

Abstract
The evolutionary origins of Madagascar's biodiversity remain mysterious despite the fact that relative to land area, there is no other place with consistently high levels of species richness and endemism across a range of taxonomic levels. Most efforts to explain diversification on the island have focused on geographical models of speciation, but recent studies have begun to address the island's accumulation of species through time, although with conflicting results. Prevailing hypotheses for diversification on the island involve either constant diversification rates or scenarios where rates decline through time. Using relative-time-calibrated phylogenies for seven endemic vertebrate clades and a model-fitting framework, I find evidence that diversification rates have declined through time on Madagascar. I show that diversification rates have clearly declined throughout the history of each clade, and models invoking diversity-dependent reductions to diversification rates best explain the diversification histories for each clade. These results are consistent with the ecological theory of adaptive radiation, and, coupled with ancillary observations about ecomorphological and life-history evolution, strongly suggest that adaptive radiation was an important formative process for one of the most species-rich regions on the Earth. These results cast the Malagasy biota in a new light and provide macroevolutionary justification for conservation initiatives.
Keywords: adaptive radiation, diversity-dependent diversification, diversification rate, island biogeography, Madagascar
1. Introduction
Madagascar accounts for less than 1% of the world's land area, but hosts nearly 3% of all plant and animal species; relative to land area, there is no other place with such consistently high levels of endemism across a range of taxonomic levels [1]. Madagascar's large size and long period of isolation from neighbouring land masses clearly contributed to this spectacular diversity [2–4], but the evolution of this diversity remains poorly understood. To understand the evolution of Madagascar's biodiversity, authors have primarily focused on historical patterns of gene flow and geographical models of speciation, such as riverine barriers or refugial isolation during periods of climate change, but support for these models is mixed [5–9]. Only recently have studies explored the temporal aspects of species accumulation through time on the island.
Recent studies exploring species diversification on Madagascar have arrived at seemingly conflicting conclusions. On the one hand, studies of vanga birds and day geckos [10–12] suggest that species accumulation declines over time, supporting the a model of adaptive radiation, where speciation and adaptation act in concert to produce distinctive new forms until ecological opportunities are exhausted [13–15]. On the other hand, a study that included the majority of Malagasy vertebrate clades suggested that diversification rates have been continuous throughout history, possibly indicating the absence of diversification limits on an island as large and diverse as Madagascar, or that Madagascan radiations have not yet reached whatever limits to their diversification may exist [3].
The possibly conflicting results of these studies are symptomatic of other studies addressing the role of constant versus declining diversification rates in island biogeography. The classic MacArthur & Wilson [16,17] model of island biogeography maintains that speciation is a negligible source of species diversity over short time scales and certain geographical contexts, and so species richness emerges as a dynamic equilibrium between the opposing processes of immigration and extinction. However, an ever-growing body of work reveals in situ speciation is often the dominant source of species on islands [18–21], and evolutionary studies of island biogeography have begun to address the impact of diversification on diversity patterns [18,20]. Yet no study has employed a comparative approach to examine temporal diversification patterns for clades that radiated on the same island and in the same habitats. Such an approach may be particularly informative, because it may be impossible to disentangle the vagaries of history for comparisons across regions.
Madagascar is an ideal setting to evaluate the importance of constant or declining diversification rates for several reasons. First, many lineages have undergone extensive in situ diversification during the island's long period of isolation from the other land masses (65–80 Ma [22]), producing endemic clades. Second, relative to many other large islands, Madagascar has remained remarkably geologically cohesive and isolated [23]. Third, many of Madagascar's endemic radiations are taxonomically and phylogenetically well characterized [3]. Simply put, Madagascar is an ideal evolutionary experiment because its endemic clades provide replication, and its relative stability and isolation may expose processes general to the diversification of life.
Here, I present the first comparative analysis of Malagasy diversification patterns using methods capable of distinguishing between alternative diversification models. I use previously published molecular phylogenetic data to quantify temporal patterns of diversification in seven species-rich in situ radiations of Malagasy amphibians and reptiles [6,24–30]. These datasets are phylogenetically well sampled for extant species and include geographically broad intraspecific sampling that facilitates identification of putatively cryptic species (table 1) [7,9], and the clades are sufficiently well studied that current estimates of species diversity are not the result of taxonomic inflation. I analyse these datasets with Bayesian relaxed clock methodology to produce trees with branch lengths proportional to time and quantified diversification patterns with maximum likelihood.
Table 1.
Summary of taxonomic sampling and MCCR analyses. Currently recognized numbers are from Crottini et al. [3]. Because I often included additional lineages, I note the number of tips used in each analysis. Note that for Brookesia, Gephyromantis and Phelsuma I present analyses including putative cryptic species that are not included in published checklists of species counts from which the final column is derived. Additionally, I agree with Main et al. [31] that there is at least one additional undescribed or unrecognized Paroedura that was not included by Crottini et al. [3], and as well as with Raxworthy et al. [26] that the taxon Uroplatus giganteus requires a more thorough systematic evaluation as it is not clearly differentiated from neighbouring populations of Uroplatus frimbriatus. MCCR analyses: γ—the value for the MCC tree with the mean γ for the posterior distribution in parentheses. p-values are for the MCC tree assuming the number of tips in the analysis is just a fraction of the true diversity for the clade. Values in parentheses are the percentage of posterior trees with a significant γ-value (i.e. p < 0.05). Bold values indicate significant results.
| summary of taxonomic sampling |
summary of MCCR analyses |
||||||
|---|---|---|---|---|---|---|---|
| clade | study | included species | total Malagasy species* | γ | p at 25% | p at 50% | p at 100% |
| Brookesia | Townsend et al. [6] | 31 | 32 | −3.899 (−3.818, −3.801) | 0.016 (86.29%) | 0.001 (100%) | < 0.001 (100%) |
| Cophylinae | Wollenberg et al. [27] | 61 | 86 | −5.763 (−5.549, −5.531) | 0.001 (99.93%) | < 0.001 (100%) | < 0.001 (100%) |
| Gephyromantis | Kaffenberger et al. [30] | 52 | 51 | −3.739 (−3.596, −3.579) | 0.124 (1.5%) | 0.006 (99.18%) | < 0.001 (100%) |
| Heterixalus | Wollenberg et al. [24] | 9 | 9 | −2.227 (−1.972, −1.942) | 0.08 (16.03%) | 0.032 (44.86%) | 0.013 (76.32%) |
| Paroedura | Jackman et al. [25] | 15 | 20 | −3.248 (−2.908, −2.890) | 0.013 (62.69%) | 0.002 (98.05%) | 0.001 (99.88%) |
| Phelsuma | Rocha et al. [28] | 32 | 34 | −3.418 (−3.380, −3.365) | 0.071 (20.23%) | 0.006 (99.78%) | < 0.001 (100%) |
| Uroplatus | Raxworthy et al. [26] | 19 | 19 | −3.375 (−3.260, −3.247) | 0.02 (90.08%) | 0.002 (100%) | < 0.001 (100%) |
2. Material and methods
(a). Focal clades
I searched the literature for molecular phylogenies of Malagasy radiations, requiring that clades be nearly completely endemic to the island and well sampled for recognized diversity, limiting myself to seven clades of Malagasy reptiles and amphibians that are both species-rich and distributed island-wide: Brookesia leaf chameleons, microhylid frogs from the subfamily Cophylinae, Gephryomantis leaf frogs, Heterixalus reed frogs, Paroedura ground geckos, Phelsuma day geckos and Uroplatus leaf-tailed geckos (table 1) [6,24–30]. Each of these studies employed geographical sampling to assess cryptic species diversity of widespread species as well as samples not readily assigned to named species. Taxonomic sampling of currently recognized diversity was no less than 70% (Cophylinae). These datasets span taxonomic orders from genera (and subgenera) to subfamilies. Ideally, I would have preferred to analyse the maximum level of inclusiveness for these groups (e.g. all Madagascan chameleon genera, or the entirely endemic family Mantellidae and not simply Gephyromantis), but the data necessary for such analyses are not available (missing data and saturation would bias branch lengths). Among the chosen datasets, only Paroedura and Phelsuma have dispersed or radiated outside of Madagascar, and biogeographic reconstructions generally reveal a Malagasy origin with clear instances of dispersal away from, and not to, Madagascar (electronic supplementary material, figures S1–S7). All datasets consisted of both nuclear and mitochondrial loci except the cophyline microhylids [27], for which only mitochondrial sequences were available.
(b). Phylogenetic analyses
To obtain trees with branch lengths scaled to reflect time, I re-analysed each published dataset using the Bayesian relaxed clock methodology implemented by the program BEAST v. 1.6.1 [32,33]. Analyses of Brookesia used the same BEAST input file from the original study. For the other clades, I generated input files with the following four-step protocol. First, I pruned sequences with incomplete data that may bias branches towards longer lengths [34], but constructed a chimeric sequence from two individuals of Uroplatus malahelo to avoid the loss of this taxon. I realigned sequences with MAFFT [35] when necessary. Second, I generated maximum-likelihood starting trees for BEAST analyses with RAxML v. 7.2.6 [36,37], in each case conducting multiple independent RAxML searches to ensure the algorithm did not become trapped on local optima. For RAxML analysis, I partitioned datasets by gene, or codon and gene where applicable, and applied the GTR+Γ model to each partition. Third, I rendered RAxML starting trees compatible with BEAST priors using the penalized likelihood function chronopl from ape [38,39], with a smoothing parameter (λ) of 1, and rescaled these with an arbitrary root age of 100 units using the geiger function rescaleTree [40]. Fourth, I partitioned the data by gene, or codon and gene where applicable, and applied a GTR+Γ model of sequence evolution to each partition. All BEAST analyses also shared a lognormal-relaxed clock with a uniform rate prior on 0–105 units, a uniform root height prior arbitrarily set to 95–105 units, and a Yule process prior on node heights. I ran each dataset for 20 000 000 generations and sampled chains every 10 000 generations. I repeated this analysis four times independently for each dataset and ultimately pooled the trees from the posterior distribution of each analysis. I assessed stationarity and convergence with TRACER v. 1.5 [41] and AWTY [42], ultimately discarding the first 10 000 000 generations from each run as burn-in. I adopted this uniform approach as I found this sample of trees sufficiently recovered the results of the previous analyses and enabled me to formulate an analytical pipeline for any number of clades (R scripts and associated functions are included in the electronic supplementary material). I calculated the maximum clade credibility (MCC) tree with node heights set to the mean of the 4004 posterior trees.
After phylogeny reconstruction, I followed standard procedures for the analysis of in situ radiations [13,15] by pruning all tips resulting from dispersal out of Madagascar (i.e. Indian Ocean Phelsuma and the Comoran Paroedura sanctijohanis), as these are not expected to influence diversification dynamics on Madagascar. I also pruned duplicate intraspecific samples from the MCC tree and the entire posterior distribution, and relied primarily on the species boundaries diagnosed by the authors of the original studies to do so. However, I also retained lineages from paraphyletic species evident on the MCC tree. These lineages are often not included in taxonomic assessments. Brookesia thieli, for instance, is rendered paraphyletic by the morphologically diagnosed species Brookesia betschi, Brookesia lineata and Brookesia vadoni, so I retained exemplars from each distinct B. thieli lineage (see electronic supplementary material, figures S1–S7 for more information). This strategy is the most conservative when it comes to rejecting rate decline models as it tends to include additional nodes between the midpoint and tips. Tips pruned for diversification analyses are illustrated in the electronic supplementary material, figures S1–S7.
(c). Diversification analyses
I tested the hypothesis that diversification rates decline throughout the history of a clade with two complementary methods. First, I implemented Pybus & Harvey's [43] Monte Carlo constant rates (MCCR) test. Second, I used a maximum-likelihood model-fitting framework to compare the fit of models having a constant diversification rate with models that assume a decline in diversification rate over time while accounting for incomplete taxonomic sampling [44]. I accounted for phylogenetic uncertainty by calculating diversification statistics for the MCC tree and a sample of the posterior distribution.
(i). Constant rates test
Pybus & Harvey's [43] constant rates test computes the γ statistic, which compares the distribution of inter-node distances between the root and the temporal midpoint of the tree to the distances between the midpoint and the tips. Nodes are distributed evenly throughout the tree and γ is normally distributed with a mean of 0 when clades evolve under the null model of a constant rate Yule process. Negative values indicate that inter-node distances between the root and the midpoint are shorter than distances closer to the tips, consistent with a decline in the rate of species accumulation over time. Assuming complete sampling of extant species, the critical γ value for identifying a significant decline in the rate of species accumulation is −1.645 [43]. Application of a pure birth model (i.e. no extinction) is unlikely to reject the null model due to a high background rate of extinction alone because extinction rates increase the expected value of γ [43]. Incomplete taxon sampling is known to increase type I error rates of this test, so I employed the MCCR test [43] and generated corrected γ distributions through simulation (with my own R code) assuming actual taxon sampling accounts for 25–75% of each clade's true diversity. I calculated γ for the MCC tree and every tree in the posterior distribution.
Recent work has shown that the MCCR test may not be sufficient if tips are randomly pruned from simulated trees because the shallowest divergences between cryptic species are mostly likely to be excluded from molecular phylogenies [45,46]. However, this bias is unlikely to characterize these datasets because the original authors included geographically broad intraspecific sampling to estimate cryptic species diversity. Species missing from these phylogenies have probably not been collected and do not reflect a systemic bias towards particular phylogenetic scales. In addition to accounting for incomplete taxon sampling, I also tested for molecular saturation to ensure the γ statistics were not biased towards negative values [47]. I examined each of the datasets for evidence of molecular saturation by plotting uncorrected distances against maximum-likelihood-corrected distances (from RAxML) for all sequence partitions, with the expectation that saturated sites will display a nonlinear, asymptotic decline. Only third position sites from the Cophylinae dataset show evidence of a nonlinear relationship, but they have not levelled off (data not shown).
(ii). Maximum-likelihood and model fitting
I used the maximum-likelihood model-fitting approach of Etienne et al. [44] to test four alternative hypotheses about species diversification over time. This approach uses a hidden Markov model that enables consideration of both missing species as well as species that went extinct but nevertheless might have influenced diversification rates, thus my results for these analyses are comparable to the MCCR test in their consideration of missing taxa. Two models assume constant rates: pure birth (Yule) and constant rate birth–death process (crBD). The remaining two assume diversity dependence and test if diversification slows as diversity increases while accounting for extinction; one models exponentially declining speciation rates as a function of lineage diversity (DDE+E) and the other is a logistic model, with linear rate declines (DDL+E). I fitted diversification models in R using functions from the DDD package [44] (all R code is available in the electronic supplementary material). Because of computational limitations, I limited my likelihood analyses to the MCC tree and 49 randomly chosen posterior trees. For each included tree, I fitted models assuming both complete sampling of extant diversity (100% completeness) and that the included species account for only half of the total extant diversity (50% completeness). I was unable to calculate these statistics for 25% completeness because of computational times due to consideration of extreme amounts of missing species.
I used the Akaike information criterion [48] to assess the fit of each model. I calculated AIC weights (wi), which are estimates of the conditional probabilities of each model [49]. I use these conditional probabilities to interpret the goodness of fit of each model relative to the whole candidate set.
3. Results
(a). Monte Carlo constant rates test
The MCCR test detected significant rate declines for all clades even when I assumed considerably more incomplete taxonomic sampling than is likely to characterize these clades (table 1). Indeed, γ values are significantly negative for each clade even after assuming that only 50% of total diversity is currently sampled. For Brookesia, Cophylinae, Paroedura and Uroplatus, γ-values remain significant even assuming that only 25% of the current species diversity has been sampled (table 1). Furthermore, with the exception of Gephyromantis, other clades are only marginally insignificant at this level of incomplete sampling (0.08 > p > 0.001; table 1). These results are consistent and expected from the highly concave lineage through time (LTT) plots that characterize the history of speciation for each clade (figure 1a).
Figure 1.
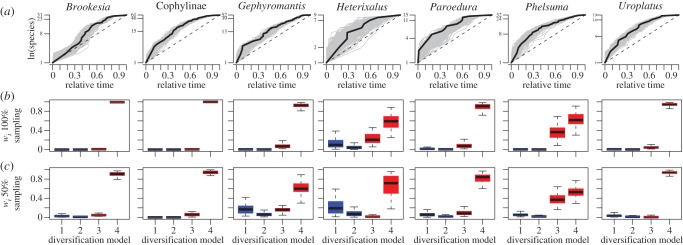
Summary of diversification analyses. (a) LTT plots for each clade; the light lines depict 200 random posterior trees, the dark line depicts the MCC tree and the dashed line depicts the Yule expectation. (b,c) Boxplots depict the AIC weights (wi) assuming different levels of taxonomic sampling for each model from 49 randomly selected posterior trees. Models 1–4 are Yule, crBD, DDE+E, DDL+E, respectively (see text for more details). Constant rate models are numbered 1 and 2, whereas models invoking declining rates are numbered 3 and 4. All box plots are drawn without outliers. (Online version in colour.)
(b). Diversification models
Maximum-likelihood diversification models strongly support rate decline models for each clade. Akaike weights for each MCC tree favour the DDL+E model, where diversification rates decline linearly as a function of increasing lineage diversity. Assuming only 50% completeness, weight values for the MCC range between 0.56 and ≈1.0 for this model (see the electronic supplementary material, table S1). These results are robust to phylogenetic uncertainty and 50% incomplete taxonomic sampling as the 49 random posterior trees show similar patterns, where Akaike weights for the DDL+E model are typically the highest, and confidence intervals for this model do not overlap with any constant rate models; the only model with any overlap is DDE+E (figure 1b,c). The only other model that received comparable levels of support in the model-fitting analyses is the DDE+E model (see the electronic supplementary material, table S1). These results are robust to 50% incomplete taxonomic sampling, and are consistent with the hypothesis that diversification on Madagascar is diversity-dependent and ultimately limited by ecological opportunity.
4. Discussion
In each of seven clades of reptiles and amphibians endemic to Madagascar, I reject the hypothesis of constant rates of species diversification over time and recover strong support for the alternative hypothesis that accumulation of species diversity has steadily declined (table 1 and figure 1a–c; electronic supplementary material, table S1). LTT plots, the MCCR test and maximum-likelihood diversification models all reveal declining diversification rates, and all analyses are robust to phylogenetic uncertainty and incomplete taxonomic sampling.
It seems unlikely that these results are due to bias stemming from phylogenetic uncertainty, saturation of DNA substitutions or incomplete taxon sampling (due to oversight or recent extinction). First, the majority of trees sampled from the posterior distribution of the Bayesian phylogenetic analyses for each clade favour the DDL+E model (see the electronic supplementary material, table S1; figure 1b,c). Although the DDE+E and DDL+E models are not strongly distinguished among posterior trees for Phelsuma, the posterior distribution marginally favours the DDL+E model (see the electronic supplementary material, table S1; figure 1b,c). Second, inspection of saturation plots reveals that the sequence data are not strongly saturated, and that potential issues with saturation are largely resolved via use of highly parametrized models of sequence evolution. Third, all of the datasets in my analyses include complete or nearly complete sampling of currently recognized species as well as numerous putative cryptic species diagnosed via extensive geographical sampling of widespread forms (table 1) [3,7], so I conjecture these trees are not biased away from shallow divergences. Moreover, the constant rates test reveals a general signature of declining rates assuming that only 25% of the current species diversity has been sampled (table 1), a pattern reflected in the computationally intensive likelihood analyses assuming 50% complete sampling (see the electronic supplementary material, table S1; figure 1b,c). Similarly, while recent extinction has undoubtedly touched many Malagasy clades, these data do not show evidence of substantial extinction, which would cause an excess of short tip lengths and impart the signature of accelerating diversification rates [43], and the likelihood methods explicitly accommodate the existence of extinct species on diversification dynamics. I also note that the evidence for diversity dependence is likely to be much greater, as these trees were formed with a Yule prior on node heights. Thus, the phylogenies themselves are already biased towards constant rate models. I also examined trees produced with a conditioned birth–death prior with computationally less intensive diversification analyses [50] and found no qualitative difference among priors (see the electronic supplementary material, table S6).
The type of diversity-dependent diversification I observed among Madagascan reptiles and amphibians is not likely from neutral processes [51], but is consistent with the ecological theory of adaptive radiation [13–15,52]. Although my study addresses only one prediction of the ecological theory of adaptive radiation (declining speciation rates), my results, along with recent studies of vanga birds [10,11], are consistent with adaptive radiation being the source of many endemic radiations on Madagascar. Although additional work is required to quantitatively test this theory's predictions about adaptation and specialization, many of the clades included in my study appear to exhibit evidence for these aspects of adaptive radiation. For instance, the cophylines have repeatedly diverged along a habitat gradient to produce fossorial to completely arboreal specialists with associated morphologies and life histories [53], and other Malagasy amphibian clades not examined here show remarkable patterns of life-history evolution consistent with adaptive radiation [54,55]. All the clades examined here show a range of body size diversity, and many contain habitat specialists (e.g. Brookesia, Phelsuma and Uroplatus [56]).
Diversity-dependent declines may not be distinguishable from time-dependent declines, as might happen through protracted speciation, environmental change or neutral processes, but time dependence does not seem supported by these data for two reasons. First, it is difficult to imagine how time-based models might consistently account for rate declines across clades of different ages and sizes, like those examined [3,4,6,26,57], through external forces (e.g. changing environments), as observed here [52,58]. Second, additional model-based analyses that do not incorporate missing taxa uniformly reject models where diversification rates change as a function of time (approximating neutral models [21,58]) and not necessarily lineage diversity (electronic supplementary material, table S6).
This study adds to a body of recent work exploring the temporal dimensions of diversification on the island. Combined with previous studies of vangas and day geckos [10–12], as well as a growing catalogue of studies showing that declining diversification rates occur across a wide range of other taxa and regions [20,59–61], my results demonstrate that diversification rate declines are a general macroevolutionary pattern and are consistent with adaptive radiation having been an important formative process for one of the most species-rich regions on Earth. The only possible Malagasy exception to this trend is a study including the majority of Malagasy vertebrate clades that favours ongoing rates of species accumulation [3] because of an association between clade age and species richness. However, this study did not explicitly test alternative models of cladogenesis over time. Indeed, an association between clade age and richness may still be evident if clades have not yet, or just recently, approached equilibrium levels of diversity. The model-fitting analyses presented here provide a finer resolution of diversification dynamics on the island and no other study has presented a comparative perspective of diversification on Madagascar coupled with the methods capable of deciphering declining diversification rates. Future investigations should apply these methods to other Malagasy radiations.
Two additional patterns warrant discussion. First, Crottini et al. [3] recovered evidence of accelerated diversification in clades that radiated within rainforests, where the majority of the included clades are found. Among the clades I analysed, only Heterixalus and Paroedura do not have the majority of their species occurring within rainforests. I believe this shared result indicates our studies contribute different perspectives of the same underlying pattern. Second, many Malagasy species, including those examined here, contain considerable levels of genetic structure which may reflect incipient species. However, the role of intraspecific divergence in clade-level dynamics remains difficult to ascertain with the data and methods used here, but diversity dependence may still underlie diversification if newly emerging, incipient species are less likely to occupy novel niches [58]. Future studies addressing the rates at which this diversity accrues, as well as its relationship to the process of speciation, might be particularly informative.
My investigation of the history of diversification on Madagascar casts the island in a paradoxical light: although its endemic diversity has led to recognition as a biodiversity hotspot [1], Madagascar does not appear to be a hotspot for ongoing evolutionary diversification. Instead, Madagascar's endemic reptile and amphibian radiations, and perhaps bird radiations as well, appear to comprise relatively ancient species. This temporal, macroevolutionary perspective on Madagascan species diversification provides evolutionary justification for efforts to conserve Madagascar's biodiversity by highlighting that human conservation efforts may affect the constitution of the Malagasy biota over the short term and the long term.
Acknowledgements
I thank Katharina C. Wollenberg, Christopher J. Raxworthy and Ted M. Townsend, who provided access to their datasets. I am grateful for critical feedback on an early version of this manuscript provided by Daniel Rabosky and Ted Townsend. Daniel Rabosky also provided informative discussions about the methods used in the study, and Rampal S. Etienne provided considerable help with implementing the maximum-likelihood analyses. Richard E. Glor provided comments to previous drafts that substantially improved the quality of the paper. Additionally, I wish to thank Julienne Ng, Anthony Geneva, Audrey Kelly, Ryane Logdson, and Robert Minckley for comments on previous drafts of the manuscript, as well as the efforts of four reviewers.
Funding statement
This research was supported by funding from the National Science Foundation (DEB-1110605 and DEB-0920892).
References
- 1.Myers N, Mittermeier RA, Mittermeier CG, da Fonseca GAB, Kent J. 2000. Biodiversity hotspots for conservation priorities. Nature 403, 853–858 (doi:10.1038/35002501) [DOI] [PubMed] [Google Scholar]
- 2.Vences M, Wollenberg KC, Vieites DR, Lees DC. 2009. Madagascar as a model region of species diversification. Trends Ecol. Evol. 24, 456–465 (doi:10.1016/j.tree.2009.03.011) [DOI] [PubMed] [Google Scholar]
- 3.Crottini A, Madsen O, Poux C, Strauss A, Vieites DR, Vences M. 2012. Vertebrate time-tree elucidates the biogeographic pattern of a major biotic change around the K–T boundary in Madagascar. Proc. Natl Acad. Sci. USA 109, 5358–5363 (doi:10.1073/pnas.1112487109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Samonds KE, Godfrey LR, Ali JR, Goodman SM, Vences M, Sutherland MR, Irwin MT, Krause DW. 2013. Imperfect isolation: factors and filters shaping Madagascar's extant vertebrate fauna. PLoS ONE 8, e62086 (doi:10.1371/journal.pone.0062086) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Raxworthy CJ, Ingram CM, Rabibisoa N, Pearson RG. 2007. Applications of ecological niche modeling for species delimitation: a review and empirical evaluation using day geckos (Phelsuma) from Madagascar. Syst. Bio. 56, 907–923 (doi:10.1080/10635150701775111) [DOI] [PubMed] [Google Scholar]
- 6.Townsend TM, Vieites DR, Glaw F, Vences M. 2009. Testing species-level diversification hypotheses in Madagascar: the case of microendemic Brookesia leaf chameleons. Syst. Biol. 58, 641–656 (doi:10.1093/sysbio/syp073) [DOI] [PubMed] [Google Scholar]
- 7.Vieites DR, Wollenberg KC, Andreone F, Kohler J, Glaw F, Vences M. 2009. Vast underestimation of Madagascar's biodiversity evidenced by an integrative amphibian inventory. Proc. Natl Acad. Sci. USA 106, 8267–8272 (doi:10.1073/pnas.0810821106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ruedi M, Weyeneth N, Goodman SM. 2011. Do diversification models of Madagascar's biota explain the population structure of the endemic bat Myotis goudoti (Chiroptera: Vespertilionidae)? J. Biogeogr. 38, 44–54 (doi:10.1111/j.1365-2699.2010.02393.x) [Google Scholar]
- 9.Yoder AD, et al. 2005. A multidimensional approach for detecting species patterns in Malagasy vertebrates. Proc. Natl Acad. Sci. USA 102(Suppl. 1), 6587–6594 (doi:10.1073/pnas.0502092102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reddy S, Driskell A, Rabosky DL, Hackett SJ, Schulenberg TS. 2012. Diversification and the adaptive radiation of the vangas of Madagascar. Proc. R. Soc. B 279, 2062–2071 (doi:10.1098/rspb.2011.2380) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jønsson KA, et al. 2012. Ecological and evolutionary determinants for the adaptive radiation of the Madagascan vangas. Proc. Natl Acad. Sci. USA 109, 6620–6625 (doi:10.1073/pnas.1115835109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harmon LJ, Melville J, Larson A, Losos JB. 2008. The role of geography and ecological opportunity in the diversification of day geckos (Phelsuma). Syst. Biol. 57, 562–573 (doi:10.1080/10635150802304779) [DOI] [PubMed] [Google Scholar]
- 13.Glor RE. 2010. Phylogenetic insights on adaptive radiation. Annu. Rev. Ecol. Evol. Syst. 41, 251–270 (doi:10.1146/Annurev.Ecolsys.39.110707.173447) [Google Scholar]
- 14.Losos JB. 2010. Adaptive radiation, ecological opportunity, and evolutionary determinism. American Society of Naturalists E. O. Wilson Award address. Am. Nat. 175, 623–639 (doi:10.1086/652433) [DOI] [PubMed] [Google Scholar]
- 15.Schluter D. 2000. The ecology of adaptive radiation, p. 288 Oxford, UK: Oxford University Press [Google Scholar]
- 16.MacArthur RH, Wilson EO. 1963. An equilibrium theory of insular zoogeography. Evolution 17, 373–387 (doi:10.2307/2407089) [Google Scholar]
- 17.MacArthur RH, Wilson EO. 1967. The theory of island biogeography, p. 203 Princeton, NJ: Princeton University Press [Google Scholar]
- 18.Losos JB, Schluter D. 2000. Analysis of an evolutionary species–area relationship. Nature 408, 847–850 (doi:10.1038/35048558) [DOI] [PubMed] [Google Scholar]
- 19.Kisel Y, Barraclough TG. 2010. Speciation has a spatial scale that depends on levels of gene flow. Am. Nat. 175, 316–334 (doi:10.1086/650369) [DOI] [PubMed] [Google Scholar]
- 20.Rabosky DL, Glor RE. 2010. Equilibrium speciation dynamics in a model adaptive radiation of island lizards. Proc. Natl Acad. Sci. USA 107, 22 178–22 183 (doi:10.1073/pnas.1007606107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rosindell J, Phillimore AB. 2011. A unified model of island biogeography sheds light on the zone of radiation. Ecol. Lett. 14, 552–560 (doi:10.1111/j.1461-0248.2011.01617.x) [DOI] [PubMed] [Google Scholar]
- 22.Ali JR, Aitchison JC. 2008. Gondwana to Asia: plate tectonics, paleogeography and the biological connectivity of the Indian sub-continent from the Middle Jurassic through latest Eocene (166–35 Ma). Earth Sci. Rev. 88, 145–166 (doi:10.1016/J.Earscirev.2008.01.007) [Google Scholar]
- 23.de Wit MJ. 2003. Madagascar: heads it's a continent, tails it's an island. Annu. Rev. Earth Planet. Sci. 31, 213–248 (doi:10.1146/Annurev.Earth.31.100901.141337) [Google Scholar]
- 24.Wollenberg KC, Glaw F, Meyer A, Vences M. 2007. Molecular phylogeny of Malagasy reed frogs, Heterixalus, and the relative performance of bioacoustics and color-patterns for resolving their systematics. Mol. Phylogenet. Evol. 45, 14–22 (doi:10.1016/j.ympev.2007.06.024) [DOI] [PubMed] [Google Scholar]
- 25.Jackman TR, Bauer AM, Greenbaum E, Glaw F, Vences M. 2008. Molecular phylogenetic relationships among species of the Malagasy-Comoran gecko genus Paroedura (Squamata: Gekkonidae). Mol. Phylogenet. Evol. 46, 74–81 (doi:10.1016/j.ympev.2007.10.018) [DOI] [PubMed] [Google Scholar]
- 26.Raxworthy CJ, Pearson RG, Zimkus BM, Reddy S, Deo AJ, Nussbaum RA, Ingram CM. 2008. Continental speciation in the tropics: contrasting biogeographic patterns of divergence in the Uroplatus leaf-tailed gecko radiation of Madagascar. J. Zool. 275, 423–440 (doi:10.1111/j.1469-7998.2008.00460.x) [Google Scholar]
- 27.Wollenberg KC, Vieites DR, van der Meijden A, Glaw F, Cannatella DC, Vences M. 2008. Patterns of endemism and species richness in Malagasy cophyline frogs support a key role of mountainous areas for speciation. Evolution 62, 1890–1907 (doi:10.1111/j.1558-5646.2008.00420.x) [DOI] [PubMed] [Google Scholar]
- 28.Rocha S, Vences M, Glaw F, Posada D, Harris DJ. 2009. Multigene phylogeny of Malagasy day geckos of the genus Phelsuma. Mol. Phylogenet. Evol. 52, 530–537 (doi:10.1016/j.ympev.2009.03.032) [DOI] [PubMed] [Google Scholar]
- 29.Rocha S, Rosler H, Gehring PS, Glaw F, Posada D, Harris DJ, Vences M. 2010. Phylogenetic systematics of day geckos, genus Phelsuma, based on molecular and morphological data (Squamata: Gekkonidae). Zootaxa 2429, 1–28 [Google Scholar]
- 30.Kaffenberger N, Wollenberg KC, Kohler J, Glaw F, Vieites DR, Vences M. 2012. Molecular phylogeny and biogeography of Malagasy frogs of the genus Gephyromantis. Mol. Phylogenet. Evol. 62, 555–560 (doi:10.1016/J.Ympev.2011.09.023) [DOI] [PubMed] [Google Scholar]
- 31.Main H, Scantlebury DP, Zarkower D, Gamble T. 2012. Karyotypes of two species of Malagasy ground gecko (Paroedura: Gekkonidae). Afr. J. Herpetol. 61, 81–90 (doi:10.1080/21564574.2012.667837) [Google Scholar]
- 32.Drummond A, Ho S, Phillips M, Rambaut A. 2006. Relaxed phylogenetics and dating with confidence. PLoS Biol. 4, e88 (doi:10.1371/journal.pbio.0040088) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Drummond AJ, Rambaut A. 2007. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7, 214 (doi:10.1186/1471-2148-7-214) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lemmon AR, Brown JM, Stanger-Hall K, Lemmon EM. 2009. The effect of ambiguous data on phylogenetic estimates obtained by maximum likelihood and Bayesian inference. Syst. Biol. 58, 130–145 (doi:10.1093/sysbio/syp017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Katoh K, Misawa K, Kuma KÄ, Miyata T. 2002. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066 (doi:10.1093/nar/gkf436) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stamatakis A, Hoover P, Rougemont J. 2008. A rapid bootstrap algorithm for the RAxML Web servers. Syst. Biol. 57, 758–771 (doi:10.1080/10635150802429642) [DOI] [PubMed] [Google Scholar]
- 37.Stamatakis A, Ludwig T, Meier H. 2005. RAxML-III: a fast program for maximum likelihood-based inference of large phylogenetic trees. Bioinformatics 21, 456–463 (doi:10.1093/bioinformatics/bti191) [DOI] [PubMed] [Google Scholar]
- 38.Paradis E, Claude J, Strimmer K. 2004. APE: Analyses of phylogenetics and evolution in R language. Bioinformatics 20, 289–290 (doi:10.1093/bioinformatics/btg412) [DOI] [PubMed] [Google Scholar]
- 39.Sanderson MJ. 2002. Estimating absolute rates of molecular evolution and divergence times: a penalized likelihood approach. Mol. Biol. Evol. 19, 101–109 (doi:10.1093/oxfordjournals.molbev.a003974) [DOI] [PubMed] [Google Scholar]
- 40.Harmon LJ, Weir JT, Brock CD, Glor RE, Challenger W. 2008. GEIGER: investigating evolutionary radiations. Bioinformatics 24, 129–131 (doi:10.1093/bioinformatics/btm538) [DOI] [PubMed] [Google Scholar]
- 41.Rambaut A, Drummond AJ. 2009. Tracer v. 1.5 See http://beast.bio.ed.ac.uk/
- 42.Nylander JA, Wilgenbusch JC, Warren DL, Swofford DL. 2008. AWTY (are we there yet?): a system for graphical exploration of MCMC convergence in Bayesian phylogenetics. Bioinformatics 24, 581–583 (doi:10.1093/bioinformatics/btm388) [DOI] [PubMed] [Google Scholar]
- 43.Pybus OG, Harvey PH. 2000. Testing macro-evolutionary models using incomplete molecular phylogenies. Proc. R. Soc. Lond. B 267, 2267–2272 (doi:10.1098/rspb.2000.1278) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Etienne RS, Haegeman B, Stadler T, Aze T, Pearson PN, Purvis A, Phillimore AB. 2012. Diversity-dependence brings molecular phylogenies closer to agreement with the fossil record. Proc. R. Soc. B 279, 1300–1309 (doi:10.1098/rspb.2011.1439) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brock CD, Harmon LJ, Alfaro ME. 2011. Testing for temporal variation in diversification rates when sampling is incomplete and nonrandom. Syst. Biol. 60, 410–419 (doi:10.1093/sysbio/syr007) [DOI] [PubMed] [Google Scholar]
- 46.Cusimano N, Renner SS. 2010. Slowdowns in diversification rates from real phylogenies may not be real. Syst. Biol. 59, 458–464 (doi:10.1093/Sysbio/Syq032) [DOI] [PubMed] [Google Scholar]
- 47.Revell LJ, Harmon LJ, Glor RE. 2005. Underparameterized model of sequence evolution leads to bias in the estimation of diversification rates from molecular phylogenies. Syst. Biol. 54, 973–983 (doi:10.1080/10635150500354647) [DOI] [PubMed] [Google Scholar]
- 48.Akaike H. 1974. A new look at the statistical model identification. Autom. Control, IEEE Trans. 19, 716–723 (doi:10.1109/TAC.1974.1100705) [Google Scholar]
- 49.Burnham KP, Anderson DR. 2002. Model selection and multimodel inference: a practical information-theoretic approach, 2nd edn, p. 488 New York, NY: Springer [Google Scholar]
- 50.Gernhard T. 2008. The conditioned reconstructed process. J. Theor. Biol. 253, 769–778 (doi:10.1016/j.jtbi.2008.04.005) [DOI] [PubMed] [Google Scholar]
- 51.Davies TJ, Allen AP, Borda-de-Agua L, Regetz J, Melian CJ. 2011. Neutral biodiversity theory can explain the imbalance of phylogenetic trees but not the tempo of their diversification. Evolution 65, 1841–1850 (doi:10.1111/j.1558-5646.2011.01265.x) [DOI] [PubMed] [Google Scholar]
- 52.Etienne RS, Haegeman B. 2012. A conceptual and statistical framework for adaptive radiations with a key role for diversity dependence. Am. Nat. 180, E75–E89 (doi:10.1086/667574) [DOI] [PubMed] [Google Scholar]
- 53.Andreone F, Vences M, Vieites DR, Glaw F, Meyer A. 2005. Recurrent ecological adaptations revealed through a molecular analysis of the secretive cophyline frogs of Madagascar. Mol. Phylogenet. Evol. 34, 315–322 (doi:10.1016/j.ympev.2004.10.013) [DOI] [PubMed] [Google Scholar]
- 54.Vences M, Andreone F, Glaw F, Kosuch J, Meyer A, Schaefer HC, Veith M. 2002. Exploring the potential of life-history key innovation: brook breeding in the radiation of the Malagasy treefrog genus Boophis. Mol. Ecol. 11, 1453–1463 (doi:10.1046/j.1365-294X.2002.01543.x) [DOI] [PubMed] [Google Scholar]
- 55.Bossuyt F, Milinkovitch MC. 2000. Convergent adaptive radiations in Madagascan and Asian ranid frogs reveal covariation between larval and adult traits. Proc. Natl Acad. Sci. USA 97, 6585–6590 (doi:10.1073/Pnas.97.12.6585) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Glaw F, Vences M. 2007. A field guide to the amphibians and reptiles of Madagascar, 3rd edn. Cologne, Germany: Vences & Glaw [Google Scholar]
- 57.van der Meijden A, Vences M, Hoegg S, Boistel R, Channing A, Meyer A. 2007. Nuclear gene phylogeny of narrow-mouthed toads (Family: Microhylidae) and a discussion of competing hypotheses concerning their biogeographical origins. Mol. Phylogenet. Evol. 44, 1017–1030 (doi:10.1016/J.Ympev.2007.02.008) [DOI] [PubMed] [Google Scholar]
- 58.Etienne RS, Rosindell J. 2012. Prolonging the past counteracts the pull of the present: protracted speciation can explain observed slowdowns in diversification. Syst. Bio. 61, 204–213 (doi:10.1093/Sysbio/Syr091) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McPeek MA. 2008. The ecological dynamics of clade diversification and community assembly. Am. Nat. 172, E270–E284 (doi:10.1086/593137) [DOI] [PubMed] [Google Scholar]
- 60.Rabosky DL, Lovette IJ. 2008. Density-dependent diversification in North American wood warblers. Proc. R. Soc. B 275, 2363–2371 (doi:10.1098/rspb.2008.0630) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rabosky DL. 2009. Ecological limits on clade diversification in higher taxa. Am. Nat. 173, 662–674 (doi:10.1086/597378) [DOI] [PubMed] [Google Scholar]