) (p. 59, ref. [48–50]) as confirmed herein with cultured primary hepatocytes. Further, non-invasive glucose nanosensors showed that nucleoplasmic and cytoplasmic glucose levels were similar and responsive to extracellular glucose in living cells [90,112]. Finally, L-FABP directly interacts with PPARα and L-FABP/PPARα binding affinity is enhanced at high glucose [29,30]. By enhancing L-FABP binding to PPARα, high glucose may further facilitate L-FABP-mediated signaling of bezafibrate to PPARα. These key differences between liver and most peripheral tissues suggest that fibrate-mediated PPARα signaling in hepatocytes may be particularly sensitive to hyperglycemia—consistent with the data presented herein. While we recognize that there are other possible events whereby high glucose could potentiate L-FABP-mediated fibrate signaling to PPARα (e.g. by post-translational modifications such as phosphorylation, ubiquitinylation, and sumoylation), there is no current evidence for L-FABP posttranslational modification by these processes [81,114]. Although increased serum insulin is known to increase PPARα phosphorylation and transcriptional activity [115], the level of insulin in the hepatocyte culture medium was not increased in the current studies. While PPARα can be targeted for degradation by ubiquitinylation, a high glucose diet in mice increased rather than decreased PPARα mRNA and protein (not shown). Likewise, although sumoylation results in transcriptional repression of PPARα [116], our studies indicate that high glucose increased rather than decreased PPARα transcriptional activity. Our finding that high glucose alone without exogenous lipidic (fibrate, LCFA) ligand did not induce PPARα transcription further suggested that potential post-translational modifications of PPARα induced by high glucose alone were insufficient to affect PPARα transcriptional activity. Thus any potential contribution of PPARα post-translational modification (especially phosphorylation) remains to be elucidated.
In summary, the present work examined the effect of glucose, L-FABP, and PPARα on the expression of PPARα target genes in cultured primary mouse hepatocytes in response to activation by bezafibrate. While bezafibrate only modestly induced PPARα transcription of CPT1A, CPT2 and ACOX1 at physiological normal (6 mM) glucose, high glucose markedly potentiated bezafibrate-mediated PPARα transcription of these enzymes and other PPARα regulated proteins (L-FABP, ACBP, and PPARα itself). These effects, especially potentiation by high glucose, required expression of both L-FABP and PPARα (or functional PPARα). Whether these findings represent a normal physiological mechanism which may be less functional in diabetic livers remains to be addressed.
Supplementary Material
Highlights.
Bezafibrate weakly activates PPARα transcription at 6mM glucose.
Bezafibrate markedly activates PPARα transcription at 20mM glucose.
Bezafibrate action (20mM glucose) required intact L-FABP/PPARα signaling pathway.
High glucose without bezafibrate was ineffective in the primary mouse hepatocytes.
Acknowledgments
The helpful assistance of Ross Payne and the facilities of the Microscopy and Imaging Center at Texas A&M University were used for steps in electron microscopy. This work was supported in part by the USPHS National Institutes of Health DK41402 (FS, ABK).
Abbreviations
- ACBP
acyl CoA binding protein
- ACOX1
acyl-CoA oxidase 1, palmitoyl
- ACAT
acyl CoA cholesterol acyltransferase
- BZ
bezafibrate
- CPT1A
carnitine palmitoyl transferase IA, liver
- CPT2
carnitine palmitoyl-CoA transferase II
- C16:0
palmitic acid
- C16:0-CoA
palmitoyl CoA
- GPAT
glycerol-3-phosphate acyltransferase
- HNF1α
hepatocyte nuclear factor 1α
- HNF4α
hepatocyte nuclear factor 4α
- L-FABP
liver fatty acid binding protein or FABP1
- LCFA
long chain fatty acids, unesterified
- LCFA-CoA
long chain fatty acid-CoA thioester
- L-FABP(−/−)
L-FABP knock out mouse genotype
- PPARα,β/δ,γ
peroxisome proliferator-activated receptors alpha, beta/delta, and gamma
- PPARα(−/−)
PPARα knock out mouse genotype
- WT
wild type mouse genotype
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ting RZW, Yang X, Yu LWL, Luk AOY, Kong APS, Tong PCY, So W-Y, Chan JCN, Ma RCW. Lipid control and use of lipid-regulating drugs for prevention of cardiovascular events in Chinese type 2 diabetic patients: a prospective cohort study. Cardiovasc Diabetol. 2010;9 doi: 10.1186/1475-2840-9-77. 10.1186/1475-2840-9-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Saha SA, Arora RR. Fibrates in the prevention of cardiovascular disease in pateints with type 2 diabetes mellitus--A pooled meta-analysis of randomized placebo-controlled clinical trials. Int J Cardiol. 2010;141:157–166. doi: 10.1016/j.ijcard.2008.11.211. [DOI] [PubMed] [Google Scholar]
- 3.Staels B, Maes M, Zambon A. Fibrates and future PPARα agonists in the treatment of cardiovascular disease. Nature Clin Pract Cardiovasc Med. 2008;5:542–553. doi: 10.1038/ncpcardio1278. [DOI] [PubMed] [Google Scholar]
- 4.Desvergne B, Michalik L, Wahli W. Be fit or be sick: peroxisome proliferator-activated receptors are down the road. Mol Endocrinology. 2004;18:1321–1332. doi: 10.1210/me.2004-0088. [DOI] [PubMed] [Google Scholar]
- 5.Sanderson LM, Degenhart T, Desvergne B, Muller M, Kersten S. The roles of PPARα and PPARβ in liver: dietary vs endogenous fat sensor. Chem Phys Lip. 2008;154:S17. [Google Scholar]
- 6.Sanderson LM, Boekschoten MV, Desvergne B, Muller M, Kersten S. Transcriptional profiling reeals divergent roles of PPARα and PPARβ/δ in regulatoin of gene expression in mouse liver. Physiol Genomics. 2010;41:42–52. doi: 10.1152/physiolgenomics.00127.2009. [DOI] [PubMed] [Google Scholar]
- 7.Kersten S, Desvergne B, Wahli W. Roles of PPARs in health and disease. Nature. 2000;405:421–424. doi: 10.1038/35013000. [DOI] [PubMed] [Google Scholar]
- 8.Escher P, Braissant O, Basu-Modak S, Michalik L, Wahli W, Desvergne B. Rat PPARs: quantitative analysis in adult rat tissues and regulation in fasting and refeeding. Endocrinology. 2001;142:4195–4202. doi: 10.1210/endo.142.10.8458. [DOI] [PubMed] [Google Scholar]
- 9.Rakhshandehroo M, Hooiveld G, Muller M, Kersten S. Comparative analysis of gene regulation by the transcription factor PPARα between mouse and human. PLoS ONE. 2009;4:e6796. doi: 10.1371/journal.pone.0006796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jump DB, Clarke SD. Regulation of gene expression by dietary fat. Annu Rev Nutr. 1999;19:63–90. doi: 10.1146/annurev.nutr.19.1.63. [DOI] [PubMed] [Google Scholar]
- 11.Wolfrum C, Spener F. Fatty acids as regulators in lipid metabolism. Eur J Lip Sci Technol. 2000;102:746–762. [Google Scholar]
- 12.Atshaves BP, Storey SM, Petrescu AD, Greenberg CC, Lyuksyutova OI, Smith R, Schroeder F. Expression of fatty acid binding proteins inhibits lipid accumulation and alters toxicity in L-cell fibroblasts. Am J Physiol. 2002;283:C688–C703. doi: 10.1152/ajpcell.00586.2001. [DOI] [PubMed] [Google Scholar]
- 13.Atshaves BP, Storey S, Huang H, Schroeder F. Liver fatty acid binding protein expression enhances branched-chain fatty acid metabolism. Mol Cell Biochem. 2004;259:115–129. doi: 10.1023/b:mcbi.0000021357.97765.f2. [DOI] [PubMed] [Google Scholar]
- 14.Atshaves BP, Payne HR, McIntosh AL, Tichy SE, Russell D, Kier AB, Schroeder F. Sexually dimorphic metabolism of branched chain lipids in C57BL/6J mice. J Lipid Res. 2004;45:812–830. doi: 10.1194/jlr.M300408-JLR200. [DOI] [PubMed] [Google Scholar]
- 15.Atshaves BP, McIntosh AL, Payne HR, Mackie J, Kier AB, Schroeder F. Effect of branched-chain fatty acid on lipid dynamics in mice lacking liver fatty acid binding protein gene. Am J Physiol. 2005;288:C543–C558. doi: 10.1152/ajpcell.00359.2004. [DOI] [PubMed] [Google Scholar]
- 16.Zomer AWM, van der Burg B, Jansen GA, Wanders RJA, Poll-The BT, van der Saag PT. Pristanic acid and phytanic acid: naturally occurring ligands for the nuclear receptor peroxisome proliferator activated receptor -α. J Lipid Res. 2000;41:1801–1807. [PubMed] [Google Scholar]
- 17.Robins SJ. Fibrates and coronary heart disease reduction. Curr Op in Endocrin & Diabetes. 2002;9:312–322. [Google Scholar]
- 18.Frederiksen KS, Wulf EM, Wassermann K, Sauerberg P, Fleckner J. Identification of hepatic transcriptional changes in insulin-resistant rats treated with peroxisome proliferator activated receptor-α agonists. J Mol Endocrinol. 2003;30:317–329. doi: 10.1677/jme.0.0300317. [DOI] [PubMed] [Google Scholar]
- 19.Otvos JD, Collins D, Freedman DS, Shaluarova I, Schaefer EJ, McNamara JR, Bloomfield HE, Robins SJ. Low-density lipoprotein and high-density lipoproteion particle subclasses predict coronary events and are favorably changed by gemfibrozil therapy in the Veterans Affairs high density lipoprotein intervention trial. Circulation. 2006;113:1556–1563. doi: 10.1161/CIRCULATIONAHA.105.565135. [DOI] [PubMed] [Google Scholar]
- 20.Oosterveer MH, Grefhourst A, van Dijk TH, Havinga R, Staels B, Kuipers F, Groen AK, Reijngoud D-J. Fenofibrate simultaneously induces hepatic fatty acid oxidation, synthesis, and elongation in mice. J Biol Chem. 2009;284:34036–34044. doi: 10.1074/jbc.M109.051052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sekiya M, Yhagi N, Matsuzaka T, Najima Y, Nakakuki M, Nagai R, Ishibashi S, Osuga J-I, Yamada N, Shimano H. Polyunsaturated fatty acids ameliorate hepatic steatosis in obese mice by SREBP-1 suppression. Hepatology. 2003;38:1529–1539. doi: 10.1016/j.hep.2003.09.028. [DOI] [PubMed] [Google Scholar]
- 22.Froyland L, Madsen L, Vaagenes H, Totland GK, Auwerx J, Kryvi H, Staels B, Berge RK. Mitochondrion is the principal target for nutritional and pharmacological control of triglyceride metabolism. J Lipid Res. 1997;38:1851–1858. [PubMed] [Google Scholar]
- 23.Bijland S, Pieterman EJ, Maas ACE, van der Hoorn JWA, van Erk MJ, van Klinken JB, Havekes LM, van Dijk KW, Princen HM, Rensen PCN. Fenofibrate increases VLDL triglyceride production despite reducing plasma triglyceride levels in APOE3-Leiden.CETP mice. J Biol Chem. 2010;285:25168–25175. doi: 10.1074/jbc.M110.123992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tailleux A, Wouters K, Staels B. Role of PPARs in NAFLD: potential therapeutic targets. Biochim Biophys Acta. 2012;1821:809–818. doi: 10.1016/j.bbalip.2011.10.016. [DOI] [PubMed] [Google Scholar]
- 25.Schroeder F, Petrescu AD, Huang H, Atshaves BP, McIntosh AL, Martin GG, Hostetler HA, Vespa A, Landrock K, Landrock D, Payne HR, Kier AB. Role of fatty acid binding proteins and long chain fatty acids in modulating nuclear receptors and gene transcription. Lipids. 2008;43:1–17. doi: 10.1007/s11745-007-3111-z. [DOI] [PubMed] [Google Scholar]
- 26.Wolfrum C, Borrmann CM, Borchers T, Spener F. Fatty acids and hypolipidemic drugs regulate PPARα and PPARγ gene expresion via L-FABP: a signaling path to the nucleus. Proc Natl Acad Sci. 2001;98:2323–2328. doi: 10.1073/pnas.051619898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McIntosh AL, Huang H, Atshaves BP, Wellburg E, Kuklev DV, Smith WL, Kier AB, Schroeder F. Fluorescent n-3 and n-6 very long chain polyunsaturated fatty acids: three photon imaging and metabolism in living cells overexpressing liver fatty acid binding protein. J Biol Chem. 2010;285:18693–18708. doi: 10.1074/jbc.M109.079897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McIntosh AL, Atshaves BP, Hostetler HA, Huang H, Davis J, Lyuksyutova OI, Landrock D, Kier AB, Schroeder F. Liver type fatty acid binding protein (L-FABP) gene ablation reduces nuclear ligand distribution and peroxisome proliferator activated receptor-α activity in cultured primary hepatocytes. Arch Biochem Biophys. 2009;485:160–173. doi: 10.1016/j.abb.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hostetler HA, McIntosh AL, Atshaves BP, Storey SM, Payne HR, Kier AB, Schroeder F. Liver type Fatty Acid Binding Protein (L-FABP) interacts with peroxisome proliferator activated receptor-α in cultured primary hepatocytes. J Lipid Res. 2009;50:1663–1675. doi: 10.1194/jlr.M900058-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hostetler HA, Balanarasimha M, Huang H, Kelzer MS, Kaliappan A, Kier AB, Schroeder F. Glucose regulates fatty acid binding protein interaction with lipids and PPARα. J Lipid Res. 2010;51:3103–3116. doi: 10.1194/jlr.M005041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Atshaves BP, Martin GG, Hostetler HA, McIntosh AL, Kier AB, Schroeder F. Liver fatty acid binding protein (L-FABP) and Dietary Obesity. Journal of Nutritional Biochemisty. 2010;21:1015–1032. doi: 10.1016/j.jnutbio.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wolfrum C, Buhlman C, Rolf B, Borchers T, Spener F. Variation of liver fatty acid binding protein content in the human hepatoma cell line HepG2 by peroxisome proliferators and antisense RNA affects the rate of fatty acid uptake. Biochim Biophys Acta. 1999;1437:194–201. doi: 10.1016/s1388-1981(99)00008-6. [DOI] [PubMed] [Google Scholar]
- 33.Kabakibi A, Morse CR, Laposata M. Fatty acid ethyl esters and HepG2 cells: intracellular synthesis and release from the cells. J Lipid Res. 1998;39:1568–1582. [PubMed] [Google Scholar]
- 34.Gao N, Qu X, Yan J, Huang Q, Yuan HY, Ouyang D-S. L-FABP T94A decreased fatty acid uptake and altered hepatic triglyceride and cholesterol accumulation in Chang liver cells stably transfected with L-FABP. Mol Cell Biochem. 2010;345:207–214. doi: 10.1007/s11010-010-0574-7. [DOI] [PubMed] [Google Scholar]
- 35.Cornu-Chagnon MC, Dupont H, Edgar A. Fenofibrate: metabolism and species differences for peroxisome proliferation in cultured hepatocytes. Fundamental Applied Toxicology. 1995;26:63–74. doi: 10.1006/faat.1995.1075. [DOI] [PubMed] [Google Scholar]
- 36.Schoonjans K, Staels B, Grimaldi P, Auwerx J. Acyl-CoA synthetase mRNA expression is controlled by fibric-acid derivatives, feeding and liver proliferation. Eur J Biochem. 1993;216:615–622. doi: 10.1111/j.1432-1033.1993.tb18181.x. [DOI] [PubMed] [Google Scholar]
- 37.Schoonjans K, Staels B, Auwerx J. Role of the peroxisome proliferator-activated receptor (PPAR) in mediating the effects of fibrates and fatty acids on gene expression. [Review] [193 refs] Journal of Lipid Research. 1996;37:907–925. [PubMed] [Google Scholar]
- 38.Garcia MA, Vazquez J, Gimenez C, Valdivieso F, Zafra F. Transcription factor AP-2 regulates human apolipoprotein E gene expression in astrocytoma cells. Journal of Neurosciences. 1996;16:7550–7556. doi: 10.1523/JNEUROSCI.16-23-07550.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tal M, Schneider DL, Thorens B, Lodish HF. Restricted expression of the erythroid/brain glucose transporter isoform to perivenous hepatocytes in rats. Modulation by glucose. J Clin Inv. 1990;86:986–992. doi: 10.1172/JCI114801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Valera A, Bosch F. Glucokinase expression in rat hepatoma cells induces glucose uptake and is rate limiting in glucose utilization. Eur J Biochem. 1994;222:533–539. doi: 10.1111/j.1432-1033.1994.tb18895.x. [DOI] [PubMed] [Google Scholar]
- 41.Bilir BM, Gong TWL, Kwasiborski V, Shen CS, Fillmore CS, Berkowitz CM, Gumucio JJ. Novel control of the position dependent expression of genes in hepatocytes: The GLUT-1 transporter. J Biol Chem. 1993;268:19776–19784. [PubMed] [Google Scholar]
- 42.Hostetler HA, Petrescu AD, Kier AB, Schroeder F. Peroxisome proliferator activated receptor alpha (PPARα) interacts with high affinity and is conformationally responsive to endogenous ligands. J Biol Chem. 2005;280:18667–18682. doi: 10.1074/jbc.M412062200. [DOI] [PubMed] [Google Scholar]
- 43.Chuang S, Velkov T, Horne J, Wielens J, Chalmers DK, Porter CJH, Scanlon MJ. Probing fibrate binding specificity of rat liver fatty acid binding protein. J Med Chem. 2009;52:5344–5355. doi: 10.1021/jm801349e. [DOI] [PubMed] [Google Scholar]
- 44.Chuang S, Velkov T, Horne J, Porter CJH, Scanlon MJ. Characterization of the drug binding specificity of rat liver fatty acid binding protein. J Med Chem. 2008;51:3755–3764. doi: 10.1021/jm701192w. [DOI] [PubMed] [Google Scholar]
- 45.Wolfrum C, Borchers T, Sacchettini JC, Spener F. Binding of fatty acids and perooxisome proliferators to orthologous fatty acid binding proteins from human, murine, and bovine liver. Biochemistry. 2000;39:1469–1474. doi: 10.1021/bi991638u. [DOI] [PubMed] [Google Scholar]
- 46.Di Pietro SM, Santome JA. Isolation, characterization, and binding properties of two rat liver fatty acid binding protein isoforms. Biochim Biophys Acta. 2000;1478:186–200. doi: 10.1016/s0167-4838(00)00042-x. [DOI] [PubMed] [Google Scholar]
- 47.Velkov T. Interactions between human liver fatty acid binding protein and peroxisome proliferator activated receptor drugs. PPAR Research. 2013 doi: 10.1155/2013/938401. org/10.1155/2013/938401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Harris RA. Carbohydrate Metabolism I: Major Metabolic Pathways and Their Control. In: Devlin TM, editor. Textbook of Biochemistry With Clinical Correlations. John Wiley and Sons, Inc; Hoboken, NJ: 2006. pp. 581–635. [Google Scholar]
- 49.Williamson DH, Brosnan JT. Concentrations of Metabolites in Animal Tissues. In: Bergmeyer HU, editor. Methods of Enzymatic Analysis. 4. Academic Press Inc; New York: 1974. pp. 2266–2302. [Google Scholar]
- 50.Garrett RH, Grisham CM. Glycolysis. In: Garrett RH, Grisham CM, editors. Biochemistry. Brooks/Cole, Cengage Learning; Boston, MA: 2010. pp. 535–562. [Google Scholar]
- 51.Fehr M, Takanaga H, Ehrhardt DW, Frommer WB. Evidence for high-capacity bidirectional glucose transport across the endoplasmic reticulum membrane by genetically encoded fluorescence resonance energy transfer nanosensors. Mol Cell Biol. 2005;25:11102–11112. doi: 10.1128/MCB.25.24.11102-11112.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Atshaves BP, McIntosh AL, Lyuksyutova OI, Zipfel WR, Webb WW, Schroeder F. Liver fatty acid binding protein gene ablation inhibits branched-chain fatty acid metabolism in cultured primary hepatocytes. J Biol Chem. 2004;279:30954–30965. doi: 10.1074/jbc.M313571200. [DOI] [PubMed] [Google Scholar]
- 53.Martin GG, Danneberg H, Kumar LS, Atshaves BP, Erol E, Bader M, Schroeder F, Binas B. Decreased liver fatty acid binding capacity and altered liver lipid distribution in mice lacking the liver fatty acid binding protein (L-FABP) gene. J Biol Chem. 2003;278:21429–21438. doi: 10.1074/jbc.M300287200. [DOI] [PubMed] [Google Scholar]
- 54.Storey SM, Atshaves BP, McIntosh AL, Landrock KK, Martin GG, Huang H, Johnson JD, Macfarlane RD, Kier AB, Schroeder F. Effect of sterol carrier protein-2 gene ablation on HDL-mediated cholesterol efflux from primary cultured mouse hepatocytes. Am J Physiol. 2010;299:244–254. doi: 10.1152/ajpgi.00446.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Storey SM, McIntosh AL, Huang H, Martin GG, Landrock KK, Landrock D, Payne HR, Kier AB, Schroeder F. Intracellular cholesterol binding proteins enhance HDL-mediated cholesterol uptake in cultured primary mouse hepatocytes. Am J Physiol Gastrointest and Liver Phys. 2012;302:G824–G839. doi: 10.1152/ajpgi.00195.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Storey SM, McIntosh AL, Huang H, Martin GG, Landrock KK, Landrock D, Payne HR, Kier AB, Schroeder F. Loss of intracellular lipid binding proteins differentially impacts saturated fatty acid uptake and nuclear targeting in mouse hepatocytes. Am J Physiol Gastrointest and Liver Phys. 2012;303:G837–G850. doi: 10.1152/ajpgi.00489.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Moya M, Gomez-Lechon MJ, Castell JV, Jover R. Enhanced steatosis by nuclear receptor ligands: a study in cultured human hepatocytes and hepatoma cells with a characterized nuclear receptor expression profile. Chem Biol Interactions. 2010;184:376–387. doi: 10.1016/j.cbi.2010.01.008. [DOI] [PubMed] [Google Scholar]
- 58.Pawar A, Jump DB. Unsaturated fatty acid regulation of peroxisome proliferator activated receptor-alpha activity in primary rat hepatocytes. J Biol Chem. 2003;278:35931–35939. doi: 10.1074/jbc.M306238200. [DOI] [PubMed] [Google Scholar]
- 59.Kubota T, Yano T, Fujisaki K, Itoh Y, Oishi R. Fenofibrate induces apoptotic injury in cultured human hepatocytes by inhibiting phosphorylation of Akt. Apoptosis. 2005;10:349–358. doi: 10.1007/s10495-005-0809-3. [DOI] [PubMed] [Google Scholar]
- 60.Spector AA, Hoak JC. An improved method for the addition of long chian free fatty acid to protein solutions. Anal Biochem. 1969;32:297–302. doi: 10.1016/0003-2697(69)90089-x. [DOI] [PubMed] [Google Scholar]
- 61.Yan J, Gong Y, Wang G, Gong Y, Burczynski FJ. Regulation of L-FABP expression by clofibrate treatment in hepatoma cells. Biochem Cell Biol. 2010;88:957–967. doi: 10.1139/O10-151. [DOI] [PubMed] [Google Scholar]
- 62.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-DDCT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 63.Zhang J-Z, Gao L, Widness M, Xi X, Kern TS. Captopril inhibits glucose accumulation in retinal cells in diabetes. Invest Ophthal Visual Science. 2003;44:4001–4005. doi: 10.1167/iovs.02-1193. [DOI] [PubMed] [Google Scholar]
- 64.Pieri C, Giuli C, Del Moro M, Piantanelli L. Electron-microscopic morphometric analysis of mouse liver. II. Effect of ageing and thymus transplantation in old animals. Mech Ageing Dev. 1980;13:275–283. doi: 10.1016/0047-6374(80)90040-8. [DOI] [PubMed] [Google Scholar]
- 65.Philimonenko AA, Janacek J, Hozak P. Statistical evaluation of colocalization patterns in immunogold labeling experiments. J Struct Biol. 2000;132:201–210. doi: 10.1006/jsbi.2000.4326. [DOI] [PubMed] [Google Scholar]
- 66.Karam WG, Ghanayem BI. Induction of replicative DNA synthesis and PPARα-dependent gene transcription by Wy-14 643 in primary rat hepatocyte and non-parenchymal cell co-cultures. Carcinogenesis. 1997;18:2077–2083. doi: 10.1093/carcin/18.11.2077. [DOI] [PubMed] [Google Scholar]
- 67.Jump DB. N-3 polyunsaturated fatty acid regulation of hepatic gene transcription. Cur Opin Lipidology. 2008;19:242–247. doi: 10.1097/MOL.0b013e3282ffaf6a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lin Q, Ruuska SE, Shaw NS, Dong D, Noy N. Ligand selectivity of the peroxisome proliferator-activated receptor a. Biochem. 1999;38:185–190. doi: 10.1021/bi9816094. [DOI] [PubMed] [Google Scholar]
- 69.Madsen L, Rustan AC, Vaagenes H, Berge K, Dyroy E, Berge RK. Eicosapentaenoic and docosahexaenoic acid affect mitochondrial and peroxisomal fatty acid oxidation in relation to substrate preference. Lipids. 1999;34:951–963. doi: 10.1007/s11745-999-0445-x. [DOI] [PubMed] [Google Scholar]
- 70.Gyamfi MA, He L, French SW, Damjanov I, Wan Y-JY. Hepatocyte retinoid X receptor alpha-dependent regulation of lipid homeostasis and inflammatory cytokine expression contributes to alcohol-induced liver injury. J Pharm Exp Ther. 2008;324:443–453. doi: 10.1124/jpet.107.132258. [DOI] [PubMed] [Google Scholar]
- 71.Hostetler HA, Lupas D, Tan Y, Dai J, Kelzer MS, Martin GG, Woldegiorgis G, Kier AB, Schroeder F. Acyl-CoA binding proteins interact with the acyl-CoA binding domain of mitochondrial carnitine palmitoyltransferase I. Mol Cell Biochem. 2011;355:135–148. doi: 10.1007/s11010-011-0847-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gossett RE, Frolov AA, Roths JB, Behnke WD, Kier AB, Schroeder F. Acyl Co A binding proteins: multiplicity and function. Lipids. 1996;31:895–918. doi: 10.1007/BF02522684. [DOI] [PubMed] [Google Scholar]
- 73.Torra ISP, Jamshidi Y, Flavell DM, Fruchart J-C, Staels B. Characterization of the human PPARα promoter: identification of a functional nuclear receptor response element. Mol Endocrinology. 2002;16:1013–1028. doi: 10.1210/mend.16.5.0833. [DOI] [PubMed] [Google Scholar]
- 74.Hong F, Radaeva S, Pan H, Tian Z, Veech R, Gao B. Interleukin 6 alleviates hepatic steatosis and ischemia/reperfusion injury in mice with fatty liver disease. Hepatology. 2004;40:933–941. doi: 10.1002/hep.20400. [DOI] [PubMed] [Google Scholar]
- 75.Akiyama TE, Ward JM, Gonzalez FJ. Regulation of liver fatty acid binding protein gene by hepatocyte nuclear factors 1a (HNF1a) J Biol Chem. 2000;275:27117–27122. doi: 10.1074/jbc.M004388200. [DOI] [PubMed] [Google Scholar]
- 76.Martinez-Jimenez CP, Kyrmizi I, Cardot P, Gonzalex FJ, Taliandis I. Hepatocyte nuclear factor 4α coordinates a transcription factor network regulating hepatic fatty acid metabolism. Mol Cell Biol. 2010;30:565–577. doi: 10.1128/MCB.00927-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Arbuckle MI, Kane S, Porter LM, Seatter MJ, Gould GW. Structure-function analysis of liver type (GLUT2) and brain type (GLUT3) glucose transporters: expression of chimeric transporters in X. oocytes suggests an important role for putative transmembrane helix 7 in determining substrate selectivity. Biochem. 1996;35:16519–16527. doi: 10.1021/bi962210n. [DOI] [PubMed] [Google Scholar]
- 78.Sanderson LM, Degenhardt T, Koppen A, Kalkhoven E, Desvergne B, Muller M, Kersten S. Peroxisome proliferator activated receptor β/δ (PPARβ/δ) but not PPARα serves as a plasma free fatty acid sensor in liver. Mol Cell Biol. 2009;29:6257–6267. doi: 10.1128/MCB.00370-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Prows DR, Murphy EJ, Schroeder F. Intestinal and liver fatty acid binding proteins differentially affect fatty acid uptake and esterification in L-Cells. Lipids. 1995;30:907–910. doi: 10.1007/BF02537481. [DOI] [PubMed] [Google Scholar]
- 80.Jolly CA, Hubbell T, Behnke WD, Schroeder F. Fatty acid binding protein: stimulation of microsomal phosphatidic acid formation. Arch Biochem Biophys. 1997;341:112–121. doi: 10.1006/abbi.1997.9957. [DOI] [PubMed] [Google Scholar]
- 81.Schroeder F, Jolly CA, Cho TH, Frolov AA. Fatty acid binding protein isoforms: structure and function. Chem Phys Lipids. 1998;92:1–25. doi: 10.1016/s0009-3084(98)00003-6. [DOI] [PubMed] [Google Scholar]
- 82.Bordewick U, Heese M, Borchers T, Robenek H, Spener F. Compartmentation of hepatic fatty-acid-binding protein in liver cells and its effect on microsomal phosphatidic acid biosynthesis. Biol Chem Hoppe-Seyler. 1989;370:229–238. doi: 10.1515/bchm3.1989.370.1.229. [DOI] [PubMed] [Google Scholar]
- 83.Nemecz G, Schroeder F. Selective binding of cholesterol by recombinant fatty acid-binding proteins. J Biol Chem. 1991;266:17180–17186. [PubMed] [Google Scholar]
- 84.Chao H, Zhou M, McIntosh A, Schroeder F, Kier AB. Acyl CoA binding protein and cholesterol differentially alter fatty acyl CoA utilization by microsomal acyl CoA: cholesterol transferase. J Lipid Res. 2003;44:72–83. doi: 10.1194/jlr.m200191-jlr200. [DOI] [PubMed] [Google Scholar]
- 85.Takeuchi K, Reue K. Biochemistry, physiology, and genetics of GPAT, AGPAT, and lipin enzymes in triglyceride synthesis. Am J Physiol Endocrinol Metab. 2012;296:E1195–E1209. doi: 10.1152/ajpendo.90958.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.O’Rourke L, Gronning LM, Yeaman SJ, Shepherd PR. Glucose dependent regulationof cholesterol ester metabolism in macrophages by insulin and leptin. J Biol Chem. 2002;277:42557–42562. doi: 10.1074/jbc.M202151200. [DOI] [PubMed] [Google Scholar]
- 87.McIntosh AL, Atshaves BP, Wellberg EKDV, Smith WL, Schroeder F. Uptake kinetics of fluorescent long chain n-3 and n-6 fatty acids in intact cells. FASEB J. 2005;19:A292. [Google Scholar]
- 88.Richert L, Lamboley C, Viollon-Abadie C, Grass P, Hartmann N, Laurent S, Heyd B, Mantion G, Chibout S-D, Staedtler F. Effects of clofibric acid on mRNA expression profiles in primary cultures of rat, mouse, and human hepatocytes. Toxicol Appl Pharmacol. 2003;191:130–146. doi: 10.1016/s0041-008x(03)00231-x. [DOI] [PubMed] [Google Scholar]
- 89.Hostetler HA, Huang H, Kier AB, Schroeder F. Glucose directly links to lipid metabolism through high-affinity interaction with peroxisome proliferator activated receptor-alpha. J Biol Chem. 2008;283:2246–2254. doi: 10.1074/jbc.M705138200. [DOI] [PubMed] [Google Scholar]
- 90.Fehr M, Lalonde S, Ehrhardt EW, Frommer WB. Live imaging of glucose homeostasis in nuclei of COS-7 cells. J of Fluorescence. 2004;14:603–609. doi: 10.1023/b:jofl.0000039347.94943.99. [DOI] [PubMed] [Google Scholar]
- 91.Cramer JA. A systematic review of adherence with medications for diabetes. Diabetes Care. 2004;27:1218–1224. doi: 10.2337/diacare.27.5.1218. [DOI] [PubMed] [Google Scholar]
- 92.Rubin RR. Adherence to pharmacologic therapy in patients with type 2 diabetes mellitus. Am J Med. 2012;118:27S–34S. doi: 10.1016/j.amjmed.2005.04.012. [DOI] [PubMed] [Google Scholar]
- 93.Schoenthaler AM, Schwartz BS, Wood C, Stewart WF. Patient and physician factors associated with adherence to diabetes medications. The Diabestes Educator. 2012;38:397–408. doi: 10.1177/0145721712440333. [DOI] [PubMed] [Google Scholar]
- 94.Sato A, Kawano H, Notsu T, Ohta M, Nakakuki M, Mizuguchi K, Itoh M, Suganami T, Ogawa Y. Antiobesity effect of eicosapentaenoic acid in high-fat/high-sucrose diet-induced obesity. Diabetes. 2010;59:2495–2504. doi: 10.2337/db09-1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ma T, Liaset B, Hao Q, Petersen RK, Fjaere E, Ngo HT, Lillefosse HH, Ringholm S, Sonne SB, Treebak JT, Pilegaard H, Freyland L, Kristiansen K, Madsen L. Sucrose counteracts the anti-inflammatory effect of fish oil in adipose tissue and increases obesity development in mice. PLoS ONE. 2011;6:21647. doi: 10.1371/journal.pone.0021647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Montori VM, Wollan PC, Farmer A, Dinneen SE. Fish oil supplementation in type 2 diabetes. A quantitative systematic review. Diabetes Care. 2000;23:1407–1415. doi: 10.2337/diacare.23.9.1407. [DOI] [PubMed] [Google Scholar]
- 97.Pownall H, Brauchi D, Kilinc C, Osmundsen K, Pao Q, Payton-Ross C, Gotto AM, Ballantyne CM. Correlation of serum triglyceride and its reduction by ω-3 fatty acids with lipid transfer activity and the neutral lipid compositions of HDL and LDL. Atherosclerosis. 1999;143:285–297. doi: 10.1016/s0021-9150(98)00301-3. [DOI] [PubMed] [Google Scholar]
- 98.Kabir M, Skurnik G, Naour N, Pechtner V, Meugnier E, Rome S, Quignard-Boulange A, Vidal H, Slama G, Clement K, Guerre-Millo M, Rizkalla SW. Treatment for 2 mo with n-3 polyunsaturated fatty acids reduces adiposity and some atherogenic fctors but doe not improve insulin sensitivity in women with type 2 diabetes: a randomized controlled study. Am J Clin Nutr. 2007;86:1670–1679. doi: 10.1093/ajcn/86.5.1670. [DOI] [PubMed] [Google Scholar]
- 99.Sirtori CR, Crepaldi G, Manzato E, Mancini M, Rivellese A, Paoletti R, Pazzucconi F, Pamparana F, Stragliotto E. One-year treatment with ethyl esters of n-3 fatty acids in patients with hypertriglyceridemia and glucose intolerance. Reduced triglyceridemia, total cholesterol and increased HDL-C without glycemic alterations. Atherosclerosis. 1998;137:419–427. doi: 10.1016/s0021-9150(97)00298-0. [DOI] [PubMed] [Google Scholar]
- 100.Erol E, Kumar LS, Cline GW, Shulman GI, Kelly DP, Binas B. Liver fatty acid-binding protein is required for high rates of hepatic fatty acid oxidation but not for the action of PPAR-a in fasting mice. FASEB J. 2004;18:347–349. doi: 10.1096/fj.03-0330fje. [DOI] [PubMed] [Google Scholar]
- 101.Newberry EP, Xie Y, Kennedy S, Buhman KK, Luo J, Gross RW, Davidson NO. Decreased hepatic triglyceride accumulation and altered fatty acid uptake in mice with deletion of the liver fatty acid binding protein gene. J Biol Chem. 2003;278:51664–51672. doi: 10.1074/jbc.M309377200. [DOI] [PubMed] [Google Scholar]
- 102.Aoyama T, Peters JM, Iritani N, Nakajima T, Furihata K, Hashimoto T, Gonzalez FJ. Altered constitutive expression of fatty acid metabolizing enzymes in mice lacking PPARα. J Biol Chem. 1998;273:5678–5684. doi: 10.1074/jbc.273.10.5678. [DOI] [PubMed] [Google Scholar]
- 103.Frolov A, Cho TH, Murphy EJ, Schroeder F. Isoforms of rat liver fatty acid binding protein differ in structure and affinity for fatty acids and fatty acyl CoAs. Biochemistry. 1997;36:6545–6555. doi: 10.1021/bi970205t. [DOI] [PubMed] [Google Scholar]
- 104.Richieri GV, Ogata RT, Kleinfeld AM. Equilibrium constants for the binding of fatty acids with fatty acid binding proteins from adipocyte, intestine, heart, and liver measured with the flourscent probe ADIFAB. J Biol Chem. 1994;269:23918–23930. [PubMed] [Google Scholar]
- 105.Maatman RG, van Moerkerk HT, Nooren IM, van Zoelen EJ, Veerkamp JH. Expression of human liver fatty acid-binding protein in Escherichia coli and comparative analysis of its binding characteristics with muscle fatty acid-binding protein. Biochim Biophys Acta. 1994;1214:1–10. doi: 10.1016/0005-2760(94)90002-7. [DOI] [PubMed] [Google Scholar]
- 106.Rolf B, Oudenampsen-Kruger E, Borchers T, Faergeman NJ, Knudsen J, Lezius A, Spener F. Analysis of the ligand binding properties of recombinant bovine liver-type fatty acid binding protein. Biochim Biophys Acta. 1995;1259:245–253. doi: 10.1016/0005-2760(95)00170-0. [DOI] [PubMed] [Google Scholar]
- 107.Mannaerts GP, Debeer LJ, Thomas J, De Schepper PJ. Mitochondrial and peroxisomal fatty acid oxidation in liver homogenates and isolated hepatocytes from control and clofibrate treated rats. J Biol Chem. 1979;254:4585–4595. [PubMed] [Google Scholar]
- 108.Nakajima T, Tanaka N, Kanbe H, Hara A, Kamijo Y, Zhang X, Gonzalez FJ, Aoyama T. Bezafibrate clinically relevantdoses decreases serum/liver triglycerides via down-regulation of SREBP-1c in mice: a novel PPARα independent mechanism. Mol Pharmacol. 2009;75:782–792. doi: 10.1124/mol.108.052928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Rioux V, Catheline D, Legrand P. In rat hepatocytes, myristic acid occurs through lipogenesis, palmitic acid shortening and lauric acid elongation. Animal. 2007;1:820–826. doi: 10.1017/S1751731107000122. [DOI] [PubMed] [Google Scholar]
- 110.McArthur MJ, Atshaves BP, Frolov A, Foxworth WD, Kier AB, Schroeder F. Cellular uptake and intracellular trafficking of long chain fatty acids. J Lipid Res. 1999;40:1371–1383. [PubMed] [Google Scholar]
- 111.John SA, Ottolia M, Weiss JN, Ribalet B. Dynamic modulation of intracellular glucose imaged in single cells using a FRET-based glucose nanosensor. Pflugers Arch -Eur J Physiol. 2008;456:307–322. doi: 10.1007/s00424-007-0395-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Takanaga H, Chaudhuri B, Frommer WB. GLUT1 and GLUT9 as major contributors to glucose influx in HepG2 cells identified by a high sensitivity intramolecular FRET glucose sensor. Biochim Biophys Acta. 2008;1778:1091–1099. doi: 10.1016/j.bbamem.2007.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Cline GW, Petersen KF, Krssak M, Shen J, Hundal RS, Trajanoski Z, Inzucchi S, Dresner A, Rothman DL, Shulman GI. Impaired glucose transport as a cause of decreased insulin-stimulated muscle glycogen synthesis in type 2 diabetes. New England J Med. 1999;341:240–246. doi: 10.1056/NEJM199907223410404. [DOI] [PubMed] [Google Scholar]
- 114.Murphy EJ, Edmondson RD, Russell DH, Schroeder F. Isolation and characterization of two distinct forms of liver fatty acid binding protein from the rat. Biochim Biophys Acta. 1999;1436:413–425. doi: 10.1016/s0005-2760(98)00150-7. [DOI] [PubMed] [Google Scholar]
- 115.Diradourian C, Girard J, Pegorier J-P. Phosphorylation of PPARs: from molecular characterization to physiological relevance. Biochemie. 2005;87:33–38. doi: 10.1016/j.biochi.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 116.Pourcet B, Pineda-Torra I, Derudas B, Staels B, Glineur C. SUMOylation of human peroxisome proliferator activated receptor- α inhibits its trans-activity through the recruitment of the nuclear corepressor NCoR. J Bio Chem. 2010;285:5983–5992. doi: 10.1074/jbc.M109.078311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Petrescu AD, Payne HR, Boedeker AL, Chao H, Hertz R, Bar-Tana J, Schroeder F, Kier AB. Physical and functional interaction of acyl CoA binding protein (ACBP) with hepatocyte nuclear factor-4α (HNF4α) J Biol Chem. 2003;278:51813–51824. doi: 10.1074/jbc.M303858200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.