

Abstract
Metoclopramide hydrochloride (MCP) is commonly used for the management of gastrointestinal disorders. Frequent administration and the undesired side effects (extra pyramidal symptoms) of the drug on the central nervous system due to the fluctuations of its plasma concentrations may lead to patient incompliance, and hence, improper therapy. Therefore, the present work will be devoted to formulate the drug in sustained release formulations. MCP was incorporated in 12 formulae containing different polymers and/or different polymer ratios. These polymers were hydroxypropylmethyl cellulose (HPMC), carboxymethylcellulose (CMC) and ethyl cellulose (EC). Sodium starch glycolate (SSG) was added to some formulae in different amounts in order to soften and/or disintegrate the tablets. Both direct compression and granulation techniques were used to prepare the tablets. The physical properties were found to be satisfactory for all the formulae. The dissolution profiles of the tablets were constructed using the change-over method. The drug release involved a combination of both diffusion and polymer-chain relaxation mechanisms. The time required to release 50% of MCP ranged from 1.2 to more than 8 h. Direct compression and dry granulation techniques produced sufficient sustaining of the drug release. However, the pellets made by wet granulation released MCP in about 2 h, i.e., pelletization spheronization technique was not effective in sustaining the drug.
Keywords: Metoclopramide, Cellulosic polymers, Super disintegrant, Sustained release, Tablet making techniques, Release kinetics
1. Introduction
Metoclopramide hydrochloride (MCP) is a freely soluble drug and is rapidly absorbed from the gastrointestinal tract (GIT) (Martindale, 1993). It has a relatively short variable biological half life (5 ± 1 h) and is usually administered in a peroral dose of 10–15 mg four times daily, as immediate release tablets. The blood level fluctuation resulting from this frequent administration leads to extrapyramidal symptoms (USP DI, 1998).
The relatively small daily dose, short half life, undesirable side effects and rapid absorption from the GIT make MCP a good candidate for formulation in a sustained release dosage form. The hepatic first pass metabolism of the drug (Desta et al., 2002) may discourage the sustaining of its release. However, it has been found that, this metabolism is linear as revealed by the bioequivalence of a sustained release dosage form with a solution of MCP (Beckett et al., 1987). This linearity does not hinder the formulation of MCP in sustained release dosage form. Such sustaining of the drug release would obviate the secondary effects of the drug on the central nervous system normally encountered with the administration of immediate release formulations. This, together with decreasing the frequency of administration, would improve the patient compliance.
Because of low costs and ease of fabrication, one of the most common approaches to get controlled release is to embed a drug in a hydrophobic matrix such as Eudragit and ethylcellulose (Lotifipour et al., 2004) or hydrophilic matrix such as hydroxypropylcellulose, hydroxypropylmethylcellulose, methylcellulose, sodium carboxymethylcellulose, alginates and scleroglucan (Hasan et al., 2003; Winterowd and Sandford, 1995; Moe et al., 1995). Dispersing the drug in cross linked chitosan, spray dried microspheres of hydrogel-forming polymers, hydrophilic polymers and alginates have been reported in the literature to sustain MCP release (Ganza-Gonzalez et al., 1999).
In the present study, MCP was formulated in matrix tablets and pellets using varying proportions of the cellulosic polymers HPMC, CMC and EC. A super disintegrant (SSG) was employed to produce softer and/or disintegrating tablets as a desired property for matrix tablets to avoid failure of release. Moreover, the influence of the technique used, namely, direct compression, dry granulation and pelletization spheronization on tablets and pellets properties was investigated. The physical properties of the tablets and the release profiles from tablets and pellets and their relevant kinetics were determined.
2. Materials
Metoclopramide (MCP) was generously obtained from Egyptian Pharmaceutical International Company (EPICO), Egypt. Carboxymethyl cellulose sodium (CMC), high viscosity grade, and sodium starch glycolate (SSG) were purchased from BDH Co, Poole, England. Hydrxypropylmethyl cellulose (HPMC K4M) was purchased from Dow Chemical Company, Midland, Michigan, USA. Ethyl cellulose (EC) was obtained from ICN Biomedicals, Inc, Ohio, USA. Avecil PH102 was obtained from Serva GmbH & Co., Heidelberg, Germany.
3. Methods
3.1. Preparation of tablets by the direct compression technique
Components of each formula as shown in Table 1 were mixed in turbula mixer (type S27, Erweka, Apparatebau, Germany) for 15 min and then directly compressed into tablets using a single punch tablet machine (type EKO, Erweka, Apparatebau, Germany) using 10 mm flat punches. Tablets hardness was kept within the range of 5–8 kp.
Table 1.
Formulae of metoclopramide tablets made by different methods and the physical properties of the produced tablets.
| Composition (mgs) of the prepared formulations |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| F1 | F2 | F3 | F4 | F5 | F6 | F7 | F8 | F9 | F10 | F11 | F12 | |
| MCP | 30 | 30 | 30 | 30 | 30 | 30 | 30 | 30 | 30 | 30 | 30 | 30 |
| HPMC | 90 | 70 | 90 | 30 | 90 | 70 | 70 | 80 | 90 | 80 | 70 | 80 |
| CMC | 50 | – | 30 | 90 | 30 | 30 | 50 | 30 | 30 | 30 | 30 | 30 |
| SSG | – | – | – | – | 20 | 20 | 20 | 30 | 10 | 30 | 40 | 30 |
| Avecil | 30 | 50 | 50 | 50 | 30 | 50 | 30 | 30 | 40 | 30 | 30 | 30 |
| Mag stearate | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| EC | – | 50 | – | – | – | – | – | – | – | – | – | – |
| Methoda | DC | DC | DC | DC | DC | DC | DC | DC | DC | DG | DG | PS |
| Tablet weight (mg) | 202 | 202 | 202 | 202 | 202 | 202 | 202 | 202 | 202 | 202 | 202 | – |
DC, DG, PS designate direct compression, dry granulation and pelletization spheronization, respectively.
3.2. Preparation of tablets by the dry granulation technique
Components of each formula as shown in Table 1 were mixed in turbula mixer (type S27, Erweka, Apparatebau, Germany) for 5 min and then compressed using a single punch tablet machine (type EKO, Erweka, Apparatebau, Germany), using 12 mm flat punches. The produced slugs were then crushed into dry granules and passed through sieve size 800 μm and retained on sieve size 250 μm. The rest of disintegrant and lubricant (one half amount of each) were added, mixed in turbula mixer for 3 min, and then compressed by a single punch tablet machine, (type EKO, Erweka, Apparatebau, Germany) using 10 mm flat punches. Tablet hardness was kept within the range of 5–8 kp.
3.3. Preparation of pellets
Components of formula10 as shown in Table 1 were milled, and mixed in mortar. Water was gradually added to the powder mixture with trituration until the formation of coherent mass. The resulting wet mass was extruded at a speed of 85 rpm (Mini Screw Extruder, Model MSE1014, Caleva, England), through 1 mm diameter die. Spheronization was performed in a spheronizer (Model 120, Caleva, England) with a rotating plate of regular cross-hatch geometry, at a speed of 500–600 rpm, for 10 min. Pellets were then dried on a tray in an hot oven at 50 °C for 12 h.
3.4. Tablet evaluation
Hardness of 10 tablets were measured using hardness tester (type TBH28, Erweka, Apparatebau, Germany) and the mean was listed in Table 2, friability of 20 tablets were measured using friability tester (type TA3R, Erweka, Apparatebau, Germany) and the mean was listed in Table 2. Weight uniformity test of 20 tablets was carried out according to BP limits and the results were listed in Table 2.
Table 2.
Physical properties of metoclopramide tablets made by different methods.
| Formula no. | Methoda | Weight uniformity: Average tablet weight (mg ± SD) |
Hardness (kp) mean ± SD |
Friability (%) |
|---|---|---|---|---|
| 1 | DC | 203 ± 1.5 | 7.2 ± 0.40 | 0.51 |
| 2 | DC | 204 ± 1.6 | 5.8 ± 0.30 | 0.42 |
| 3 | DC | 203 ± 1.4 | 6.3 ± 0.31 | 0.48 |
| 4 | DC | 202 ± 1.6 | 6.1 ± 0.32 | 0.46 |
| 5 | DC | 203 ± 1.4 | 6.5 ± 0.29 | 0.42 |
| 6 | DC | 202 ± 1.6 | 5.9 ± 0.30 | 0.51 |
| 7 | DC | 205 ± 1.6 | 6.2 ± 0.32 | 0.43 |
| 8 | DC | 203 ± 1.4 | 6.0 ± 0.29 | 0.44 |
| 9 | DC | 204 ± 1.4 | 6.1 ± 0.32 | 0.42 |
| 10 | DG | 202 ± 1.3 | 7.5 ± 0.41 | 0.40 |
| 11 | DG | 203 ± 1.4 | 7.3 ± 0.40 | 0.39 |
DC, DG designate direct compression and dry granulation, respectively.
3.5. In vitro release studies
The dissolution runs were carried out by the change-over methods originally used in the USP for enteric coated tablets (USP DI, 1998). In each of the flasks of the USP apparatus 1 (Caleva Ltd., Model 85T) 750 ml of 0.1 N HCl (pH 1.2) were equilibrated to 37 ± 0.5 °C at 50 rpm using a continuous automated monitoring system. This system consists of an IBM computer PK8620 series and PU 8605/60 dissolution test software, Philips VIS/UV/NIR single beam eight-cell spectrophotometer Model PU 8620, Epson FX 850 printer, and Watson–Marlow peristaltic pump. An accurately weighed tablet of each of the prepared formulations was added to each flask. Samples were withdrawn at time intervals for 2 h, and then the pH was changed to 6.8 by adding 250 ml of 0.2 M trisodium phosphate. For each formula, release runs were performed in triplicate and absorbance was recorded automatically at 309 nm up to 8 h. The cumulative percentage of drug released was determined as a function of time.
3.6. Statistical analysis
The results were analyzed by using the software graph pad prism5 applying one-way ANOVA. Differences between formulations were considered to be significant at p ⩽ 0.05.
4. Results and discussion
The MCP controlled release matrix tablets were prepared using different techniques and different polymers. As shown in Table 1, the tablets gave satisfactory physical properties concerning the hardness and friability. The values of the latter lay quite below the USP maximum limits. The weight uniformity of the tablets was within the BP limits, Table 2.
4.1. Dissolution profiles of MCP from different polymer matrices
As shown in Figs. 1–3, the steepest part for all the dissolution curves is the first 2-h-segment which represents the MCP dissolution in the acid medium. This relatively fast dissolution rate may be attributed to the relatively low viscosity of celluloseic polymers at acidic pH which enhances the acid penetration into the matrix tablet, and hence, increasing MCP release. On raising the pH from 1.2 to 6.8, the penetrability of the dissolution medium decreases leading to slower drug release.
Figure 1.
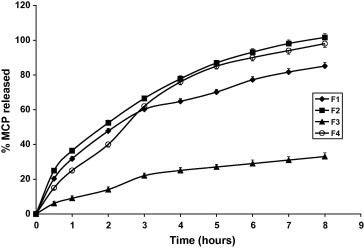
Dissolution profiles of MCP tablets in 0.1 N HCl (pH 1.2) and phosphate buffer (pH 6.8) at 37 ± 0.5 °C.
Figure 2.
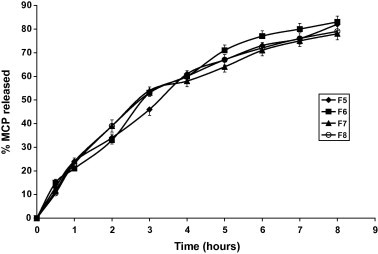
Dissolution profiles of MCP tablets in 0.1 N HCl (pH 1.2) and phosphate buffer (pH 6.8) at 37 ± 0.5 °C.
Figure 3.
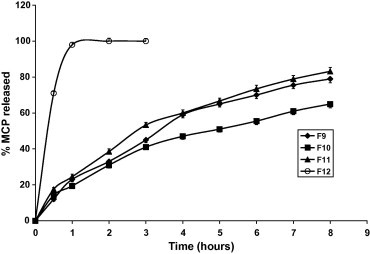
Dissolution profiles of MCP tablets and pellets in 0.1 N HCl pH (1.2) and phosphate buffer (pH 6.8) at 37 ± 0.5 °C.
Figs. 1–3 and Table 3 reveal that MCP release was the fastest from F12 (pellets) and the slowest from F3 made by direct compression and contains relatively high proportion of HPMC in absence of the disintegrant SSG. Replacing the hydrophilic polymer CMC in F1 by the hydrophobic polymer EC (F2) resulted in a significant increase in MCP release (p < 0.05) compared with F1, Table 4. This could be due to the effect of EC on the compressibility of the tablet matrix as reflected by the lower hardness value, Table 2. Comparing the values of T50% and T75% (time for 50% and 75% drug to be released) for the Formulae F3, F5 and F9 having the same composition except for SSG and made by the same technique (direct compression) reveals that the absence of SSG (F3) has drastically sustained the release (Fig. 4). Despite F5 contains double the amount of SSG present in F9, the release from F5 was non significantly increased (p > 0.05) as shown by the release rates in Table 4. This may indicate that increasing SSG above 10% would not affect anymore the release of MCP. However, it was visually observed that as the percentage of SSG in the formula is increased the tablet erosion in the dissolution medium was more apparent. For matrix tablets, more erosion is preferable (keeping the sustaining of release) because it may be helpful in avoiding release failure. If SSG helps the matrix tablet to disintegrate into granules under the hydro-dynamic conditions of the stomach, the tablet would gain the known advantages of a multi-unit dosage form (e.g. microcapsules or microspheres) including the ease of granule transfer to the intestine regardless of food content of the stomach (Gibaldi et al., 1991).
Table 3.
T50% and T75% for different formulations.
| Formula no. | T50% (h)a | T75% (h)a |
|---|---|---|
| F1 | 2.2 | 5.44 |
| F2 | 1.24 | 3.20 |
| F3 | >8 | >8 |
| F4 | 2.44 | 3.76 |
| F5 | 2.24 | 5.28 |
| F6 | 1.84 | 4.32 |
| F7 | 2.64 | 6.36 |
| F8 | 2.76 | 6.32 |
| F9 | 2.80 | 6.48 |
| F10 | 4.40 | >8 |
| F11 | 2.48 | 5.8 |
| F12 | 0.28 | 0.5 |
T50% and T75% (the time required for 50% and 75% drug to be released).
Table 4.
Kinetic modeling of MCP release from different formulations.
| Formula no. | Zero order model |
First order model |
Higuchi diffusion model |
n-value | |||
|---|---|---|---|---|---|---|---|
| r | Ko | r | K1 | r | D | ||
| F1 | 0.942 | 9.53 | 0.942 | 0.23 | 0.996 | 0.931 | 0.712 |
| F2 | 0.940 | 11.39 | 0.970 | 1.54 | 0.994 | 1.39 | 0.738 |
| F3 | 0.961 | 3.95 | 0.970 | 0.046 | 0.992 | 0.136 | 0.583 |
| F4 | 0.963 | 12.31 | 0.980 | 0.46 | 0.988 | 1.31 | 0.758 |
| F5 | 0.978 | 10.18 | 0.990 | 0.21 | 0.989 | 0.87 | 0.722 |
| F6 | 0.968 | 10.35 | 0.991 | 0.23 | 0.990 | 0.92 | 0.725 |
| F7 | 0.953 | 9.21 | 0.990 | 0.184 | 0.993 | 0.78 | 0.720 |
| F8 | 0.954 | 9.51 | 0.992 | 0.184 | 0.992 | 0.825 | 0.728 |
| F9 | 0.959 | 7.38 | 0.990 | 0.185 | 0.959 | 0.78 | 0.720 |
| F10 | 0.966 | 7.38 | 0.980 | 0.07 | 0.998 | 0.491 | 0.671 |
| F11 | 0.967 | 9.66 | 0.980 | 1.25 | 0.997 | 0.838 | 0.712 |
r = correlation coefficient, Ko is the zero order release rate constant (mg%/h), K1 is the first order release rate constant (h−1), D is the diffusion coefficient (mg%/h½), and n is the release exponent.
Figure 4.
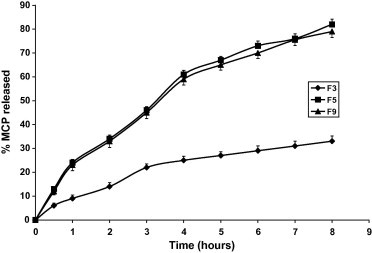
Effect of disintegrant (SSG) amount on metoclopramide release. (F3: no SSG, F5: 5% SSG, F9: 10% SSG).
The effect of HPMC/CMC ratio on MCP release is represented by Formulae F3, F4. Fig. 1 and Tables 3 and 4 show that when the ratio was 3:1 (F3) a pronounced slowing of release was observed. On reversing this ratio (F4), the rate of MCP release was 10-fold increased. This may be attributed to the effect of pH on the viscosity of CMC which is less ionized in acidic medium, and hence less viscous.
The effect of the method of preparation of the tablets and pellets is represented by F8, F10 and F12 as shown in Tables 3 and 4 and Fig. 5. Despite these three formulae have the same composition, the release is more sustained by using the dry granulation method, F10. This may be interpreted by the fact that in this method the ingredients have been subjected twice for compression. The influence of this excessive compression is also reflected in the higher hardness value of F10 compared to that of F8. The release from pellets, F12, was the highest as shown in Table 3, and Fig. 5 compared to the tablets having the same composition. This is due to the higher surface area exposed to the dissolution medium. It is worthy to mention that the pelletization method gave too hard pellets that were incompressible.
Figure 5.
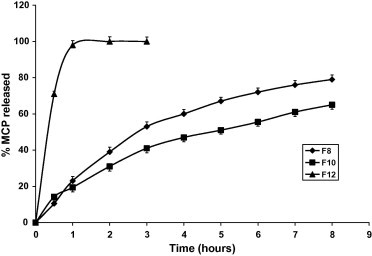
Effect of method of preparation on metoclopramide release. (F8: DC, F10: DG, F12: PS).
Both F10 and F11 were made by dry granulation. The release from F11 is quicker than that from F10, Tables 3 and 4 and Fig. 3, because of decreased weight of HPMC and increased weight of SSG in the latter formula. F9 contains more HPMC and less SSG compared to F11. However, the release of MCP is almost the same from either formulae, Fig. 3 and Table 3. This means that the dry granulation method used for F11 has cancelled the effect of decreasing of HPMC and increasing of SSG amounts in sustaining the drug release.
In general, Formulae 7–11 were successful in sustaining MCP release since T75% ranged from 5.8 to >8 h, Table 3. However, Formulae 3 and 10 have excessively retarded the drug release. Formulae F2 and F12 have released MPC too fast; T75% was 3.2 and 0.5 h, respectively.
4.2. Kinetic assessment of the in vitro release of MCP from the prepared tablets and pellets
In order to determine the release model which best describes the pattern of drug release, the in vitro release data were fitted to zero order, first order and diffusion controlled release mechanisms according to the simplified Higuchi model (Higuchi, 1963).
The preference of a certain mechanism was based on the correlation coefficient (r) for the parameters studied, where the highest correlation coefficient is preferred for the selection of mechanism of release. Successive evidence of the relative validity of diffusion and first order models obtained by analyzing the data using the following equation of Korsmeyer and Peppas (1983)
where Mt/M∞ is the fraction released by the drug at time t, K is a constant incorporating structural and geometric characteristic and n is the release exponent characteristic for the drug transport mechanism. When n = 0.5 fickian diffusion is observed and the release rate in dependent on t, while 0.5 < n < 1.0 indicate anomalous (non-fickian) transport and when n = 1, the release is zero order.
In swellable systems, factors affecting the release kinetics are liquid diffusion rate and polymeric chain relaxation rate. When the liquid diffusion is slower than the relaxation rate of the polymeric chains, the diffusion is Fickian, whereas when the relaxation process is very slow compared with the diffusion rate, the case II transport occurs. When liquid diffusion rate and polymer relaxation rate are of the same order of magnitude, anomalous or non-fickian diffusion is observed (Korsmeyer and Peppas, 1983).
The value of n as estimated by linear regression of log Mt/M∞ vis log t of different formulations are shown in table (Beckett et al., 1987), the obtained values of n ranges between 0.583 and 0.758 for MCP release for all the prepared tablets which indicates drug release mechanisms involving a combination of both diffusion and chain relaxation mechanisms. Therefore, the release of MCP from the prepared tablets is controlled by the swelling of the polymer followed by drug diffusion through the swelled polymer and slow erosion of the tablet. On the other hand, Pellets did not show disintegration but slight swelling and erosion during the dissolution test, the authors concluded that MCP release form pellets was mainly a polymer controlled diffusion and surface erosion process. This is in accordance with a previously published data (Mehata et al., 2001). The higher release rate of pellets is due to higher surface area of pellets and high water solubility of the drug.
5. Conclusions
Sustained release matrix tablets of metoclopramide hydrochloride were prepared using different ratios of hydroxypropylmethylcellulose and sodium carboxymethylcellulose. Direct compression, dry granulation and pelletiazation techniques were employed to prepare the tablets. The comparative metoclopramide release studies revealed that the release rate was dependent on the polymers relative amounts, the technique used to prepare the tablets, and the absence or presence of the super disintegrant sodium starch glycolate. The tablets made by direct compression and dry granulation were found to satisfy the compendial requirements of hardness and friability. Pellets were incompressible due to their excessive hardness on drying and were the weakest in sustaining the release. Most of the formulae prepared by direct compression and dry granulation have satisfactory sustained the drug release with the later being more effective. The drug release for all the prepared tablets has shown mechanisms involving a combination of both diffusion and chain relaxation. Future studies will be conducted to evaluate these matrix tablets in vivo in the case of developing a sustained release dosage form of the drug for better therapy and more patient compliance.
Acknowledgments
The authors would like to acknowledge Kayyali Chair for Pharmaceutical Technology, King Saud University, Saudi Arabia, for providing the facilities to carry out this study (Grant PTC 025 P 08).
References
- Beckett A.H., Behrendt W.A., Hadzija B.W. Bioavailability of controlled-release metoclopramide. 1st communication: single dose study. Arzneimettelforschung. 1987;37:221–224. [PubMed] [Google Scholar]
- Desta Z., Wu G.M., Morocho A.M., Flockhart D.A. The gastroprokinetic and antiemetic drug metoclopramide is a substrate and inhibitor of cytochrome p 450 2D6. Drug Metab. Dispos. 2002;30:336–343. doi: 10.1124/dmd.30.3.336. [DOI] [PubMed] [Google Scholar]
- Ganza-Gonzalez A., Anguiano-Igea S., Otero-Espinar F.J., Blanco-Mendez J. Chitosan and chondroitin microspheres for oral administration controlled release of metoclopramide. Eur. J. Pharm. Biopharm. 1999;48:149–155. doi: 10.1016/s0939-6411(99)00040-5. [DOI] [PubMed] [Google Scholar]
- Gibaldi, M., 1991. Biopharmaceutices and Clinical Pharmacokinetics, 4th ed. Lea & Febiger, London, England, pp. 61–79.
- Hasan E.I., Amro B.I., Arafat T., Badwan A.A. Assessment of controlled release of hydrophillic matrix formulation for metoclopramide HCL. Eur. J. Pharm. Biopharm. 2003;55:339–344. doi: 10.1016/s0939-6411(03)00022-5. [DOI] [PubMed] [Google Scholar]
- Higuchi T. Mechanism of sustained-action medication. Theoretical analysis of rate of release of solid drug dispersed in solid matrices. J. Pharm. Sci. 1963;52:1145–1149. doi: 10.1002/jps.2600521210. [DOI] [PubMed] [Google Scholar]
- Korsmeyer, R.W., Peppas, N.A., 1983. Macromolecular and modeling aspects of swelling-controlled systems. In: Roseman, T.J., Mansdorf, S.Z. (Ed.), Controlled Release Delivery Systems. Dekker, New York, NY, pp. 77–101.
- Lotifipour F., Nokhodchi A., Saeedi M., Norouzi-Sani S., Sharbafi J., Siahi-Shadbad M.R. The effect of hydrophilic and lipophillic polymers and fillers on the release rate of atenolol from HPMC matrices. IL FARM-ACO. 2004;59:819–825. doi: 10.1016/j.farmac.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Martindale, W., Reynolds, J.E.F. (Eds.), 1993. The Extra Pharmacopoeia, 30th ed. Pharmaceutical Press, London, p. 892.
- Mehata A.K., Kislalioglu M.S., Phuapardit W., Malick A.W., Shah N.H. Release performance of a poorly soluble drug from a novel Eudragit based multi-unit erosion matrix. Int. J. Pharm. 2001;213:7–12. doi: 10.1016/s0378-5173(00)00594-9. [DOI] [PubMed] [Google Scholar]
- Moe, S.T., Draget, K.I., Skjak-Braek, G., Smidsrod, Alginiates, O., 1995. In: Alistair, S.M. (Ed.), Food Polysaccharides and their Applications, 1st ed. Marcel Dekker, New York, NY, pp. 245–286.
- USP DI, 1998. Drug Information for the Health Care Professional, vol. 1, 18th ed. United States Pharmacopoeial Convention, Rockville, MD, USA, pp. 2009–2013.
- Winterowd, J.G., Sandford, P.A., 1995. Chitin and chitosan. In: Alistair, S.M. (Ed.), Food Poly-saccharides and their Applications, 1st ed. Marcel Dekker, New York, NY, pp. 441–462.