

Abstract
Hydrogen sulfide is an important biological signalling molecule and an important environmental target for detection. A major challenge in developing H2S detection methods is separating the often similar reactivity of thiols and other nucleophiles from H2S. To address this need, the nucleophilic aromatic substitution (SNAr) reaction of H2S with electron-poor aromatic electrophiles was developed as a strategy to separate H2S and thiol reactivity. Treatment of aqueous solutions of nitrobenzofurazan (7-nitro-1,2,3-benzoxadiazole, NBD) thioethers with H2S resulted in thiol extrusion and formation of nitrobenzofurazan thiol (λmax = 534 nm). This reactivity allows for unwanted thioether products to be converted to the desired nitrobenzofurazan thiol upon reaction with H2S. The scope of the reaction was investigated using a Hammett linear free energy relationship study, and the determined ρ = +0.34 is consistent with the proposed SN2Ar reaction mechanism. The efficacy of the developed probes was demonstrated in buffer and in serum with associated sub-micromolar detection limits as low as 190 nM (buffer) and 380 nM (serum). Furthermore, the sigmoidal response of nitrobenzofurazan electrophiles with H2S can be fit to accurately quantify H2S. The developed detection strategy offers a manifold for H2S detection that we foresee being applied in various future applications.
Introduction
Hydrogen sulfide (H2S), although historically recognized as a toxic gas, plays important roles in the global sulfur cycle and is now accepted as an important, endogenously-produced signaling molecule. Biological H2S exhibits significant effects in both the cardiovascular and neuronal systems and is produced endogenously from cystathionine-β-synthase (CBS), cystathionine-γ-lyase (CSE), and 3-mercaptopyruvate sulfur transferase (3MST), as well as from non-enzymatic processes.1–4 One unique aspect of H2S by comparison to other gasotransmitters, such as NO and CO, is that its different protonation states facilitate lipid and water solubility in the diprotic (H2S) and monoanionic (SH−) forms, respectively. The diprotic nature of H2S also has important reactivity consequences because both the reduction potential and nucleophilicity can be modulated by deprotonation.5 These properties not only highlight the diverse biological reactivity of H2S, but also provide viable strategies for intercepting and detecting this important biological messenger.
Coinciding with the discovery of emerging biological roles of H2S has been the development of reaction-based chemical methods for H2S detection. Such methods have typically used H2S either as a reductant to reduce azides or nitro groups,6–11 as a nucleophile to attack activated electrophiles12–18 or react with and remove metals from coordination environments.19–22 Although these systems have provided methods for H2S detection with moderate to good selectivity, the development of innovative chemistry that clearly differentiates thiols from H2S is still needed. For example, many nucleophile-based approaches suffer from irreversible probe deactivation upon reaction with thiols, thereby greatly diminishing the H2S detection capacity. Current strategies for addressing this problem include the use of reversible Michael acceptors or disulfide-exchange reactions, which allow for chemically-reversible reactions with thiols prior to reaction with H2S.13,23 For systems that use H2S as a reductant, the amine reaction product obtained from H2S- or thiol-mediated reduction is identical, thus greatly complicating detection or quantification unless the exact concentrations and reaction rates of competing thiols are known.
Most recent reports of reaction-based H2S detection have focused on fluorescent reporters to detect H2S in live cells or tissues, but other detection methods, especially those with low instrumentation requirements, are also needed. Classical methods of H2S detection and quantification, including gas chromatography, polarography, or sulfide-selective electrodes, share the high instrumentation requirements with fluorimetry.24–28 Colorimetric detection methods, by comparison, have relatively undemanding requirements.10,21,29,30 Additionally, suitably large spectral changes between the off and on state of a colorimetric system allow for unassisted visual detection. This low instrumentation requirement not only makes such colorimetric detection methods viable for high throughput detection and quantification in biological media or homogenates, but also provides access to simple detection methods for applications in which instrumentation or laboratory costs are often prohibitive or otherwise unavailable. For example, a major problem in remote or developing areas is contamination of improperly treated drinking water by pathogenic sulfur-reducing bacteria. Because laboratory tests for these types of organisms are often prohibitively expensive or time consuming, elevated H2S levels in water have been used as a marker for potential pathogenic bacterial contaminations in different water sources.31,32
To address both the need for simple and selective H2S detection methods, we report here the development and scope of a method for H2S detection based on nucleophilic aromatic substitution. This method allows for separation of the reactivity of H2S with other biologically-relevant nucleophiles. UV-vis and NMR spectroscopic studies of the different reaction pathways of the developed platform are presented. The strategy for selective H2S detection was then applied to develop a sensitive and selective colorimetric probe for H2S that is unaffected by the presence of other biologically relevant nucleophiles.
Results and Discussion
Electron-deficient arenes with suitable leaving groups can undergo nucleophilic aromatic substitution (SNAr) reactions. We reasoned that such scaffolds could function as a versatile platform for H2S detection that would allow for differentiation of H2S from thiols based on the different products formed from these two reaction pathways. Reaction of an electrophilic aryl-halide with H2S, for example, would afford a thiophenol, whereas reaction with a thiol would form a thioether. If the resultant thioether product remained suitably electrophilic, it could further react with H2S to generate the thiophenol product (Scheme 1). This reaction manifold would allow for thiol attack on the probe without compromising reactivity toward H2S, thus creating a thiol-insensitive platform for H2S detection. For such a platform to be feasible, the intermediate thioether generated from thiol attack must (i) react irreversibly with H2S and (ii) have different photophysical properties from the thiophenol H2S reaction product.
Scheme 1.
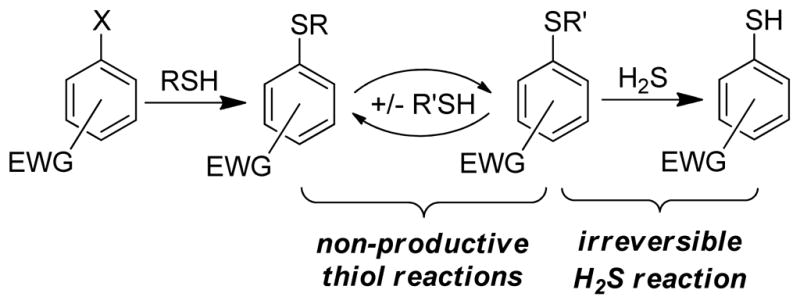
Thiol-insensitive H2S sensing platform based on the non-productive, chemically-reversible formation of thioethers that maintain reactivity toward H2S.
To test this hypothesis, we investigated the reactivity of H2S with 4-chloro-7-nitrobenzofurazan (NBD-Cl, 1), an electron-deficient small molecule that reacts with thiols to afford a fluorescent thioether (NBD-SR) product. Nucleophilic attack on 1 proceeds through a stepwise SN2Ar mechanism, often with reversible addition of the nucleophile to formation of the intermediate σ-complex (Meisenheimer complex) and subsequent final product.33 Although 1 and other electrophilic benzofurazan derivatives have been used extensively for labeling, modifying, and quantifying endogenous thiols,34 they can also react with other nucleophiles including cysteine sulfenic acids,35 amino groups,36 and tyrosine hydroxyl groups37 at neutral and basic pH ranges. In certain cases, if suitably nucleophilic groups are present near an NBD-labeled residue, the ligated NBD group can be transferred intramolecularly to adjacent nucleophilic residues.38,39 This NBD group transfer reaction suggested to us that an NBD-thioether might maintain the required electrophilicity to react with H2S. In such a scheme, 1 would react with HS− to form 4-thio-7-nitrobenzofurzan (NBD-SH, 2), which could be converted to the corresponding (NBD)2S thioether (3) in the presence of excess 1. If 3 remained sufficiently electrophilic to react with H2S, then further reaction with H2S would regenerate 2 (Scheme 2). This reaction manifold provides an efficient route to generate H2S-derived benzene thiols, which can be used for sensing applications based on their photophysical properties.
Scheme 2.
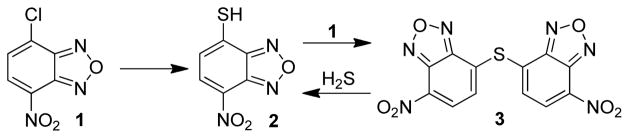
Proposed reactivity of 1 with H2S to form the corresponding thiol (2) and thioether (3).
To test the proposed reactivity of 1 toward H2S, we titrated 1 with NaSH, a common H2S source, in PIPES buffer (50 mM PIPES, 100 mM KCl, pH 7.4).40 During the titration, the characteristic 343 nm absorbance of 1 decreased with the concomitant growth of a new absorbance at 534 nm corresponding to nitrobenzofurzan thiol 2 (vide infra) (Figure 1a). During the initial portion of the titration, formation of a second species with absorbances at 298 nm and 413 nm was observed; however, this intermediate was completely consumed during the titration to afford 2, as evidenced by the clean isosbestic points at 256 nm, 371 nm, and 445 nm. Neither 2 nor 3 are fluorescent, but the bathochromic shift and characteristic absorbance of 2 (λmax 534 nm, ε534 = 19,000 ± 600 M−1 cm−1) allows for visual detection. Additionally, isolated 2 does not react with other nucleophiles, such as cysteine or glutathione (GSH), and it does not extrude HS− by purging with nitrogen. The 534 nm absorbance of 2 can be abolished, however, by treatment with excess 1. This reaction proceeds through clean isosbestic points at 256 nm, 371 nm, and 445 nm, resulting in the characteristic absorbances of thioether 3 at 413 nm (ε413 = 11,700 ± 100 M−1 cm−1) and 298 nm. These studies establish that the reaction pathway outlined in Scheme 2 is operable in solution.
Figure 1.

UV-vis spectra of (a) 1 upon reaction with NaSH. Conditions: 66 μM of 1, 3.3 μM increments of NaSH, 50 mM PIPES, 100 mM KCl, pH 7.4, 37 °C. (b) UV-vis spectra of isolated 1 (λmax = 343 nm), 3 (λmax = 413 nm), and 2 (λmax = 534 nm) for comparison.
The absorbance of 2 is uncommonly red-shifted for nitrobenzofurazan-derived compounds, which prompted our investigation to the source of the 534 nm absorbance. Based on the electron-withdrawing nitro and furazan moieties in 2, we surmised that the thiol proton would be highly acidic. Acidification of a solution of 2 extinguished the 534 nm absorbance and resulted in a new absorbance at 400 nm, suggesting that electron delocalization of the deprotonated thiol over the nitrobenzofurazan ring accounts for the purple color. By monitoring the absorbance of 2 at 534 nm as a function of pH, an apparent pKa of 2.6(1) for the thiol was determined by fitting the titration data (Figure 2), which is consistent with the high reported acidity of NBD-OH.41
Figure 2.
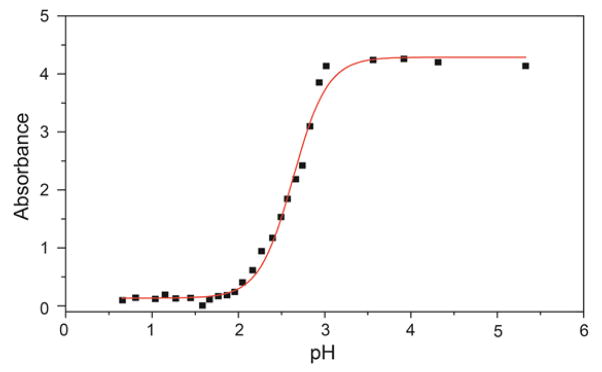
pH titration of 2 (111 μM) in 100 mM KCl at 25 °C. The pH was adjusted by addition of aliquots of 10 M, 5 M, 1 M, 50 mM, or 1 mM solutions of HCl. All absorbance data are corrected for dilution.
Thioether Reactivity
Thioether 3 was isolated by addition of one half of one equiv of NaSH to a DMF solution of 1. The resultant UV-vis spectrum with λmax at 413 nm and at 298 nm (Figure 1b) matched that of the intermediate formed during the titration shown in Figure 1a. Similarly, the 8.57 ppm and 7.85 ppm resonances in the 1H NMR spectrum matched those generated in situ during NMR titrations of 1 with NaSH (Figure S1). Although the downfield 1H NMR resonances of 3 suggested that the nitro groups were intact, recent reports on the H2S-mediated reduction of nitro groups to amines prompted unambiguous structural determination.8,42,43 Single crystals of 3 suitable for X-ray diffraction were grown from CHCl3/hexanes and confirmed no reduction of NO2 groups of compound 3 occurred (Figure 3).
Figure 3.
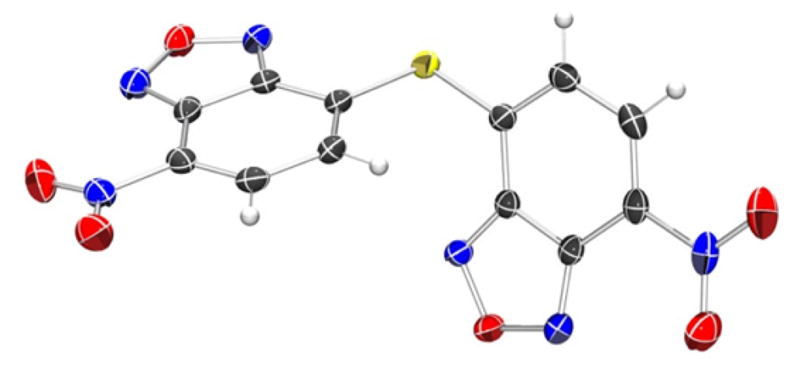
ORTEP diagram of the X-ray crystal structure of 3. Thermal ellipsoids are shown at 50% probability levels. Selected crystallographic data and metrics: P21/c, a = 13.1415(13) Å, b = 11.4080(11) Å, c = 9.3240(9) Å; β = 107.601(2)°; GOF = 1.054; R1 = 0.0360; NBD-NBD planes: 61.1°; C-S-C: 101.5°.
Based on the intermediate formation of 3 during the titration of 1 with H2S, we postulated that HS− is sufficiently nucleophilic to attack the ipso carbon of 3 to yield 2. Using isolated 3, we investigated this reactivity directly by titrating NaSH to a solution of 3 in pH 7.4 PIPES buffer at 37 °C. The reaction was monitored by both UV-vis (Figure 4) and 1H NMR spectroscopy (see Supporting Information). As expected from the titration of 1 with NaSH, addition of 1 equiv of NaSH to a solution of 3 resulted in formation of two equiv of 2, confirming both the overall stoichiometry of the reaction and the high electrophilicity of thioether 3.44
Figure 4.
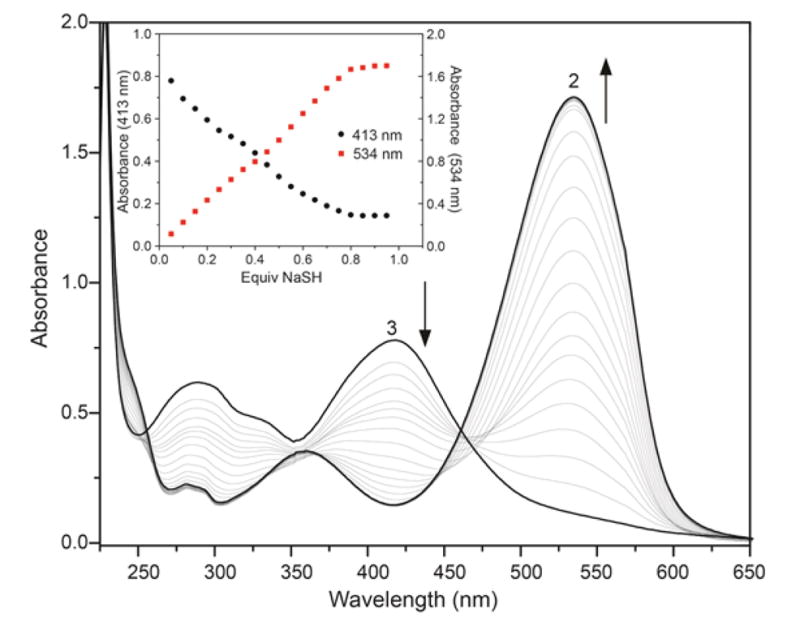
UV-vis absorption spectra of the reaction of 3 with NaSH to generate 2. Conditions: 66 μM 3, 50 mM PIPES, 100 mM KCl, pH 7.4, 37 °C, titration up to 1.0 equiv of NaSH. Insert: UV-vis absorbances at 413 nm and 534 nm during the course of the titration.
Scope and Mechanistic Investigations
To further understand the mechanism of reaction towards H2S, the electrophilicity of NBD-thioethers, and to determine the generality of this reaction platform for H2S detection, we prepared a variety of differently-substituted NBD-thioethers and investigated their reactivity with H2S. Treatment of 1 with different thiols in DMF with excess K2CO3 afforded NBD-thioether adducts 4–7 (Scheme 3). The resultant thioethers (4–7) all reacted cleanly with NaSH in pH 7.4 PIPES buffer to afford 2 and one equiv of the extruded thiol (Scheme 2), which demonstrated the tolerance for both electron donating and withdrawing groups. Kinetic data from the reaction of 4–7 with 20 equiv of NaSH under pseudo first-order conditions were used to construct a Hammett plot. Based on the proposed SN2Ar mechanism of the reaction, we expected that electron withdrawing groups appended to the benzene ring would facilitate the nucleophilic attack of HS− on 3. Construction of a Hammett plot using σp values45 revealed a positive slope with ρ = +0.34, consistent with the proposed SN2Ar mechanism (Figure 5). This value demonstrates that although the reaction is facilitated by electron withdrawing groups, different substitutions on the arene are readily tolerated while maintaining reaction rates amenable to real-time H2S detection.
Scheme 3.
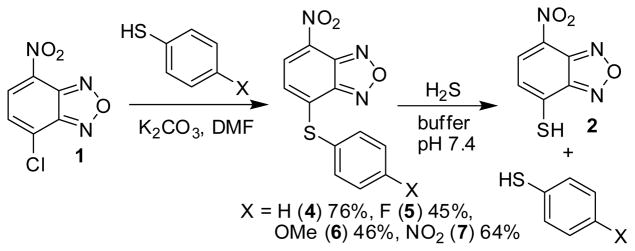
Reactivity of NBD-thioethers with H2S generates 2 and the associated starting thiol.
Figure 5.
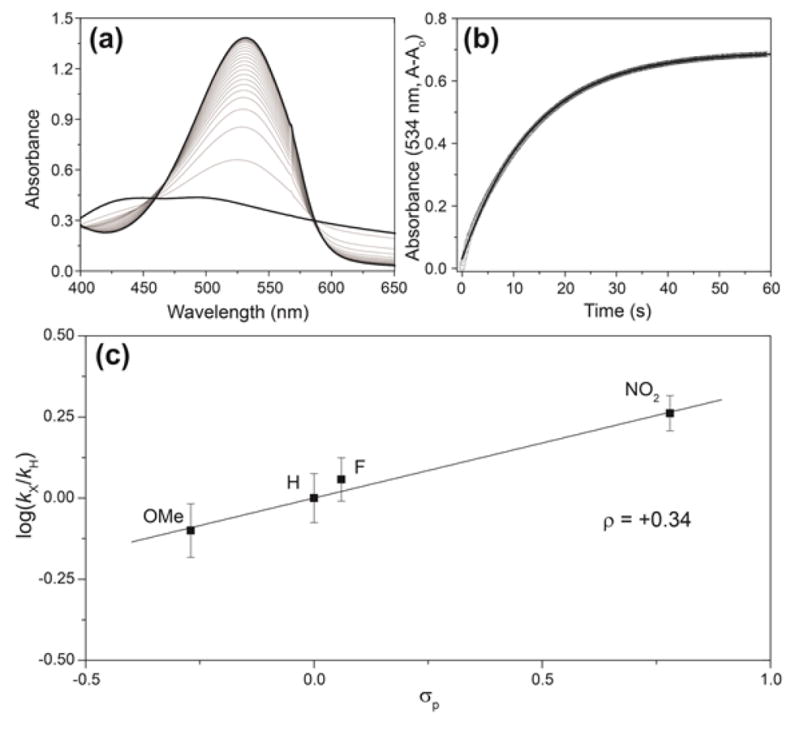
(a) Representative example of the reaction of thioether with excess NaSH. (b) Time course data of the absorbance at 534 nm fit directly to the first-order rate equation. (c) Hammett plot of the reaction of NBD-thioether adducts 4–7 with NaSH under pseudo first order conditions. Conditions: 67 μM thioether, 20 equiv of NaSH, 50 mM PIPES, 100 mM KCl, pH 7.4, 25.0 °C. Each point on the plot represents the average of at least five independent kinetic experiments.
The overall reactivity of 1 toward both H2S and thiols is summarized in Scheme 4. Treatment of 1 with sub-stoichiometric H2S forms a mixture of 2 and 3, consistent with the titration results shown in Figure 1a. This mixture can be completely converted to 2 by stoichiometric treatment with H2S. Similarly, thioethers such as 4–7 react with H2S to generate 2 with concomitant extrusion of one equiv of the corresponding aryl thiol. Thioether 3 also reacts with thiols, such as PhSH, to generate one equiv of a nonsymmetric thioether and one equiv of 2. This reactivity of benzofurazan sulfides is consistent with the recent report of sulfide-thiol exchange between NBD-sulfides and N-acetylcysteine methyl ester as a probe for cysteine residues in proteins.46 Although the NBD thioethers react readily with thiols or H2S at physiological pH, no reaction with amine or alcohol nucleophiles is observed, thus highlighting the tolerance of this reaction platform for thiol/H2S reactions over other potential biological nucleophiles.
Scheme 4.

Overall reactivity of 1 with NaSH and RSH compounds. The resultant thioether (3) formed after reaction with NaSH is sufficiently electrophilic to react further with NaSH or other thiols.
Colorimetric H2S Detection and Quantification
Having characterized the reaction chemistry of both 1 and NBD thioethers 3–7 toward H2S, we viewed the characteristic color change upon treatment with H2S as a potential platform for colorimetric H2S detection and quantification. Although the methylene blue assay,27,29 a colorimetric method for H2S detection and quantification, has been used extensively to quantify biological H2S, recent studies suggest that the initially reported detection limit (~10 nM) is erroneously low and may actually be closer to 2 μM.16 Most new colorimetric methods for H2S detection have used similar strategies to those used for fluorescence H2S detection. For example, the reduction of azide or nitro groups, precipitation of Cu(II), or attack on activated electrophiles has been employed to generated ratiometric colorimetric probes.10,21,30,47–49 Although additional methods are beginning to emerge,50,51 new selective H2S detection strategies are needed. Based on the fast response of 1 toward H2S and the characteristic color of 2, we sought to establish 1 as a colorimetric probe for H2S. To establish the ability of 1 to act as a ‘naked-eye’ H2S detector, we treated 1 with 50 equiv of different amino acids or reactive sulfur, oxygen, and nitrogen species. Although reaction of 1 with cysteine or GSH results in a light yellow color due to formation of NBD-SR thioethers, only H2S generates the characteristic purple color corresponding to 2 (Figure 6). These results demonstrate that 1 can be utilized to visually detect H2S without the aid of any instrumentation.
Figure 6.
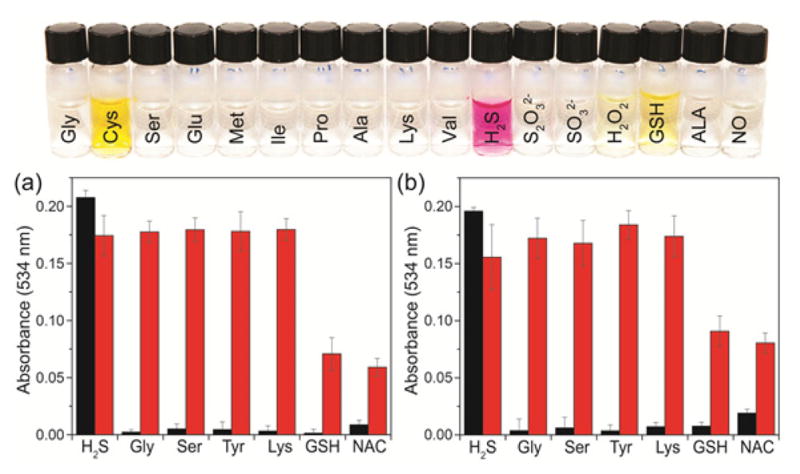
Top: Colorimetric response of 1 toward H2S over other amino acids and reactive sulfur, oxygen, and nitrogen species (RSONS). From left to right: L-glycine, L-cysteine, L-serine, L-glutamic acid, L-methionine, L-isoleucine, L-proline, L-alanine, L-lysine, L-valine, NaSH (H2S donor), Na2S2O3, Na2SO3, glutathione, H2O2, α-lipoic acid, S-nitroso-N-acetyl-DL-penicillamine (SNAP, an NO donor). Conditions: 50 μM 1, 50 equiv of RSONS and amino acids, 50 mM PIPES, 100 mM KCl, pH 7.0, 37 °C. Bottom: Colorimetric response of 1 (a) and 7 (b) for H2S after pre-treatment of the probes with biological nucleophiles. Bars show 3 μM probe after 10 equiv of analyte addition (red), followed by 30 equiv of H2S and incubation at 45 °C for 8 min (red). Conditions: 50 mM PIPES, 100 mM KCl, pH 7.4, 37 °C.
To test the selectivity of both 1 and 7 for H2S over other biologically-relevant nucleophiles, 3 μM of each probe was pre-incubated with 10 equiv of H2S, glycine, serine, tyrosine, lysine, glutathione, and N-acetyl-L-cysteine. After this initial incubation, only the H2S samples showed the characteristic absorbance peak at 534 nm, which was consistent with the visual H2S detection studies. To test whether the biologically-relevant nucleophiles deactivated the probes toward H2S, we then added 30 equiv of H2S and incubated the probes for 45 min.52 In all cases, a robust colorimetric response to H2S was observed even after incubation with biological nucleophiles (Figure 7). Although the H2S response of probes 1 and 7 was somewhat eroded after incubation of GSH and N-acetylcysteine, these studies demonstrate a proof of principle for nucleophilic displacement ligated biologically-relevant thiols by H2S. Similarly, the selectivity for H2S over other biologically-relevant nucleophiles, either separately or in competition, demonstrates the high fidelity of the nucleophilic aromatic substitution method for H2S detection.
Figure 7.
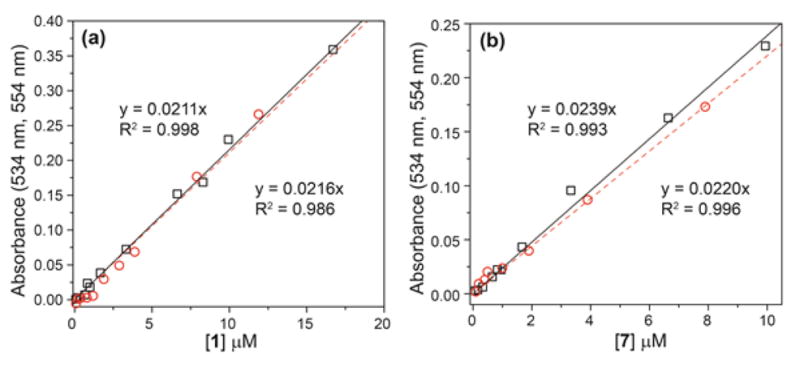
Linearity of the colorimetric response to H2S for 1 (a) and 7 (b). Detection of H2S in buffer (□, solid line) and in serum (○, dashed line).
Based on the characteristic absorbance of 2 at 534 nm, we determined the detection limit of 1 for H2S in PIPES buffer. Because of the sigmoidal response of electrophilic nitrobenzofurazans due to initial formation of 3 prior to formation of 2, as demonstrated in Figure 1a, we added a 5-fold excess of H2S to each sample. Under these conditions, a highly linearity response of 1 with H2S was observed, with a corresponding detection limit (3σ) of 210 ± 40 nM H2S (Figure 5a). Similarly, we tested the detection limit of 7 for H2S under identical conditions, which revealed a H2S detection limit of 190 ± 60 nM H2S (Figure 5b). We also tested the efficacy of 1 and 7 to detect H2S in biological media by testing the reactivity and detection limit of both probes toward H2S in fetal bovine serum (FBS). In FBS, reaction of 2 or 7 toward H2S was identical to that observed in buffer, but the λmax of the H2S reaction product 2 shifted to 554 nm. We attribute this shift in λmax to association of 2 with proteins present in FBS. Titration of FBS into a solution of 2 demonstrated that this bathochromic shift is complete after approximately 3% FBS by volume (Figure S2). Following the same procedure as in PIPES buffer, we determined the H2S detection limits of 1 and 7 in FBS to be 380 ± 60 nM and 440 ± 40 nM, respectively. Importantly, both of these ranges are well below the reported range of biologically-relevant H2S concentrations including reported levels of 5–100 μM in blood and 50–160 μM in brain homogenates.53–56 The low detection limits, fast reaction times, and characteristic product absorbance make both 1 and 7 robust compounds for colorimetric H2S detection that can be easily adapted to use with a plate reader for both H2S detection and quantification.
In addition to the low detection limits of H2S detection, the reaction mechanism of H2S with 1 or 7, which proceeds through initial formation of thioether 3 before formation of 2, allows for H2S quantification. If the concentration of H2S is below that of the NBD electrophile, then thioether 3 is the main species in solution. If the concentration of H2S is above that of the NBD electrophile, however, then 2 is the major species in solution. This dichotomy results in a sigmoidal colorometric response to H2S under conditions in which the concentration of probe is constant and the concentration of H2S is modulated. As a proof of principle for this quantification strategy, we held the concentration of 1 constant at 16 μM and added varying amounts of H2S and measured the resultant absorbance of each sample at 534 nm (Figure 8). The resultant data was fit to a sigmoidal curve to determine the H2S concentration. In all cases excellent recovery was obtained and the H2S concentrations calculated from the sigmoidal fit matched well with the known concentrations of H2S thus demonstrating the efficacy of H2S quantification with the developed probes.
Figure 8.
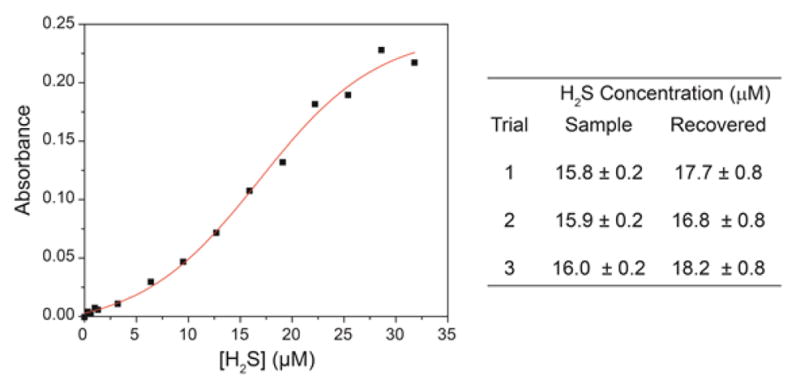
Treatment of 1 with varying amounts of H2S results in a sigmoidal generation of 2. Fitting the sigmoidal curve allows for H2S concentrations to be quantified.
Conclusions
We have shown that NBD-Cl, as well as other NBD-derived electrophiles, reacts with H2S to form both NBD thioether 3 and NBD thiol 2 and have characterized the mechanism and generality of the reaction. Based on the strong colorimetric response of 1 to H2S, we established that 1 or thioether 7 can be used for the colorimetric detection of H2S. The ease of use, fast reaction times, and characteristic absorbance of 2 makes this method attractive for both biological and environmental applications. Significantly, this H2S detection strategy offers a platform on which the reactivity of H2S and thiols can be separated, and allows for facile H2S detection in the presence of biologically-relevant nucleophiles. We are currently expanding the analytical capacity of this method with modified systems, which will be reported in due course.
Experimental Section
Materials and Methods
Flash chromatography was performed using silica gel and an automated flash chromatography instrument. Thin-layer chromatography (TLC) was performed on silica gel plates (250 μm thickness) and viewed by UV illumination. NMR spectra were acquired on either a 500 or 600 MHz spectrometer at 25.0 °C. Chemical shifts are reported in parts per million (δ) and are referenced to residual protic solvent resonances. The following abbreviations are used in describing NMR couplings: (s) singlet, (d) doublet, (t) triplet, (m) multiplet and (b) broad. NMR spectra of NaSH-containing solutions were prepared under nitrogen in sealable J-Young NMR tubes. UV–visible spectra were acquired on a UV-vis spectrophotometer equipped with a dual cuvette temperature controller and fluorescence spectra were obtained on a spectrofluorimeter equipped with a cuvette temperature controller. All spectroscopic measurements were made under anaerobic conditions, with solutions prepared under an inert atmosphere in septum-sealed cuvettes obtained from Starna Scientific.
Spectroscopic Materials and Methods
Piperazine-N,N′-bis(2-ethanesulfonic acid) and KCl were used to prepare buffered solutions (50 mM PIPES, 100 mM KCl, pH 7.4) with Millipore water. Buffered solutions were deoxygenated by vigorous sparging with nitrogen for at least two hours and were stored in an inert atmosphere glove box. DMSO was degassed by three freeze-pump-thaw cycles and stored under nitrogen. All samples for spectroscopic measurements were prepared in an N2-filled glove box with O2 levels less than 1.0 ppm. Anhydrous sodium hydrogen sulfide (NaSH, purity ~98%, Strem) was handled under nitrogen. Note: Hydrogen sulfide and its salts are highly toxic and should be handled carefully to avoid exposure. A recent review of safe handling procedures for H2S is cited.57 S-Nitroso-N-acetyl-DL-penicillamine (SNAP) was stored at −30 °C prior to use. Stock solutions of NBD-containing compounds were prepared in deoxygenated DMSO and stored in aliquots at −25 °C under nitrogen until immediately prior to use.
General procedure for Hammett rate studies
Stock solutions of each thioether (10 mM) in DMSO and cuvettes containing 3.0 mL of pH 7.4 PIPES buffer, a stir bar, and a septum cap were prepared in a glove box. Prior to each UV-vis experiment, the cuvette was allowed to equilibrate to 25.0 °C for 5 min in the sample holder. After equilibration, 20 equiv of NaSH from a 10 mM NaSH stock solution was added to the cuvette by syringe. The reaction progress was monitored by collection of UV-vis absorption data at the λmax of the either the reactant or the product in 0.1 second intervals. The raw data were fit directly to a first-order decay to obtain pseudo first-order rate constants for the reactions. All fits maintained R2 > 0.99, and the rates reported are the average of at least five independent experiments.
General procedure for detection and quantification studies
Stock solutions of each probe (100 mM, 10 mM, and 1 mM) in DMSO and cuvettes containing 3.0 mL of pH 7.4 PIPES buffer, a stir bar, and a septum cap were prepared in a glove box. Different concentrations of 1 or 4 were added to each cuvette and an initial UV-vis spectrum was recorded, after which H2S was added. After incubation for 30 min at room temperature, the absorbance spectrum was acquired. For quantification studies, H2S was added to a series of cuvettes containing a range of concentrations of 1. After incubation for 30 min at room temperature, the absorbance spectrum was recorded. Plotting the absorbance as a function of probe concentration allowed for H2S quantification after fitting to a sigmoidal curve.
General procedure for pH titration
A 15 mL solution of 2 (111 μM) in 100 mM KCl and 10 mM KOH Millipore water was prepared. The pH of the solution was adjusted with 10 M, 5 M, 1 M, 50 mM, or 1 mM solutions of HCl and the pH recorded. At each pH value, an aliquot of the solution was transferred to a cuvette and the UV-vis spectrum was measured. After measurement, the aliquot was returned to the stock solution and the pH was adjusted to the next point in the titration.
General procedure for selectivity studies
Stock solutions of each probe (10 mM) in DMSO, amino acids (10 mM) in pH 7.4 PIPES, cuvettes containing 3.0 mL of pH 7.4 PIPES buffer, a stir bar, and a septum cap were prepared in a glove box. Amino acids were added to individual cuvettes containing 3.0 mL of pH 7.4 PIPES buffer (50 mM PIPES, 100 mM KCl), stirred/shook for 45 min at 37 °C. H2S was added to cuvettes by syringe and cuvettes were stirred/shook for 8 min at 45 °C, then at 37 °C for 37 min. Data was acquired before amino acid addition, 45 min after amino acid addition, and 45 min after H2S addition.
X-ray Data Collection and Structure Solution Refinement Single crystals of 3 suitable for X-ray diffraction were grown by layering hexanes onto a CHCl3 solution of 3. Diffraction data were collected using Mo Kα radiation (λ = 0.710 73 Å) at 173(2) K. Data reduction was performed with SAINT and empirical absorption corrections were applied with SADABS.58 All refinements were performed using the SHELXTL (6.10) software package. The molecular structure was solved by direct methods and was refined using full-matrix least-squares procedures on F2.59,60 All non-hydrogen atoms were located and their positions refined anisotropically. Hydrogen atoms were found from the residual density map and were refined with isotropic thermal parameters.
Syntheses
7-Nitrobenzo[c][1,2,5]oxadiazole-4-thiol (2). NBD-Cl (1) (50.0 mg, 0.251 mmol) was dissolved in 2.0 mL of degassed MeOH. NaSH (38.0 mg, 0.501 mmol) was dissolved in 2.0 mL of degassed MeOH and added to the solution of 1. The reaction mixture was stirred at room temperature under nitrogen for 15 min and then purged with nitrogen to remove any unreacted H2S. The MeOH was removed under vacuum, to afford the desired product as a dark purple powder (46 mg, 92% yield). TLC Rf = 0.67 (SiO2, 85:15 CH2Cl2:MeOH). 1H NMR (600 MHz, MeOD) δ: 8.18 (d, J = 8.7 Hz, 1H), 7.21 (d, J = 8.7 Hz, 1H) 13C{1H} NMR (150 HMz, MeOD) δ: 175.6, 154.1, 143.5, 130.7, 128.1, 122.9. MS-ESI (m/z): [M-H]− calcd for [C6 H2 N3 O3 S]−, 196.0; found 196.2.
bis(7-Nitrobenzo[c][1,2,5]oxadiazol-4-yl)sulfane (3). NBD-Cl (1) (300 mg, 1.50 mmol) was dissolved in 2.5 mL of degassed MeOH. NaSH (42.1 mg, 0.752 mmol) was dissolved in 2.5 mL of degassed MeOH and added drop-wise to the solution of 1. The reaction mixture was stirred at room temperature under nitrogen for 3 h and then purged with nitrogen to remove any unreacted H2S. The MeOH was removed under vacuum, and the product was purified by SiO2 chromatography (100% CH2Cl2) to afford the desired product as a yellow powder (80.3 mg, 30% yield). TLC Rf = 0.61 (SiO2, CH2Cl2). 1H NMR (500 MHz, 3:2 CD2Cl2:MeOD) δ: 8.54 (d, J = 7.5 Hz, 1H), 7.82 (d, J = 7.5 Hz, 1H). 13C{1H} NMR (125 MHz, DMSO) δ: 150.3, 143.7, 136.7, 133.8, 132.2, 129.9. MS-ESI (m/z): [M+Cl]− calcd for [C12H4ClN6O6S]−, 395.0; found 395.0.
General procedure for the synthesis of NBD-thioether adducts (4–7). NBD-Cl (30.0 mg, 0.150 mmol) and K2CO3 (20.7 mg, 0.150 mmol) were added to 3.0 mL of degassed DMF. The desired substituted benzenethiol (0.150 mmol) was added to the reaction mixture, and the resultant reaction mixture was stirred at room temperature for 16 h under nitrogen. The reaction mixture was diluted with 5 mL of H2O, and the crude product was extracted with Et2O (3 × 15 mL), dried over MgSO4, and the solvent was removed under vacuum. If required, the final product was purified by chromatography on SiO2.
4-Nitro-7-(phenylthio)benzo[c][1,2,5]oxadiazole (4). Purified by SiO2 chromatography (100% CH2Cl2) to yield a dark orange powder. Yield: 31.0 mg, 76%. TLC Rf = 0.71 (SiO2, CH2Cl2). 1H NMR (500 MHz, CDCl3) δ: 8.24 (d, J = 8.3 Hz, 1H), 7.68 (d, J = 7.8 Hz, 2H), 7.60 (m, 3H), 6.64 (d, J = 7.8 Hz, 1H). 13C{1H} NMR (125 MHz, CDCl3) δ: 148.4, 142.8, 142.5, 135.7, 131.3, 130.7, 130.7, 126.2, 121.3. Elemental Analysis: calcd (%) for C12H7N3O3S: C, 52.74; H, 2.58; N, 15.38. Found: C, 52.51; H, 2.80, N 15.09.
4-(4-Fluorophenylthio)-7-nitrobenzo[c][1,2,5]oxadiazole (5). Purified by SiO2 chromatography (100% CH2Cl2) to yield a yellow powder. Yield: 19.5 mg, 45%. TLC Rf = 0.72 (SiO2, 3:1 hexanes:EtOAc). 1H NMR (500 MHz, CDCl3) δ: 8.29 (d, J = 8.3 Hz, 1H), 7.72 (m, 2H), 7.34 (m, 2H), 6.67 (d, J = 8.3 Hz, 1H). 13C{1H} NMR (125 MHz, CDCl3) δ: 164.6 (1JCF = 249 Hz), 148.4 142.5, 138.1 (4JCF = 8.3 Hz), 133.1, 130.6, 128.3, 121.4, 121.3, 118.2 (3JCF = 25.3 Hz). Elemental Analysis: calcd (%) for C12H6FN3O3S·0.5H2O: C, 48.00; H, 2.35; N, 13.99. Found: C, 48.42; H, 2.18, N 14.13.
4-(4-Methoxyphenylthio)-7-nitrobenzo[c][1,2,5]oxadiazole (6), a dark orange powder. Yield: 20.8 mg, 46%. TLC Rf = 0.51 (SiO2, 3:1 hexanes:EtOAc). 1H NMR (500 MHz, 3:1 MeOD:CDCl3) δ: 8.31 (d, J = 7.8 Hz, 1H), 7.59 (d, J = 7.3 Hz, 2H), 7.11 (d, J = 8.8 Hz, 2H), 6.67 (d, J = 7.8 Hz, 1H), 3.89 (s, 3H, OMe). 13C{1H} NMR (125 MHz, CDCl3) δ: 162.1, 148.4, 144.1, 142.5, 137.4, 132.7, 130.8, 121.0, 116.3, 116.2, 55.6. Elemental Analysis: calcd (%) for C13H9N3O4S: C, 51.48; H, 2.99; N, 13.85. Found: C, 51.39; H, 3.00, N 13.79
4-Nitro-7-(4-nitrophenylthio)benzo[c][1,2,5]oxadiazole (7), a yellow powder. Yield: 30.0 mg, 64%. TLC Rf = 0.73 (SiO2, CH2Cl2). 1H NMR (500 MHz, CDCl3) δ: 8.39 (d, J = 8.3 Hz, 2H), 8.32 (d, J = 7.8 Hz, 1H), 7.85 (d, J = 8.8 Hz, 2H), 6.98 (d, J = 7.8 Hz, 1H). 13C{1H} NMR (125 MHz, CDCl3) δ: 149.1, 148.8, 142.6, 138.5, 135.5, 130.3, 126.4, 125.3, 124.5, 124.1. Elemental Analysis: calcd (%) for C12H6N4O5S: C, 45.29; H, 1.90; N, 17.60. Found: C, 44.90; H, 1.99, N 17.35.
Supplementary Material
Acknowledgments
This work was supported by the National Institute of General Medical Sciences (R00 GM092970 to MDP) and funding from the University of Oregon. The NMR facilities at the University of Oregon are supported by NSF/ARRA CHE-0923589.
Footnotes
Supporting Information. NMR spectra of H2S reaction, FBS titration, NMR spectra of new compounds, X-ray crystallographic data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Li L, Rose P, Moore PK. In: Annual Review of Pharmacology and Toxicology. Cho AK, editor. Vol. 51. 2011. p. 169. [DOI] [PubMed] [Google Scholar]
- 2.Olson KR. Antioxid Redox Signaling. 2012;17:32. doi: 10.1089/ars.2011.4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang R. Curr Opin Nephrol Hy. 2011;20:107. doi: 10.1097/MNH.0b013e3283430651. [DOI] [PubMed] [Google Scholar]
- 4.Wang R. Physiol Rev. 2012;92:791. doi: 10.1152/physrev.00017.2011. [DOI] [PubMed] [Google Scholar]
- 5.Kabil O, Banerjee R. J Biol Chem. 2010;285:21903. doi: 10.1074/jbc.R110.128363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen S, Chen ZJ, Ren W, Ai HW. J Am Chem Soc. 2012;134:9589. doi: 10.1021/ja303261d. [DOI] [PubMed] [Google Scholar]
- 7.Lippert AR, New EJ, Chang CJ. J Am Chem Soc. 2011;133:10078. doi: 10.1021/ja203661j. [DOI] [PubMed] [Google Scholar]
- 8.Montoya LA, Pluth MD. Chem Commun. 2012;48:4767. doi: 10.1039/c2cc30730h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xuan WM, Sheng CQ, Cao YT, He WH, Wang W. Angew Chem Int Ed. 2012;51:2282. doi: 10.1002/anie.201107025. [DOI] [PubMed] [Google Scholar]
- 10.Yu FBA, Li P, Song P, Wang BS, Zhao JZ, Han KL. Chem Commun. 2012;48:2852. doi: 10.1039/c2cc17658k. [DOI] [PubMed] [Google Scholar]
- 11.Lin VS, Lippert AR, Chang CJ. Proc Natl Acad Sci USA. 2013;110:7131. doi: 10.1073/pnas.1302193110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu CR, Pan J, Li S, Zhao Y, Wu LY, Berkman CE, Whorton AR, Xian M. Angew Chem Int Ed. 2011;50:10327. doi: 10.1002/anie.201104305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu CR, Peng B, Li S, Park CM, Whorton AR, Xian M. Org Lett. 2012;14:2184. doi: 10.1021/ol3008183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qian Y, Karpus J, Kabil O, Zhang SY, Zhu HL, Banerjee R, Zhao J, He C. Nat Commun. 2011;2:495. doi: 10.1038/ncomms1506. [DOI] [PubMed] [Google Scholar]
- 15.Wintner EA, Deckwerth TL, Langston W, Bengtsson A, Leviten D, Hill P, Insko MA, Dumpit R, VandenEkart E, Toombs CF, Szabo C. Br J Pharmacol. 2010;160:941. doi: 10.1111/j.1476-5381.2010.00704.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shen XG, Pattillo CB, Pardue S, Bir SC, Wang R, Kevil CG. Free Radical Biol Med. 2011;50:1021. doi: 10.1016/j.freeradbiomed.2011.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qian Y, Zhang L, Ding S, Deng X, He C, Zheng XE, Zhu HL, Zhao J. Chem Sci. 2012;3:2920. [Google Scholar]
- 18.Chen YC, Zhu CC, Yang ZH, Chen JJ, He YF, Jiao Y, He WJ, Qiu L, Cen JJ, Guo ZJ. Angew Chem Int Ed. 2013;52:1688. doi: 10.1002/anie.201207701. [DOI] [PubMed] [Google Scholar]
- 19.Choi MG, Cha S, Lee H, Jeon HL, Chang SK. Chem Commun. 2009:7390. doi: 10.1039/b916476f. [DOI] [PubMed] [Google Scholar]
- 20.Sasakura K, Hanaoka K, Shibuya N, Mikami Y, Kimura Y, Komatsu T, Ueno T, Terai T, Kimura H, Naganot T. J Am Chem Soc. 2011;133:18003. doi: 10.1021/ja207851s. [DOI] [PubMed] [Google Scholar]
- 21.Zhang DQ, Jin WS. Spectrochim Acta A. 2012;90:35. doi: 10.1016/j.saa.2012.01.013. [DOI] [PubMed] [Google Scholar]
- 22.Lin VS, Chang CJ. Curr Opin Chem Biol. 2012;16:595. doi: 10.1016/j.cbpa.2012.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu Z, Xu L, Zhou J, Xu YF, Zhu WP, Qian XH. Chem Commun. 2012;48:10871. doi: 10.1039/c2cc36141h. [DOI] [PubMed] [Google Scholar]
- 24.Doeller JE, Isbell TS, Benavides G, Koenitzer J, Patel H, Patel RP, Lancaster JR, Darley-Usmar VM, Kraus DW. Anal Biochem. 2005;341:40. doi: 10.1016/j.ab.2005.03.024. [DOI] [PubMed] [Google Scholar]
- 25.Garcia-Calzada M, Marban G, Fuertes AB. Anal Chim Acta. 1999;380:39. [Google Scholar]
- 26.Wardencki W. J Chromatogr A. 1998;793:1. [Google Scholar]
- 27.Lawrence NS, Davis J, Compton RG. Talanta. 2000;52:771. doi: 10.1016/s0039-9140(00)00421-5. [DOI] [PubMed] [Google Scholar]
- 28.Ubuka T. J Chromatogr B. 2002;781:227. doi: 10.1016/s1570-0232(02)00623-2. [DOI] [PubMed] [Google Scholar]
- 29.Fogo JK, Popowsky M. Anal Chem. 1949;21:732. [Google Scholar]
- 30.Zhao Y, Zhu X, Kan H, Wang W, Zhu B, Du B, Zhang X. Analyst. 2012;137:5576. doi: 10.1039/c2an36106j. [DOI] [PubMed] [Google Scholar]
- 31.Bain R, Bartram J, Elliott M, Matthews R, McMahan L, Tung R, Chuang P, Gundry S. In J Environ Res Publ. 2012;9:1609. doi: 10.3390/ijerph9051609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McMahan L, Grunden AM, Devine AA, Sobsey MD. Water Res. 2012;46:1693. doi: 10.1016/j.watres.2011.12.037. [DOI] [PubMed] [Google Scholar]
- 33.Terrier F. Chem Rev. 1982;82:77. [Google Scholar]
- 34.Uchiyama S, Santa T, Okiyama N, Fukushima T, Imai K. Biomed Chromatogr. 2001;15:295. doi: 10.1002/bmc.75. [DOI] [PubMed] [Google Scholar]
- 35.Ellis HR, Poole LB. Biochemistry. 1997;36:15013. doi: 10.1021/bi972191x. [DOI] [PubMed] [Google Scholar]
- 36.Hao F, Lwin T, Bruckard WJ, Woodcock JT. J Chromatogr A. 2004;1055:77. doi: 10.1016/j.chroma.2004.08.130. [DOI] [PubMed] [Google Scholar]
- 37.Sigg RH, Luisi PL, Aboderin AA. J Biol Chem. 1977;252:2507. [PubMed] [Google Scholar]
- 38.Ferguson SJ, Lloyd WJ, Radda GK. Eur J Biochem. 1975;54:127. doi: 10.1111/j.1432-1033.1975.tb04121.x. [DOI] [PubMed] [Google Scholar]
- 39.Sutton R, Ferguson SJ. Eur J Biochem. 1984;142:387. doi: 10.1111/j.1432-1033.1984.tb08299.x. [DOI] [PubMed] [Google Scholar]
- 40.All titrations were performed under an anaerobic conditions to prevent H2S oxidation and to ensure proper stoichiometry, but it is important to note that the described reactivity occurs equally well under standard aerobic conditions.
- 41.Ahnoff M, Grundevik I, Arfwidsson A, Fonselius J, Persson BA. Anal Chem. 1981;53:485. doi: 10.1021/ac00226a024. [DOI] [PubMed] [Google Scholar]
- 42.Gronowitz S, Westerlund C, Hornfeldt AB. Acta Chem Scand A. 1975;B 29:224. [Google Scholar]
- 43.Nickson TE. J Org Chem. 1986;51:3903. [Google Scholar]
- 44.During the initial phase of the reaction, the isosbestic point at 447 nm shifts to 463 nm, which we attribute to partial formation of the -complex of HS– or liberated 2 with 3 during the titration.
- 45.Hansch C, Leo A, Taft RW. Chem Rev. 1991;91:165. [Google Scholar]
- 46.Li Y, Yang Y, Guan X. Anal Chem. 2012;84:6877. doi: 10.1021/ac301306s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu MY, Li K, Hou JT, Huang Z, Yu XQ. Org Biomol Chem. 2012;10:8342. doi: 10.1039/c2ob26235e. [DOI] [PubMed] [Google Scholar]
- 48.Gu XF, Liu CH, Zhu YC, Zhu YZ. Tetrahedron Lett. 2011;52:5000. [Google Scholar]
- 49.Liu J, Sun YQ, Zhang J, Yang T, Cao J, Zhang L, Guo W. Chem Eur J. 2013;19:4717. doi: 10.1002/chem.201300455. [DOI] [PubMed] [Google Scholar]
- 50.Wallace KJ, Cordero SR, Tan CP, Lynch VM, Anslyn EV. Sensor Actuat B-Chem. 2007;120:362. [Google Scholar]
- 51.Zhang J, Xu X, Yang X. Analyst. 2012;137:1556. doi: 10.1039/c2an16307a. [DOI] [PubMed] [Google Scholar]
- 52.Samples were incubated for 45 min at 37 °C prior to analysis for the best reproducibility. We found that increasing the incubation temperature to 45 °C during the first 8 min of incubation resulted in greater signal and reproducibility.
- 53.Abe K, Kimura H. J Neurosci. 1996;16:1066. doi: 10.1523/JNEUROSCI.16-03-01066.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Goodwin LR, Francom D, Dieken FP, Taylor JD, Warenycia MW, Reiffenstein RJ, Dowling G. J Anal Toxicol. 1989;13:105. doi: 10.1093/jat/13.2.105. [DOI] [PubMed] [Google Scholar]
- 55.Richardson CJ, Magee EAM, Cummings JH. Clin Chim Acta. 2000;293:115. doi: 10.1016/s0009-8981(99)00245-4. [DOI] [PubMed] [Google Scholar]
- 56.Yusuf M, Huat BTK, Hsu A, Whiteman M, Bhatia M, Moore PK. Biochem Biophys Res Commun. 2005;333:1146. doi: 10.1016/j.bbrc.2005.06.021. [DOI] [PubMed] [Google Scholar]
- 57.Hughes MN, Centelles MN, Moore KP. Free Radical Biol Med. 2009;47:1346. doi: 10.1016/j.freeradbiomed.2009.09.018. [DOI] [PubMed] [Google Scholar]
- 58.Sheldrick GM. University of Gttingen; Gttingen, Germany: 2008. [Google Scholar]
- 59.Sheldrick GM. University of Gttingen; Gttingen, Germany: 2000. [Google Scholar]
- 60.Sheldrick GM. Acta Crystallogr, Sect A. 2008;64:112. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.