

Abstract
Purpose
Furosemide is a commonly used diuretic which is used in the treatment of edema, congestive heart failure, hypertension and renal failure. Its absorption exhibits inter- and intra-subject variability that can be attributed to many factors including the intestinal efflux pumps such as the P-glycoprotein (P-gp). This study was done due to the great disagreement between what is published in the literature regarding the influence of P-gp on furosemide and at the same time due to the importance of this drug in the treatment of different conditions as described above. In addition, an investigation of the effect of two of the commonly used pharmaceutical excipients (hydroxypropyl β-cyclodextrin [HPβCD] and Tween 80) and also a P-gp inhibitor (verapamil hydrochloride) on the intestinal absorption of this drug were also done.
Methods
The study utilized the everted intestinal sacs technique to investigate both the effect of the efflux transporter (P-gp) on furosemide absorption and also the effect of the chosen excipients.
Results
The absorption of furosemide was significantly influenced by the P-gp as confirmed by the everted vis the non-everted sacs together with the verapamil study in which the transport of furosemide was inhibited by verapamil. In addition, Tween 80 was also shown to inhibit the P-gp pump whereas the HPβCD did not significantly influence the efflux of furosemide in this study.
Conclusions
P-glycoprotein and some of the used excipients in the formulation play a very important role in the transport of furosemide and other drugs. Thus excipients that affect the activity of P-gp should be avoided when formulating drugs that are substrate for the P-gp or other efflux pumps.
Keywords: Intestinal efflux, Furosemide intestinal absorption, Furosemide efflux
1. Introduction
Furosemide is a loop diuretic which is used in the treatment of edema, congestive heart failure and hypertension. It is also used for the treatment of both acute and chronic renal failure (Berko et al., 2003). Furosemide is a weak acid (pKa = 3.9) that is incompletely absorbed from the gastrointestinal tract after oral administration and it is available in the form of both oral and parenteral formulations (Berko et al., 2003). It’s absorption exhibit inter- and intra-subject variability. This variability can be attributed to many factors such as pH, food intake, drug metabolizing enzymes and efflux pumps in the intestine, dosage form administered, etc. (Chungi et al., 1979; Hammarlund et al., 1984). The influence of intestinal efflux transporters such as P-gp (P-glycoprotein), MRP (multidrug resistance protein), etc. on furosemide absorption is controversial. Some studies have reported a negative impact of these transporters on its absorption (Polli et al., 2001; Verma et al., 2004; Verma and Panchagnula, 2005a,b; Bansal et al., 2007) whereas other studies have reported positive effect for the P-gp on its intestinal absorption (Pade and Stavchansky, 1998; Rege et al., 2001; Pole, 2008).
Accordingly, the objectives of this paper were to study the intestinal absorption of furosemide and determine the exact influence of the intestinal efflux transporter (P-gp) on its intestinal absorption. Also, to investigate the effect of two of the commonly used pharmaceutical excipients hydroxypropyl β-cyclodextrin (HPβCD) and Tween 80 on the intestinal absorption and transport of this drug. This is based on what was shown in the literatures that some pharmaceutical excipients have either positive or negative influence on the absorption and transport of some drugs (Lo, 2003; Wagner et al., 2001).
Several techniques have been reported in the literature to investigate the effect of intestinal efflux pumps on drug transport. These techniques include the in situ intestinal perfusion (Verma et al., 2004; Verma and Panchagnula, 2005a,b), the Caco-2 cell model (Bansal et al., 2007; Pade and Stavchansky, 1998; Rege et al., 2001) and the everted intestinal sacs model (Barthe et al., 1998). This study employed rat everted intestinal sacs and compared the permeation of the drug through everted versus non-everted sacs. In addition, the effect of co-incubation of furosemide and verapamil, a well known P-gp inhibitor was studied.
2. Materials and methods
The animal experiments and the study protocol were approved by the ethics committee of King Saud University.
2.1. Materials
Furosemide was purchased from Co. Pharmaceutica Milanese, SPA (Italy). Hydroxypropyl β-cyclodextrin, verapamil hydrochloride and carbamazepine were purchased from Sigma Chemicals Co. (USA). Acetonitrile (HPLC grade) was purchased from BDH (England).
2.2. Preparation of intestinal sacs
Male Sprague–Dawley rats (250–300 g) were obtained from the Animal Care Center at King Saud University. Rats were given free access to food and water and on the night before the experiment the animals were fasted with free access to water. At the day of experiments, the rats were anesthetized by ether inhalation. After verification of loss of the pain reflex, a midline abdominal incision of 4–5 cm was made and the jejunum was separated and washed with an ice cold oxygenated Krebs solution (pH 6.5) containing 7 g/l sodium chloride, 0.34 g/l potassium chloride, 1.8 g/l glucose, 0.251 g/l disodium hydrogen phosphate, 0.207 g/l sodium dihydrogen phosphate and 46.8 mg/l magnesium chloride. The washed intestine was then divided into approximately 6 cm long sacs. Two sacs were taken from each rat, one of which was gently everted over a glass rod and the other was left non-everted. The sacs were tied from one end using braided suture silk, weighted, filled with the drug solution from the other end, and weighted again.
2.3. Determination of drug transport across the rat intestine
The study investigated the drug transport across everted and non-everted intestinal sacs and monitored drug transfer in presence and absence of verapamil hydrochloride. This design allowed the investigation of the influence of P-gp on the absorption and transport by two methods. The first method involved the comparison of the permeation of the drug through everted and non-everted sacs and the second method compared drug permeation in presence and absence of the P-gp inhibitor (verapamil hydrochloride).
The sacs were tied from one end, loaded with 1 ml drug solution (10 μg/ml in oxygenated Krebs solution) in presence or absence of verapamil hydrochloride (200 μg/ml) and then tied from the other end. The sacs were immersed in a 15 ml-size test tubes containing 10 ml of Krebs solution maintained at 37 °C and continuously bubbled with oxygen. Samples from the outside solution were taken at regular intervals for 60 min and the drug content in each sample was determined by HPLC.
2.4. Effect of HPβCD and Tween 80 on furosemide transport across the rat intestine
The intestinal transport of furosemide was monitored in presence of either Tween 80 (3 mg/ml) or HPβCD (1:1 ratio with the drug solution) together with the drug solutions (1 ml containing 10 μg of the drug). The mixture (drug + Tween 80 or drug + HPβCD) was loaded into the intestinal sacs and the experiment was conducted as described in the above section.
2.5. Chromatography
The study utilized a high pressure liquid chromatograph (HPLC) from Jasco (Japan) equipped with quaternary pump, a variable wavelength detector (UV/vis detector) and an automatic sampling system (auto-sampler). Separation was accomplished using a reversed phase C18, μ-Bondapak™ column 15 cm × 3.9 mm (i.d.) from Waters (USA) with an average particle size of 10 μm. The mobile phase was a mixture of acetonitrile and 20 mM phosphate buffer (35:65) running at a flow rate of 1 ml/min and Carbamazepine was used as an internal standard. The column effluent was monitored at 240 nm and the chromatographic data analysis was performed with Chrompass software (Jasco, Japan).
The samples were transferred to test tubes, spiked with the internal standard in an amount sufficient to produce a concentration of 1 μg/ml. The tubes were vortex mixed for 2 min before loading into the HPLC vials. The injection volume was 30 μl.
2.6. Calculation of the apparent permeability
The apparent permeability coefficient (Papp) was calculated from the cumulative amount permeated versus time profile according the following equation:
where dQ/dt is the steady state rate of drug appearance in the receptor, A is the surface area of the intestinal sac and C0 is the initial concentration of the drug in the sac.
The Papp through the non-everted sac was taken as a measure for the transfer from mucosal to serosal side (M-to-S) and the Papp through the everted sac was taken as a measure for the transfer from serosal to mucosal side (S-to-M).
2.7. Statistical analysis
One way ANOVA followed by Tukey test was done to evaluate the presence of significant differences between different groups at a p-value ⩽ 0.05. This was carried out using Prism Software version 3.02 (GraphPad Software Inc., San Diego, CA).
3. Results
3.1. Intestinal transport and absorption of furosemide
The intestinal permeation parameters of furosemide were studied using the rat intestinal sacs model with the intestinal transport of the drug being evaluated by two approaches. The first compared trans-intestinal transport from everted versus non-everted intestinal sacs. The second approach investigated the effect of P-gp inhibitor (verapamil hydrochloride) on the membrane transport of furosemide. Fig. 1 shows the cumulative amount of furosemide with time in presence and absence of the P-gp inhibitor (verapamil hydrochloride). Table 1 shows the intestinal permeability of furosemide through non-everted and everted rat intestinal sacs in presence or absence of the P-gp inhibitor (verapamil) or the excipients (Tween 80 or HPβCD). It showed a very low permeability in the non-everted rat intestinal sacs. This result reflects drug transfer from the mucosal to serosal side (M-to-S) which mimics the in vivo conditions. However, in the everted sacs, the drug showed significantly higher transport compared to its permeation through the non-everted intestinal sac. This confirms that the P-gp is involved in the transport of the drug. The ratio of S-to-M to M-to-S permeability was found to be 5.6 and this finding suggested a possible role for the P-gp efflux pump in the intestinal absorption of furosemide. A higher permeability in the S-to-M direction than in the M-to-S direction has generally been considered as a prove that active transport (e.g., P-gp efflux) is involved (Polli et al., 2001). In addition, the presence of verapamil increased the M-to-S significantly (Table 1). The ratio of S-to-M to M-to-S permeability was decreased from 5.6 in absence of verapamil to 1.28 in presence of verapamil. This finding further confirms the effect of P-gp efflux mechanism on the intestinal absorption.
Figure 1.
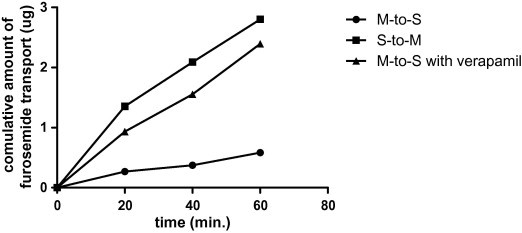
Time profile of the transport of furosemide between the two sides of the intestinal scas in presence and absence of verapamil.
Table 1.
Intestinal permeability of furosemide through non-everted and everted rat intestinal sacs in presence or absence of the P-gp inhibitor verapamil, Tween 80 or hydroxypropyl β-cyclodextrin (HPβCD). Value are presented as mean (STD).
| Applied solution |
Papp (cm/s) × 106 |
||
|---|---|---|---|
| Non-everted sac (M-to-S) | Everted sac (S-to-M) | Ratio (S-to-M)/(M-to-S) | |
| Drug solution | 0.636 (0.093) | 3.54 (0.67) | 5.6 |
| Drug + verapamil | 4.81 (2.2) | 6.17 (1.75) | 1.28⁎ |
| Drug + Tween 80 | 3.8 (1.1) | 4.12 (0.8) | 1.08⁎ |
| Drug + HPβCD | 0.703 (0.12) | 3.22 (0.48) | 4.58 |
M-to-S, the mucosal to serosal transport.
S-to-M, the serosal to mucosal transport.
Significantly different than control (drug solution alone).
3.2. Effect of excipients on the intestinal permeability to furosemide
The study investigated the effect of Tween 80 and HPβCD as two of the most commonly used pharmaceutical excipients on the intestinal permeability and absorption of furosemide. Table 1 shows the effects of excipients on the M-to-S and S-to-M intestinal permeability of the drug. The apparent M-to-S permeability of the drug which was obtained in presence of the excipient was compared to that obtained in absence of the excipient and the results are presented in Fig. 2. The calculated M-to-S transport indicated a permeation enhancing effect for Tween 80 but not for the HPβCD. This implied significant increase in the M-to-S permeability of the drug compared to the control. The presence of HPβCD resulted in marginal reduction in the S-to-M to M-to-S permeability ratio.
Figure 2.
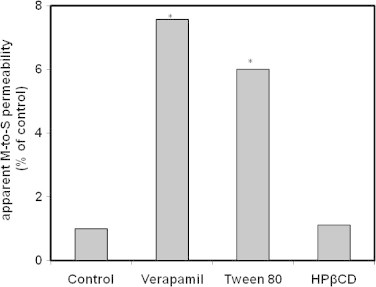
Effect of verapamil and two excipients on the intestinal permeability of furosemide (M-to-S). *Significantly different than control (drug solution alone).
4. Discussion
4.1. Intestinal transport and absorption of furosemide
The intestinal permeation parameters of furosemide were studied in the presence and absence of verapamil using the rat intestinal sacs model and the results of these investigations are presented in Table 1. These results showed low permeability in the non-everted rat intestinal sac. This result reflects drug transfer from the mucosal to serosal side (M-to-S) which mimics the in vivo conditions. Furosemide has been classified as low permeable drug (Rege et al., 2001). In the everted rat intestinal sacs, the drug showed significantly higher transport (5.5-folds) compared to its permeation through the non-everted intestinal sac (Table 1). This confirm that the P-gp is involved and since the function of the P-gp pump is to pump back the drug to the intestine after being absorbed, these results suggested higher trans-intestinal permeation from the serosal to the mucosal side (S-to-M). The ratio of S-to-M to M-to-S permeability was found to be 5.6 and this finding suggested a possible role for the P-gp efflux pump in the intestinal absorption of furosemide. To further confirm this finding the study was extended to investigate the effect of verapamil Hydrochloride on the intestinal permeability of furosemide. The presence of verapamil increased the M-to-S significantly as compared to the case with no verapamil (Table 1). This is due to the inhibition of the P-gp pump by the verapamil which will prevent the drug from being pumped back to the mucosal side. Thus, more drug will be accumulated in the serosal side (Fig. 1). The ratio of S-to-M to M-to-S permeability was decreased from 5.6 in absence of verapamil to 1.28 in presence of verapamil. This finding further confirms the effect of P-gp efflux mechanism on the intestinal absorption. Similar findings were reported for furosemide by other researchers (Verma and Panchagnula, 2005b; Pade and Stavchansky, 1998; Rege et al., 2001). Contrary to this, some researchers employed the drug as an intestinal permeability marker which is not affected by intestinal efflux (P-gp) (Verma et al., 2004; Verma and Panchagnula, 2005a,b; Bansal et al., 2007; Pole, 2008).
4.2. Effect of excipients on the intestinal permeability to furosemide
The study investigated the effect of Tween 80 and HPβCD as two of the most commonly used pharmaceutical excipients on the intestinal permeability and absorption of furosemide. The study evaluated the effects of excipients on the M-to-S and S-to-M intestinal permeability of the drug. The results are presented in Table 1. The apparent M-to-S permeability of the drug which was obtained in presence of the excipient was compared to that obtained in absence of the excipient and the results are presented in Fig. 2. The results indicated a permeation enhancing effect for Tween 80 but not for the HPβCD. This implied significant increase in the M-to-S permeability of the drug compared to the control. Tween 80 was able to reduce the ratio of S-to-M to M-to-S permeability from 5.6 to 1.08. This effect provides an indication that Tween 80 is an inhibitor of the P-gp efflux pump. Similar findings were reported by other researchers for Tween 80 with other drugs (Zhang et al., 2003). The presence of HPβCD resulted in marginal reduction in the S-to-M to M-to-S permeability ratio. This suggests a minor (non-significant) negative effect for the HPβCD on the P-gp efflux mechanism.
5. Conclusion
The study confirmed the effect of the P-gp efflux mechanism on the intestinal transport and absorption of furosemide. This effect can be inhibited by verapamil hydrochloride or Tween 80. The use of HPβCD did not significantly influence the efflux of furosemide using our model of everted sacs. The results of this study stress on the importance of choosing the right excipients during formulation of each drugs to avoid interfering with the absorption and transport of the drugs being formulated.
References
- Bansal T., Singh M., Mishra G., Talegaonkar S., Khar R.K., Jaggi M., Mukherjee R. Concurrent determination of topotecan and model permeability markers (atenolol, antipyrine, propranolol and furosemide) by reversed phase liquid chromatography: utility in caco-2 intestinal absorption studies. J. Chromatogr. B. 2007;859:261–266. doi: 10.1016/j.jchromb.2007.09.025. [DOI] [PubMed] [Google Scholar]
- Barthe L., Bessouet M., Woodley J.F., Houin G. The improved everted gut sac: a simple method to study intestinal P-glycoprotein. Int. J. Pharm. 1998;173:255–258. [Google Scholar]
- Berko S., Regdon G., Jr., Ducza E., Falkay G., Eros I. In vitro and in vivo study in rats of rectal suppositories containing furosemide. Eur. J. Pharm. Biopharm. 2003;53:311–315. doi: 10.1016/s0939-6411(02)00005-x. [DOI] [PubMed] [Google Scholar]
- Chungi V.S., Dittert L.W., Smith R.B. Gastrointestinal sites of furosemide absorption in rats. Int. J. Pharm. 1979;4:27–38. [Google Scholar]
- Hammarlund M.M., Paalzow L.K., Odlind B. Pharmacokinetics of furosemide in man after intravenous and oral administration. Application of moment analysis. Eur. J. Clin. Pharmacol. 1984;26:197–207. doi: 10.1007/BF00630286. [DOI] [PubMed] [Google Scholar]
- Lo Y. Relationship between the hydrophilic–lipophilic balance values of pharmaceutical excipients and their multidrug resistance modulating effect in caco-2 cells and rat intestines. J. Control. Release. 2003;90:37–48. doi: 10.1016/s0168-3659(03)00163-9. [DOI] [PubMed] [Google Scholar]
- Pade V., Stavchansky S. Link between drug absorption solubility and permeability measurements in caco-2 cells. J. Pharm. Sci. 1998;87:1604–1607. doi: 10.1021/js980111k. [DOI] [PubMed] [Google Scholar]
- Pole D.L. Physical and biological considerations for the use of nonaqueous solvents in oral bioavailability enhancement. J. Pharm. Sci. 2008;97:1071–1088. doi: 10.1002/jps.21060. [DOI] [PubMed] [Google Scholar]
- Polli J.W., Wring S.A., Humphreys J.E., Huang L., Morgan J.B., Webster L.O., Serabjit-Singh C.S. Rational use of in vitro P-glycoprotein assays in drug discovery. J. Pharmacol. Exp. Ther. 2001;299:620–628. [PubMed] [Google Scholar]
- Rege B.D., Yu L.X., Hussain A.S., Polli J.E. Effect of common excipients on caco-2 transport of low permeability drugs. J. Pharm. Sci. 2001;90:1776–1786. doi: 10.1002/jps.1127. [DOI] [PubMed] [Google Scholar]
- Verma M.V.S., Panchagnula R. Prediction of in vivo intestinal absorption enhancement on P-glycoprotein inhibition from rat in situ permeability. J. Pharm. Sci. 2005;94:1694–1704. doi: 10.1002/jps.20309. [DOI] [PubMed] [Google Scholar]
- Verma M.V.S., Panchagnula R. pH-dependent functional activity of P-glycoprotein in limiting intestinal absorption of protic drugs: kinetic analysis of quinidine efflux in situ. J. Pharm. Sci. 2005;94:2632–2643. doi: 10.1002/jps.20489. [DOI] [PubMed] [Google Scholar]
- Verma M.V.S., Kapoor N., Sarkar M., Panchagnula R. Simultaneous determination of digoxin and permeability markers in rat in situ intestinal perfusion samples by RP-HPLC. J. Chromatogr. B. 2004;813:352–374. doi: 10.1016/j.jchromb.2004.09.047. [DOI] [PubMed] [Google Scholar]
- Wagner D., Spahn-Langguth H., Hanafy A., Koggel A., Langguth P. Intestinal drug efflux: formulation and food effects. Adv. Drug Deliv. Rev. 2001;50:S13–S31. doi: 10.1016/s0169-409x(01)00183-1. [DOI] [PubMed] [Google Scholar]
- Zhang H., Yao M., Morrison R.A., Chong S. Commonly used surfactant, Tween 80, improves absorption of P-glycoprotein substrate, digoxin, in rats. Arch. Pharm. Res. 2003;26(9):768–772. doi: 10.1007/BF02976689. [DOI] [PubMed] [Google Scholar]