

Abstract
Most biopsy and autopsy tissues are formalin-fixed and paraffin-embedded (FFPE), but this process leads to RNA degradation that limits gene expression analysis. The RNA genome of the 1918 pandemic influenza virus was previously determined in a 9-year effort by overlapping RT-PCR from post-mortem samples. Here, the full genome of the 1918 virus at 3000× coverage was determined in one high-throughput sequencing run of a library derived from total RNA of a 1918 FFPE sample after duplex-specific nuclease treatments. Bacterial sequences associated with secondary bacterial pneumonias were also detected. Host transcripts were well represented in the library. Compared to a 2009 pandemic influenza virus FFPE post-mortem library, the 1918 sample showed significant enrichment for host defence and cell death response genes, concordant with prior animal studies. This methodological approach should assist in the analysis of FFPE tissue samples isolated over the past century from a variety of diseases.
Keywords: influenza A virus, pandemic, 1918 influenza virus, formalin-fixed, paraffin-embedded tissue, NGS
Introduction
Influenza A viruses (IAVs) are important human pathogens, with a significant morbidity and mortality impact in annual epidemics and unpredictable pandemics [1,2]. The worst pandemic in history, the ‘Spanish’ H1N1 influenza pandemic of 1918–1919, killed approximately 50 million people worldwide [3], while the most recent pandemic, the ‘Swine-origin’ 2009 H1N1 pandemic, caused far fewer deaths [4,5]. Even though case fatality rates can vary dramatically between pandemics, severe disease and death following influenza infection are correlated with viral virulence, secondary bacterial infections, and host inflammatory responses [6–10]. Since IAVs could not be isolated in 1918, the pandemic virus could only be characterized archaevirologically and reconstructed decades later, when RT-PCR was used to amplify viral RNA fragments in the post-mortem tissues of 1918 victims [9,11].
In contrast, viral sequence data from the 2009 pandemic became very rapidly available and played a crucial role in studying the virus’s origin, spread, and evolution [4,5]. However, complete IAV genome sequencing by traditional methods is time- and labour-intensive. For example, the initial characterization of the 2009 H1N1 IAV involved Sanger sequencing 46 overlapping amplicons generated by RT-PCR with degenerate primers that bound approximately every 200–250 nucleotides along the segmented RNA genome [12]. Similarly, in the first large-scale IAV sequencing project, 95 overlapping RT-PCR reactions were performed for each viral sample [13].
Since their commercialization in 2005, ‘high-throughput’ DNA sequencers, which can determine megabases of DNA sequence per run [14], have provided researchers and public health officials with another powerful tool. Pyrosequencing has been used to detect IAV and norovirus infections [15], to detect IAV mixed infections and quasi-species [16], and to characterize antiviral resistance [17]. Recent sequence analyses of 2009 pandemic infections suggest that deep-sequencing metagenomics-based strategies can augment conventional methodologies for IAV detection and surveillance [18]. All of these studies have, however, depended on high-quality viral RNA.
Most biopsy or autopsy tissue samples are formalin-fixed and paraffin-embedded (FFPE). A huge number of FFPE archival materials from the end of the 19th century to the present [19] are stored in hospitals, tissue banks, and laboratories. These samples could potentially provide a wealth of information in retrospective molecular genetic studies of diseased tissues [20]. Different analysis platforms, including microarray [21,22], mass spectrometry [23], and high-throughput sequencing [24–26], have been used on FFPE samples. There are undoubtedly many archival FFPE tissues available from autopsies of pandemics and annual IAV epidemics at least as far back as the 1918 pandemic [27–30], and including the 2009 pandemic [10,31]. Such materials have already proven invaluable in the study of influenza pandemic pathophysiology [10,31,32]. A significant challenge to the use of these archival samples, however, is the degradation and modification of RNA that occurs during fixation and embedding [33–38].
In this study, we performed high-throughput sequencing (Illumina GAIIx) on total RNA of FFPE autopsy lung tissue samples from the 1918 and 2009 influenza pandemics. Duplex-specific nuclease (DSN) was used to reduce ribosomal RNA (rRNA) representation in the sequencing libraries. Using the methodology described here, full-length IAV genomes were recovered from both the 1918 and the 2009 samples. In contrast to the initial effort to sequence the genome of the 1918 influenza virus which took 9 years [11], we obtained the complete 1918 viral genome at approximately 3000-fold base coverage in a single sequencing run over the course of a few days. Multiple mutations and minor variants were identified. We also identified genetic signatures of different co-infecting bacteria, including streptococcal species from both autopsy tissues, correlating with their clinical records. Host mRNA was also well represented in both libraries, allowing assessment of gene expression related to immune response and inflammation. Interestingly, the 1918 case showed a significant enrichment of expressed genes related to pro-inflammatory and cell death responses compared with the 2009 case, consistent with the previous animal experiments [39]. These findings demonstrate that direct and unbiased high-throughput RNA sequencing can be applied to FFPE samples almost 100 years old to yield biologically relevant data.
Materials and methods
Autopsy samples
The FFPE sample from the 1918 influenza pandemic was derived from lung tissue from a patient who died in Camp Upton, NY in 1918 (previously reported in a published case series as case 19180924d [29]). The FFPE sample from the 2009 influenza pandemic was also derived from lung tissue from a patient who died in New York City, NY in 2009 (previously reported in a published case series as case 1 [10]). RNA was extracted from the FFPE autopsy sections and RT-PCR performed as previously described [29] (see Supplementary materials and methods). These samples were considered exempt for human subjects review under US Government guidelines (http://www.hhs.gov/ohrp/policy/checklists/decisioncharts.html#c5.)
Library preparation and deep sequencing
Total RNA library construction from FFPE samples was performed according to the mRNA-seq sample preparation kit (Illumina, San Diego, CA, USA) eliminating the poly-A selection and RNA fragmentation steps and sequenced on an Illumina GAIIx with a single-end run (see supplementary materials and methods). From the 2009 pandemic sample, more than 290 million reads (of a total of >22 GB of sequences) were generated. From the 1918 pandemic sample, more than 235 million reads (of a total of ~7.8 GB of sequences) were generated. From uninfected A549 cells, more than 42 million reads (of a total of >3.3 GB of sequences) were generated in one sequencing lane. All sequences from this study have been deposited as a series at NCBI’s SRA database (accession number SAR061036).
Sequence analysis
The sequence analysis steps were as follows: (1) Reads were mapped to bowtie indexed 1918 pandemic influenza genome [A/Brevig_Mission/1/18(H1N1)] or 2009 pandemic influenza genome [A/California/04/2009(H1N1)] downloaded from GenBank as well as the pre-built index of the human genome (UCSC hg19) [40]. (2) Reads that were mapped to 1918 or 2009 influenza pandemic genomes and the human genome were screened out. (3) The rest of the sequences were assembled de novo by ABySS [41] and the resulting contigs were searched against the NCBI non-redundant nucleotide (nt) database (downloaded on 15 August 2010) and the NCBI bacteria genome database by blastn program (blastall 2.2.23) [42]. (4) The blast outputs were analysed by metagenomic analyser software, MEGAN [43,44], and taxonomical names were assigned to each contig (see Supplementary materials and methods).
Results
Case reports
Both the 1918 and the 2009 pandemic influenza pneumonia autopsy cases have been described in published case series [10,29]. Briefly, the 1918 patient was a 22-year-old male who died of influenza and pneumonia (previously reported as case 19180924d [29]). At autopsy, lung cultures were positive for Streptococcus pneumoniae. Lung sections showed acute pneumonia, bronchiolitis, acute diffuse alveolar damage, acute pulmonary oedema, and acute pleuritis. Immunohistochemistry for viral antigens was positive in bronchiolar epithelial cells, alveolar epithelial cells, and macrophages (Supplementary Figures 1A and 1B). The partial influenza viral haemagglutinin (HA) gene sequence was previously reported as A/New York/3/1918 (H1N1) [29]. The 2009 patient was a 55-year-old female with underlying comorbidities who died of influenza and pneumonia (previously reported as case 1 [10]). At autopsy, lung cultures were positive for Streptococcus pneumoniae. Sections showed acute pneumonia, tracheitis, bronchitis, and acute diffuse alveolar damage. Immunohistochemistry for viral antigens was positive in bronchiolar epithelial cells, alveolar epithelial cells, and macrophages (Supplementary Figures 1C and 1D).
Duplex-specific nuclease (DSN) normalization of cDNA libraries from FFPE samples
Because RNA extracted from FFPE samples is highly degraded [36], cDNA libraries based on poly(A) primed reverse transcription would be biased to the 3′ end of transcripts. In contrast, random primed cDNA libraries would contain a preponderance of ribosomal and transfer RNAs since in most cells they represent 60–90% of the RNA transcripts [45]. The RNA integrity numbers (RIN) for both 1918 and 2009 samples were only ~2.6. To reduce rRNA representation, two rounds of DSN normalization [46] were applied to the 1918 and 2009 libraries. Real-time RT-PCR assessment of the libraries following DSN normalization showed a strong reduction in human 18s rRNA transcripts and increased representation of human β2 microglobulin (β2M) transcripts (Table 1).
Table 1.
Real-time PCR results of the effect of DSN treatments for the library of the 2009 autopsy sample
| Gene | Before DSN treatment (CT mean) | After 1st DSN treatment (CT mean) | After 2nd DSN treatment (CT mean) |
|---|---|---|---|
| β2M | 24·912 | 20·579 | 18·927 |
| 18s rRNA | 11·103 | 11·177 | 14·965 |
Complete IAV genomes compiled from the 2009 and 1918 FFPE samples
The cDNA libraries from the 1918 and 2009 pandemic samples were sequenced on an Illumina Genome Analyzer IIx, and 7.8 GB (235 586 783 reads) and 22.0 GB (290 556 262 reads) of data were obtained, respectively. Using Tophat [40], 0.5% (1 171 876) of the reads from the 1918 sample aligned with the 1918 reference viral genome [A/Brevig_Mission/1/18(H1N1)] and 1.12% (3 246 610) of the reads from the 2009 sample aligned with the prototype 2009 pandemic IAV genome [A/California/04/2009 (H1N1)]. All eight viral genome segments were determined for both the 1918 and the 2009 samples, at 3000- and 19 000-fold average depth of coverage, respectively (Table 2).
Table 2.
Mapping result of the reads to each segments of the 2009 pandemic flu virus genome
| Influenza virus | Segments | Length | No of reads aligned | Covered length | Fraction of bases covered |
|---|---|---|---|---|---|
| A/California/04/2009 (H1N1) | FJ969516_PB2 | 2280 | 384 138 | 2276 | 0.9982456 |
| GQ377049_PB1 | 2274 | 1 045 617 | 2272 | 0.9991205 | |
| FJ969515_PA | 2151 | 338 426 | 2151 | 1 | |
| GQ117044_HA | 1701 | 544 342 | 1698 | 0.9982364 | |
| FJ969512_NP | 1497 | 374 983 | 1497 | 1 | |
| FJ969517_NA | 1410 | 255 942 | 1409 | 0.9992908 | |
| FJ969513_MP | 982 | 349 366 | 979 | 0.996945 | |
| FJ969514_NS | 863 | 147 821 | 848 | 0.9826187 | |
| A/Brevig Mission/1/1918 (H1N1) | DQ208309_PB2 | 2280 | 437685 | 2280 | 1 |
| DQ208310_PB1 | 2274 | 131 395 | 2274 | 1 | |
| DQ208311_PA | 2151 | 160 936 | 2150 | 0.9995351 | |
| AF117241_HA | 1701 | 115 462 | 1701 | 1 | |
| AY744935_NP | 1497 | 179 050 | 1493 | 0.997328 | |
| AF250356_NA | 1410 | 34 306 | 1397 | 0.9907801 | |
| AY130766_MP | 982 | 79 997 | 979 | 0.996945 | |
| AF333238_NS | 838 | 33 045 | 829 | 0.9892601 |
Sequence comparison to reference influenza viral genomes
After aligning reads from both sequencing runs to their respective reference genomes, single-nucleotide polymorphisms (SNPs) were identified [47]. We arbitrarily categorized SNPs as either a ‘mutation’ (> 90% of aligned bases did not match the reference nucleotide) or a ‘minor variant’ (> 10% to <90% of aligned bases did not match the reference nucleotide). From the 2009 library, we identified 32 SNPs (25 mutations and 7 minor variants). Among these 25 mutations, there were 10 non-synonymous and 15 synonymous changes. Among the seven minor variants, there were five non-synonymous and two synonymous changes (Supplementary Table 1). Most of the changes (26/32) were represented in viral strains reported in the NCBI IAV database [48]. Similarly, from the 1918 pandemic case, we identified 25 SNPs (18 mutations and 7 minor variants). Seven non-synonymous and 13 synonymous mutations were observed with five non-synonymous and two synonymous minor variants (Supplementary Table 2). Sequences of the 1918 HA receptor-binding domain have demonstrated variation between cases [27–29]. The 1918 HA sequence determined in this case showed a polymorphism at codon 239 (HA1 domain codon 222) from the consensus aspartic acid (D) predominantly to asparagine (N), with a minority (8%) of glycine (G).
We also identified the complete or partial 3′ and 5′ terminal non-coding regions (NCRs) of each of the eight gene segments from both the 2009 and the 1918 cases (Figures 1A and 1B). From the 1918 sample, we identified complete or partial 3′ and 5′ NCRs from all segments except the 3′ NCR of the non-structural (NS) segment. These sequences matched the UTR sequences recently determined from the A/Brevig Mission/1/1918 (H1N1) strain using a rapid amplification of cDNA ends (RACE)-based approach (unpublished data). In addition, reads spanning the spliced junctions of the matrix segment (M2 and M3) and the NS segment (NS2, or NEP) spliced open reading frames were recovered from both the 1918 and the 2009 samples.
Figure 1.

Influenza viral genome segment terminal non-coding regions (NCR) identified in the libraries. (A) NCR sequences from the 2009 influenza pandemic sample. (B) NCR sequences from the 1918 influenza pandemic autopsy sample. Sequences are presented in sense (mRNA) orientation. Start and stop codons are boxed. NCR sequences are underlined and ‘?’ represents undetermined NCR sequences. Variant sequences are identified by their degenerate nucleotide codes: MA/C; R =A/G; W =A/T; S =C/G; Y =C/T; and K =G/T.
Differential expression of host genes in the 1918 and 2009 samples
To assess human gene expression, sequences obtained from both samples were mapped to the human genome (UCSC hg19) using Tophat [40]. A total of 109 944 742 (46.67%) reads from the 1918 sample and 211 722 556 (72.87%) reads from the 2009 sample were mapped to the hg19 genome. As a control, we sequenced cDNA obtained from mock-infected cultured human lung alveolar epithelial cells (A549) and mapped the resultant reads (22 384 397, 53.07%) to hg19. We identified 878 immune and inflammatory response genes that were commonly more than two-fold up-regulated relative to the mock-infected cells (Table 3 and Supplementary Table 3) in both the 1918 and the 2009 samples, including chemokines and cytokines, such as CXCL1, CXCL10, CCL2–5, IL1B, IL6, and IL8; cellular defence response genes, such as TLR2, TLR4, IFIH1, IFIT1–3, IFITM1, and IFITM3; and death receptor pathways, including FAS and TNFSF10 (TRAIL). Functional analysis of genes induced in the two autopsy samples showed they were associated with disease states including inflammation, infection (bacterial and influenza), and pneumonia (Table 3). Using Cufflink [49], a direct comparison of the 1918 and 2009 libraries identified 566 differentially expressed human genes between these two samples (Supplementary Table 4) and revealed a significant enrichment in genes related to host defence and cell death responses in the 1918 sample (as shown in Figure 2 and Supplementary Figures 2 and 3), in agreement with previously published experimental animal model results [39].
Table 3.
Functional analysis of 878 genes induced more than two-fold in both the 1918 and the 2009 samples compared with uninfected lung epithelial cells (see Materials and methods section)
| Biological process (GO) | p value |
|---|---|
| Immune system process | 3.52E-49 |
| Immune response | 1.07E-48 |
| Defence response | 4.45E-44 |
| Response to stress | 5.00E-35 |
| Inflammatory response | 1.74E-23 |
| Innate immune response | 1.84E-21 |
| Response to cytokine stimulus | 8.91E-19 |
|
| |
| Diseases (Genomatix GGA) | p value |
|
| |
| Inflammation | 1.50E-31 |
| Infection | 8.59E-25 |
| Pneumonia | 9.32E-25 |
| Bacterial infections | 1.87E-22 |
| Unusual interstitial pneumonia | 1.67E-17 |
| Sepsis | 1.58E-17 |
| Influenza, human | 6.40E-16 |
Figure 2.

Host response in the 1918 autopsy sample is associated with increased defence and cell death responses. Network diagrams are shown that were derived from functional Gene Ontology analysis of genes with statistically significant differential expression patterns in the 1918 and 2009 samples as identified using Cufflink. Functional analysis was performed using the Genomatix Genome Analyzer (as described in the Supplementary materials and methods). Genes shown in red indicate transcripts with more than four-fold increased relative expression in the 1918 autopsy sample, and genes shown in blue indicate transcripts with more than four-fold increased relative expression in the 2009 autopsy sample.
Analysis of the bacterial sequences in the 1918 and 2009 autopsy cases
Reads of both data sets, not mapping to the IAV and human genomes, were assembled de novo into contigs, compared with the NCBI non-redundant and bacterial genome nucleotide databases, and the results taxonomically assessed using the MEtaGenome Analyzer (MEGAN) [43,44] (Figure 3 and Supplementary Figures 4 and 5). Taxonomic assignments for the 2009 sample compared with the nt database were 8245 eukaryota (82.96%), 161 bacteria (1.62%), and 80 viruses (0.80%) (Figure 3A and Supplementary Figure 4A). Similarly, in comparison to the bacterial genome database, the family was assigned to 182 contigs: 48 Lactobacillaceae, 22 Streptococcaceae, 21 Pasteurellaceae, 19 Moraxellaceae, 17 Enterobacteriaceae, and 8 Sphingomonadaceae (Figure 3B and Supplementary Figure 4B). Taxonomic assignments for the 1918 contigs compared with the nt database were 19 768 eukaryota (69.38%), 3376 bacteria (11.85%), 28 viruses (< 0.01%) (Figure 3C and Supplementary Figure 5A). In both samples, all viral assignments were to influenza A viruses (Supplementary Figures 4 and 5). In the comparison of the 1918 contigs with the bacterial genome database, 4238 were assigned to 20 bacterial families, with Streptococcaceae being most highly represented (1020) (Figure 3D and Supplementary Figure 5B). Streptococcus pneumoniae was isolated from post-mortem lung cultures from both the 2009 and the 1918 cases. From the 2009 dataset, 20 out of 22 of the contigs assigned to Streptococcaceae were assigned to the genus Streptococcus unambiguously. Similarly, from the 1918 dataset, within the most highly represented bacterial family, Streptococcaceae, 966 out of 1020 contigs were unambiguously assigned to the genus Streptococcus, with eight contigs assigned to Streptococcus pneumoniae.
Figure 3.
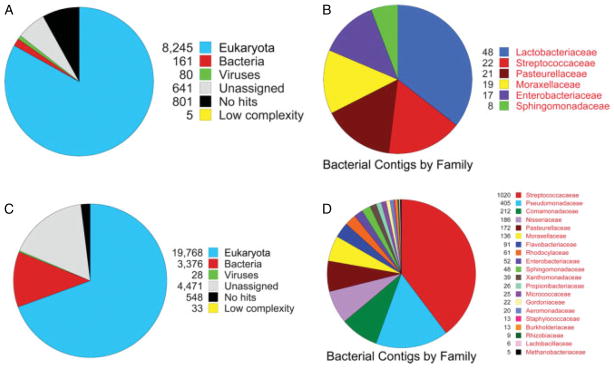
Taxonomic analysis of the 2009 and 1918 influenza pandemic samples. (A) From the 2009 influenza pandemic sample, a pie chart was generated by MEGAN based on blastn output of the NCBI nt database from contigs of at least 76 bp. (B) From the 2009 influenza pandemic sample, a pie chart was generated by MEGAN based on blastn output of the NCBI bacterial genome database from contigs of at least 76 bp. (C) From the 1918 influenza pandemic sample, a pie chart was generated by MEGAN based on blastn output of the NCBI nt database from contigs of at least 48 bp. (D) From the 1918 influenza pandemic sample, a pie chart was generated by MEGAN based on blastn output of the NCBI bacterial genome database from contigs of at least 48 bp.
Discussion
Formalin-fixed, paraffin-embedded (FFPE) tissues reflect the most widely practised method for clinical sample preservation and archiving [50], making them invaluable resources for different disease studies, but their potential for RNA analysis is hampered by RNA degradation [51–53]. The genome of the 1918 influenza virus was sequenced using overlapping RT-PCR from post-mortem tissue samples over a 9-year period [9,11]. In this study, the complete genome of the 1918 virus was determined in a single sequencing run, with an average per base sequence coverage of ~3000. In addition, both host mRNA expression and bacterial RNA could also be analysed. Thus, the method described here will facilitate additional studies from FFPE archival tissues for many important biomedical questions.
Duplex-specific nuclease (DSN) isolated from the Kamchatka crab can specifically cleave dsDNA and has been used to normalize cDNA libraries [46]. It has been used to preferentially degrade highly represented rRNA and thus reduce the range spanned from the most to the least abundant RNA species [54,55]. After two rounds of DSN treatment in the 1918 and 2009 samples, the difference in representation of 18s rRNA and β2M transcripts was greatly reduced (Table 1), and both IAV genomes were sequenced with a high base coverage.
The IAV RNA-dependent RNA polymerase error-rate [56,57] contributes to the rapid evolution of these viruses. It has also been reported that the error rate of the Illumina platform generally increases towards the end of the read, reaching as high as 5% [58]. Conservatively, here we only reported nucleotide polymorphisms occurring with a frequency greater than 10% different from the reference genomes and observed in reads from both directions. We also eliminated the variant calls at the first or last mapping positions [59,60]. Over 3000 complete genomes are published for the 2009 pandemic H1N1 influenza virus, allowing us to compare identified mutations and polymorphisms. From the 2009 case examined here, we found ten non-synonymous mutations in NP, NS, PA, PB2, NA, and HA segments compared with the A/California/04/2009 (H1N1) virus with five in HA (Supplementary Table 1), with most of these polymorphisms already present in the influenza database [48]. Previous studies on the 2009 pandemic influenza virus have suggested that the HA D222G mutation was associated with severe or fatal infections [61]. However, in the 2009 sample examined here, no polymorphisms were detected at this codon, supporting the view that the D222G mutation is not crucial for virulence or pulmonary tropism, in contrast to the suggestion of recent studies [62]. Interestingly, at the same HA position from the 1918 sample, it is predominantly N222 with 8% of G222. HA S220T and NA N248D mutations have been used to distinguish early- and peak-phase pandemic isolates in Japan [63]. The 2009 viral sequences determined here possess both changes, consistent with a peak-phase virus. Another mutation recovered from our 2009 case is nucleoprotein V100I, also associated with 2009 pandemic peak isolates [64] and previously associated with human adaptation [65,66]. Further work will be required to evaluate the significance of the seven non-synonymous mutations in the 1918 genome determined here in relation to A/Brevig Mission/1/1918 (H1N1).
The eight negative sense RNA gene segments comprising the influenza A virus genome contain open reading frames flanked at both ends by short NCRs [67]. The NCRs are conserved and are critical for viral polymerase binding, cap-snatching, transcription initiation, and replication [68,69]. Most published IAV NCRs are derived from the conserved ‘universal’ primer sequences used to reverse transcribe and/or amplify the viral RNA segments [70], but these sequences may not always match the native sequences of the viral RNA; for example, at the known fourth base polymorphism at the 3′ end of the vRNA [71]. Although the RNA from both FFPE samples was highly degraded, sequence reads covering both the 3′ and the 5′ NCRs from both 1918 and 2009 influenza viruses were recovered (Figure 1), without a primer-based sequence bias.
Experimental animal models have been used to understand the host response to both 1918 and 2009 pandemic influenza virus infection [39,72–74]. These studies have shown that the 1918 virus results in significantly more severe disease, host inflammatory, and cell death responses in mice and non-human primates. In the present study, we did not have access to normal human FFPE lung samples to use as a control; therefore, we chose to use mock-infected human alveolar epithelial cells (A549) as a reference. As expected, we observed significant expression of immune response-related responses in the FFPE autopsy samples when compared with uninfected A549 cells. Furthermore, these gene expression changes are associated with infectious lung disease states such as pneumonia, influenza, and bacterial infection, and these results were concordant with the medical diagnoses of these patients at time of death and provide validity for additional analyses comparing the host response between the 1918 and 2009 autopsies. Accordingly, when the host gene expression responses in the 1918 and 2009 FFPE samples were compared, the 1918 sample was associated with significantly increased inflammatory and cell death responses, concordant with results seen in experimental animal 1918 influenza virus pathogenesis studies [39,73]. While this study was limited to examination of the host immune response in single cases from the 1918 and 2009 pandemics, expression data here add support to the hypothesis that the pandemic virus that killed the 22 year old, otherwise healthy soldier in 1918 [29] was intrinsically more virulent than the pandemic virus that killed a 55 year old with underlying comorbidities in 2009 [10]. Additional studies using more diverse samples will be required to determine if the differences in gene expression observed are broadly characteristic of host response to the two pandemic influenza virus infections.
Metagenomic analysis was performed on the assembled contigs to evaluate bacterial sequences. From the 2009 dataset, the bacterial family with the most contigs was Lactobacillaceae and most were identified as genus Lactobacillus (Figure 3B), which are mostly present in the human gastrointestinal tract [75], and although lung abscess and pleuritis have been reported with Lactobacillus rhamnosus [76], its presence could be post-mortem contamination. Alternatively, aspiration may also explain these sequences as nausea and vomiting [77] were commonly observed in patients infected by the 2009 pandemic influenza virus. The Streptococcaceae had the next most contigs assigned, consistent with a positive post-mortem lung culture of Streptococcus pneumoniae in this case [10]. Secondary streptococcal infections have long been associated with severe disease in influenza pneumonia [7], as was also observed in the 2009 pandemic [78]. Streptococcus pnemoniae and S. pyogenes were the two major species identified in the blast results. Pasteurellaceae and Moraxellaceae were other predominant bacterial families identified. Within the Pasteurellaceae family, most sequences were identified as Haemophilus influenzae, which was also detected in many 2009 pandemic influenza patients [78]. Most of the sequence assignments within the Moraxellaceae were to Moraxella caterrhalis, also reported in 2009 influenza pandemic cases [79].
More diverse bacterial taxonomies were assigned from the 1918 dataset (Figure 3D). This may be due in part to contamination introduced during sample preparation and archival preservation for more than 90 years. Nevertheless, the bacterial family with the most representation was Streptococcaceae, and most of them were Streptococcus. Similarly, the 1918 case also had a positive post-mortem lung culture for Streptococcus pneumoniae [29], with eight contigs unambiguously assigned to S. pneumoniae (Supplementary Figure 5B). The next most common bacterial family identified was Pseudomonadaceae and the majority were assigned as Pseudomonas (Supplementary Figure 5B). In vitro, Pseudomonas aeruginosa can attach to tracheal epithelial cells injured by primary influenza virus infection [80]. While bacterial family Pasteurellaceae (mainly Haemophilus influenzae) were also identified and were commonly cultured from 1918 influenza patients [7], many bacterial families represented in the sample might represent environmental contaminants, including Comamonadaceae, Neisseriaceae, and Acinetobacter.
Overall, we have demonstrated the success of unbiased high-throughput sequencing of DSN-treated cDNA libraries made from highly degraded RNA isolated from FFPE autopsy samples from fatal cases of the 1918 and 2009 influenza pandemics. We recovered the complete genomic sequences of both pandemic influenza viruses, identified co-infecting bacterial sequences, and were able to examine host gene expression in both cases. This methodological approach should assist in the analysis of preserved tissue samples over the past century on a variety of human or animal diseases. The rich information potentially contained within such archival material will likely provide important insights into different diseases and lead to improvements in human health.
Supplementary Material
Acknowledgments
This work was supported by the Intramural Research Program of the NIH and the NIAID.
Footnotes
No conflicts of interest were declared.
Author contribution statement
JKT conceived the experiments. YLX, JCK, SBB, ZMS, JMM, and JKT carried out experiments and analysed data. All authors were involved in writing the paper and had final approval of the submitted version.
SUPPORTING INFORMATION ON THE INTERNET
The following supporting information may be found in the online version of this article.
Supplementary materials and methods.
Table S1. Identified SNPs from 2009 autopsy sample.
Table S2. Identified SNPs from 1918 autopsy sample.
Table S3. Common up-regulated host genes in 1918 and 2009 autopsy samples versus mock A549 cells.
Table S4. Differentially expressed host genes in 1918 autopsy versus 2009 autopsy samples.
Figure S1. Immunohistochemistry for influenza viral antigens in sections from the 1918 and 2009 pandemic influenza post-mortem FFPE lung tissues.
Figure S2. Genomatix gene ontology analysis showing cellular response to cytokine stimulus genes (GO: 0071345) with differential expression in the 1918 sample versus the 2009 sample.
Figure S3. Genomatix gene ontology analysis showing immune system process genes (GO: 0002376) with differential expression in the 1918 sample versus the 2009 sample.
Figure S4. Taxonomic trees from the 2009 influenza pandemic sample.
Figure S5. Taxonomic trees from the 1918 influenza pandemic sample.
References
- 1.Taubenberger JK, Morens DM. Pandemic influenza – including a risk assessment of H5N1. Rev Sci Tech. 2009;28:187–202. doi: 10.20506/rst.28.1.1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morens DM, Taubenberger JK. Pandemic influenza: certain uncertainties. Rev Med Virol. 2011;21:262–284. doi: 10.1002/rmv.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Johnson NP, Mueller J. Updating the accounts: global mortality of the 1918–1920 “Spanish” influenza pandemic. Bull Hist Med. 2002;76:105–115. doi: 10.1353/bhm.2002.0022. [DOI] [PubMed] [Google Scholar]
- 4.Fraser C, Donnelly CA, Cauchemez S, et al. Pandemic potential of a strain of influenza A (H1N1): early findings. Science. 2009;324:1557–1561. doi: 10.1126/science.1176062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garten RJ, Davis CT, Russell CA, et al. Antigenic and genetic characteristics of swine-origin 2009 A(H1N1) influenza viruses circulating in humans. Science. 2009;325:197–201. doi: 10.1126/science.1176225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kuiken T, Taubenberger JK. The pathology of human influenza revisited. Vaccine. 2008;26:D59–D66. doi: 10.1016/j.vaccine.2008.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morens DM, Taubenberger JK, Fauci AS. Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: implications for pandemic influenza preparedness. J Infect Dis. 2008;198:962–970. doi: 10.1086/591708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Taubenberger JK, Kash JC. Influenza virus evolution, host adaptation, and pandemic formation. Cell Host Microbe. 2010;7:440–451. doi: 10.1016/j.chom.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Taubenberger JK, Kash JC. Insights on influenza pathogenesis from the grave. Virus Res. 2011;162:2–7. doi: 10.1016/j.virusres.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gill JR, Sheng ZM, Ely SF, et al. Pulmonary pathologic findings of fatal 2009 pandemic influenza A/H1N1 viral infections. Arch Pathol Lab Med. 2010;134:235–243. doi: 10.5858/134.2.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Taubenberger JK, Hultin JV, Morens DM. Discovery and characterization of the 1918 pandemic influenza virus in historical context. Antivir Ther. 2007;12:581–591. [PMC free article] [PubMed] [Google Scholar]
- 12.Dawood FS, Jain S, Finelli L, et al. Emergence of a novel swine-origin influenza A (H1N1) virus in humans. N Engl J Med. 2009;360:2605–2615. doi: 10.1056/NEJMoa0903810. [DOI] [PubMed] [Google Scholar]
- 13.Ghedin E, Sengamalay NA, Shumway M, et al. Large-scale sequencing of human influenza reveals the dynamic nature of viral genome evolution. Nature. 2005;437:1162–1166. doi: 10.1038/nature04239. [DOI] [PubMed] [Google Scholar]
- 14.Service RF. Gene sequencing. The race for the $1000 genome. Science. 2006;311:1544–1546. doi: 10.1126/science.311.5767.1544. [DOI] [PubMed] [Google Scholar]
- 15.Nakamura S, Yang CS, Sakon N, et al. Direct metagenomic detection of viral pathogens in nasal and fecal specimens using an unbiased high-throughput sequencing approach. PLoS One. 2009;4:e4219. doi: 10.1371/journal.pone.0004219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ramakrishnan MA, Tu ZJ, Singh S, et al. The feasibility of using high resolution genome sequencing of influenza A viruses to detect mixed infections and quasispecies. PLoS One. 2009;4:e7105. doi: 10.1371/journal.pone.0007105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ghedin E, Laplante J, DePasse J, et al. Deep sequencing reveals mixed infection with 2009 pandemic influenza A (H1N1) virus strains and the emergence of oseltamivir resistance. J Infect Dis. 2011;203:168–174. doi: 10.1093/infdis/jiq040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Greninger AL, Chen EC, Sittler T, et al. A metagenomic analysis of pandemic influenza A (2009 H1N1) infection in patients from North America. PLoS One. 2010;5:e13381. doi: 10.1371/journal.pone.0013381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fox CH, Johnson FB, Whiting J, et al. Formaldehyde fixation. J Histochem Cytochem. 1985;33:845–853. doi: 10.1177/33.8.3894502. [DOI] [PubMed] [Google Scholar]
- 20.Tang W, David FB, Wilson MM, et al. DNA extraction from formalin-fixed, paraffin-embedded tissue. Cold Spring Harb Protoc 2009. 2009 doi: 10.1101/pdb.prot5138. pdb prot5138. [DOI] [PubMed] [Google Scholar]
- 21.April C, Klotzle B, Royce T, et al. Whole-genome gene expression profiling of formalin-fixed, paraffin-embedded tissue samples. PLoS One. 2009;4:e8162. doi: 10.1371/journal.pone.0008162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mittempergher L, de Ronde JJ, Nieuwland M, et al. Gene expression profiles from formalin fixed paraffin embedded breast cancer tissue are largely comparable to fresh frozen matched tissue. PLoS One. 2011;6:e17163. doi: 10.1371/journal.pone.0017163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.MacConaill LE, Campbell CD, Kehoe SM, et al. Profiling critical cancer gene mutations in clinical tumor samples. PLoS One. 2009;4:e7887. doi: 10.1371/journal.pone.0007887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beck AH, Weng Z, Witten DM, et al. 3′-end sequencing for expression quantification (3SEQ) from archival tumor samples. PLoS One. 2010;5:e8768. doi: 10.1371/journal.pone.0008768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schweiger MR, Kerick M, Timmermann B, et al. Genome-wide massively parallel sequencing of formaldehyde fixed-paraffin embedded (FFPE) tumor tissues for copy-number- and mutation-analysis. PLoS One. 2009;4:e5548. doi: 10.1371/journal.pone.0005548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wood HM, Belvedere O, Conway C, et al. Using next-generation sequencing for high resolution multiplex analysis of copy number variation from nanogram quantities of DNA from formalin-fixed paraffin-embedded specimens. Nucleic Acids Res. 2010;38:e151. doi: 10.1093/nar/gkq510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reid AH, Fanning TG, Hultin JV, et al. Origin and evolution of the 1918 “Spanish” influenza virus hemagglutinin gene. Proc Natl Acad Sci U S A. 1999;96:1651–1656. doi: 10.1073/pnas.96.4.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reid AH, Janczewski TA, Lourens RM, et al. 1918 influenza pandemic caused by highly conserved viruses with two receptor-binding variants. Emerg Infect Dis. 2003;9:1249–1253. doi: 10.3201/eid0910.020789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sheng ZM, Chertow DS, Ambroggio X, et al. Autopsy series of 68 cases dying before and during the 1918 influenza pandemic peak. Proc Natl Acad Sci U S A. 2011;108:16416–16421. doi: 10.1073/pnas.1111179108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Taubenberger JK, Reid AH, Krafft AE, et al. Initial genetic characterization of the 1918 “Spanish” influenza virus. Science. 1997;275:1793–1796. doi: 10.1126/science.275.5307.1793. [DOI] [PubMed] [Google Scholar]
- 31.Denison AM, Blau DM, Jost HA, et al. Diagnosis of influenza from respiratory autopsy tissues: detection of virus by real-time reverse transcription-PCR in 222 cases. J Mol Diagn. 2011;13:123–128. doi: 10.1016/j.jmoldx.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Taubenberger JK, Morens DM. The pathology of influenza virus infections. Annu Rev Pathol. 2008;3:499–522. doi: 10.1146/annurev.pathmechdis.3.121806.154316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Evers DL, Fowler CB, Cunningham BR, et al. The effect of formaldehyde fixation on RNA: optimization of formaldehyde adduct removal. J Mol Diagn. 2011;13:282–288. doi: 10.1016/j.jmoldx.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Evers DL, He J, Kim YH, et al. Paraffin embedding contributes to RNA aggregation, reduced RNA yield, and low RNA quality. J Mol Diagn. 2011;13:687–694. doi: 10.1016/j.jmoldx.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Foss RD, Guha-Thakurta N, Conran RM, et al. Effects of fixative and fixation time on the extraction and polymerase chain reaction amplification of RNA from paraffin-embedded tissue. Comparison of two housekeeping gene mRNA controls. Diagn Mol Pathol. 1994;3:148–155. doi: 10.1097/00019606-199409000-00003. [DOI] [PubMed] [Google Scholar]
- 36.Krafft AE, Duncan BW, Bijwaard KE, et al. Optimization of the isolation and amplification of RNA From formalin-fixed, paraffin-embedded tissue: the Armed Forces Institute of Pathology Experience and Literature Review. Mol Diagn. 1997;2:217–230. doi: 10.1054/MODI00200217. [DOI] [PubMed] [Google Scholar]
- 37.McKinney MD, Moon SJ, Kulesh DA, et al. Detection of viral RNA from paraffin-embedded tissues after prolonged formalin fixation. J Clin Virol. 2009;44:39–42. doi: 10.1016/j.jcv.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 38.Masuda N, Ohnishi T, Kawamoto S, et al. Analysis of chemical modification of RNA from formalin-fixed samples and optimization of molecular biology applications for such samples. Nucleic Acids Res. 1999;27:4436–4443. doi: 10.1093/nar/27.22.4436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kash JC, Tumpey TM, Proll SC, et al. Genomic analysis of increased host immune and cell death responses induced by 1918 influenza virus. Nature. 2006;443:578–581. doi: 10.1038/nature05181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Simpson JT, Wong K, Jackman SD, et al. ABySS: a parallel assembler for short read sequence data. Genome Res. 2009;19:1117–1123. doi: 10.1101/gr.089532.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Altschul SF, Madden TL, Schaffer AA, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huson DH, Auch AF, Qi J, et al. MEGAN analysis of metagenomic data. Genome Res. 2007;17:377–386. doi: 10.1101/gr.5969107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huson DH, Mitra S, Ruscheweyh HJ, et al. Integrative analysis of environmental sequences using MEGAN4. Genome Res. 2011;21:1552–1560. doi: 10.1101/gr.120618.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tariq MA, Kim HJ, Jejelowo O, et al. Whole-transcriptome RNAseq analysis from minute amount of total RNA. Nucleic Acids Res. 2011;39:e120. doi: 10.1093/nar/gkr547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhulidov PA, Bogdanova EA, Shcheglov AS, et al. Simple cDNA normalization using Kamchatka crab duplex-specific nuclease. Nucleic Acids Res. 2004;32:e37. doi: 10.1093/nar/gnh031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li H, Handsaker B, Wysoker A, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bao Y, Bolotov P, Dernovoy D, et al. The influenza virus resource at the National Center for Biotechnology Information. J Virol. 2008;82:596–601. doi: 10.1128/JVI.02005-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Trapnell C, Williams BA, Pertea G, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Klopfleisch R, Weiss AT, Gruber AD. Excavation of a buried treasure – DNA, mRNA, miRNA and protein analysis in formalin fixed, paraffin embedded tissues. Histol Histopathol. 2011;26:797–810. doi: 10.14670/HH-26.797. [DOI] [PubMed] [Google Scholar]
- 51.Farragher SM, Tanney A, Kennedy RD, et al. RNA expression analysis from formalin fixed paraffin embedded tissues. Histochem Cell Biol. 2008;130:435–445. doi: 10.1007/s00418-008-0479-7. [DOI] [PubMed] [Google Scholar]
- 52.Frank M, Doring C, Metzler D, et al. Global gene expression profiling of formalin-fixed paraffin-embedded tumor samples: a comparison to snap-frozen material using oligonucleotide microarrays. Virchows Arch. 2007;450:699–711. doi: 10.1007/s00428-007-0412-9. [DOI] [PubMed] [Google Scholar]
- 53.Hewitt SM, Lewis FA, Cao Y, et al. Tissue handling and specimen preparation in surgical pathology: issues concerning the recovery of nucleic acids from formalin-fixed, paraffin-embedded tissue. Arch Pathol Lab Med. 2008;132:1929–1935. doi: 10.5858/132.12.1929. [DOI] [PubMed] [Google Scholar]
- 54.Yi H, Cho YJ, Won S, et al. Duplex-specific nuclease efficiently removes rRNA for prokaryotic RNA-seq. Nucleic Acids Res. 2011;39:e140. doi: 10.1093/nar/gkr617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Christodoulou DC, Gorham JM, Herman DS, et al. Construction of normalized RNA-seq libraries for next-generation sequencing using the crab duplex-specific nuclease. Curr Protoc Mol Biol. 2011;Ch 4(Unit 4):12. doi: 10.1002/0471142727.mb0412s94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen R, Holmes EC. Avian influenza virus exhibits rapid evolutionary dynamics. Mol Biol Evol. 2006;23:2336–2341. doi: 10.1093/molbev/msl102. [DOI] [PubMed] [Google Scholar]
- 57.Jenkins GM, Rambaut A, Pybus OG, et al. Rates of molecular evolution in RNA viruses: a quantitative phylogenetic analysis. J Mol Evol. 2002;54:156–165. doi: 10.1007/s00239-001-0064-3. [DOI] [PubMed] [Google Scholar]
- 58.Kao WC, Chan AH, Song YS. ECHO: a reference-free short-read error correction algorithm. Genome Res. 2011;21:1181–1192. doi: 10.1101/gr.111351.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kircher M, Stenzel U, Kelso J. Improved base calling for the Illumina Genome Analyzer using machine learning strategies. Genome Biol. 2009;10:R83. doi: 10.1186/gb-2009-10-8-r83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nakamura K, Oshima T, Morimoto T, et al. Sequence-specific error profile of Illumina sequencers. Nucleic Acids Res. 2011;39:e90. doi: 10.1093/nar/gkr344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chan PK, Lee N, Joynt GM, et al. Clinical and virological course of infection with haemagglutinin D222G mutant strain of 2009 pandemic influenza A (H1N1) virus. J Clin Virol. 2011;50:320–324. doi: 10.1016/j.jcv.2011.01.013. [DOI] [PubMed] [Google Scholar]
- 62.Memoli MJ, Bristol T, Proudfoot KE, et al. In vivo evaluation of pathogenicity and transmissibility of influenza A(H1N1)pdm09 hemagglutinin receptor binding domain 222 intrahost variants isolated from a single immunocompromised patient. Virology. 2012;428:21–29. doi: 10.1016/j.virol.2012.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Morlighem JE, Aoki S, Kishima M, et al. Mutation analysis of 2009 pandemic influenza A(H1N1) viruses collected in Japan during the peak phase of the pandemic. PLoS One. 2011;6:e18956. doi: 10.1371/journal.pone.0018956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pan C, Cheung B, Tan S, et al. Genomic signature and mutation trend analysis of pandemic (H1N1) 2009 influenza A virus. PLoS One. 2010;5:e9549. doi: 10.1371/journal.pone.0009549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Miotto O, Heiny AT, Albrecht R, et al. Complete-proteome mapping of human influenza A adaptive mutations: implications for human transmissibility of zoonotic strains. PLoS One. 2010;5:e9025. doi: 10.1371/journal.pone.0009025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Reid AH, Fanning TG, Janczewski TA, et al. Novel origin of the 1918 pandemic influenza virus nucleoprotein gene. J Virol. 2004;78:12462–12470. doi: 10.1128/JVI.78.22.12462-12470.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liang Y, Huang T, Ly H, et al. Mutational analyses of packaging signals in influenza virus PA, PB1, and PB2 genomic RNA segments. J Virol. 2008;82:229–236. doi: 10.1128/JVI.01541-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Neumann G, Brownlee GG, Fodor E, et al. Orthomyxovirus replication, transcription, and polyadenylation. Curr Top Microbiol Immunol. 2004;283:121–143. doi: 10.1007/978-3-662-06099-5_4. [DOI] [PubMed] [Google Scholar]
- 69.Fodor E, Pritlove DC, Brownlee GG. Characterization of the RNA-fork model of virion RNA in the initiation of transcription in influenza A virus. J Virol. 1995;69:4012–4019. doi: 10.1128/jvi.69.7.4012-4019.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hoffmann E, Stech J, Guan Y, et al. Universal primer set for the full-length amplification of all influenza A viruses. Arch Virol. 2001;146:2275–2289. doi: 10.1007/s007050170002. [DOI] [PubMed] [Google Scholar]
- 71.Lee KH, Seong BL. The position 4 nucleotide at the 3′ end of the influenza virus neuraminidase vRNA is involved in temporal regulation of transcription and replication of neuraminidase RNAs and affects the repertoire of influenza virus surface antigens. J Gen Virol. 1998;79:1923–1934. doi: 10.1099/0022-1317-79-8-1923. [DOI] [PubMed] [Google Scholar]
- 72.Kash JC, Qi L, Dugan VG, et al. Prior infection with classical swine H1N1 influenza viruses is associated with protective immunity to the 2009 pandemic H1N1 virus. Influenza Other Respir Viruses. 2010;4:121–127. doi: 10.1111/j.1750-2659.2010.00132.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kobasa D, Jones SM, Shinya K, et al. Aberrant innate immune response in lethal infection of macaques with the 1918 influenza virus. Nature. 2007;445:319–323. doi: 10.1038/nature05495. [DOI] [PubMed] [Google Scholar]
- 74.Safronetz D, Rockx B, Feldmann F, et al. Pandemic swine-origin H1N1 influenza A virus isolates show heterogeneous virulence in macaques. J Virol. 2011;85:1214–1223. doi: 10.1128/JVI.01848-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Moore WE, Holdeman LV. Human fecal flora: the normal flora of 20 Japanese-Hawaiians. Appl Microbiol. 1974;27:961–979. doi: 10.1128/am.27.5.961-979.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shoji H, Yoshida K, Niki Y. Lung abscess and pleuritis caused by Lactobacillus rhamnosus in an immunocompetent patient. J Infect Chemother. 2010;16:45–48. doi: 10.1007/s10156-009-0004-5. [DOI] [PubMed] [Google Scholar]
- 77.Chan PA, Mermel LA, Andrea SB, et al. Distinguishing characteristics between pandemic 2009–2010 influenza A (H1N1) and other viruses in patients hospitalized with respiratory illness. PLoS One. 2011;6:e24734. doi: 10.1371/journal.pone.0024734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Palacios G, Hornig M, Cisterna D, et al. Streptococcus pneumoniae coinfection is correlated with the severity of H1N1 pandemic influenza. PLoS One. 2009;4:e8540. doi: 10.1371/journal.pone.0008540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Skarbinski J, Jain S, Bramley A, et al. Hospitalized patients with 2009 pandemic influenza A (H1N1) virus infection in the United States – September–October 2009. Clinical Infect Dis. 2011;52 (Suppl 1):S50–S59. doi: 10.1093/cid/ciq021. [DOI] [PubMed] [Google Scholar]
- 80.Ramphal R, Small PM, Shands JW, Jr, et al. Adherence of Pseudomonas aeruginosa to tracheal cells injured by influenza infection or by endotracheal intubation. Infection Immun. 1980;27:614–619. doi: 10.1128/iai.27.2.614-619.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Runstadler JA, Happ GM, Slemons RD, et al. Using RRT-PCR analysis and virus isolation to determine the prevalence of avian influenza virus infections in ducks at Minto Flats State Game Refuge, Alaska, during August 2005. Arch Virol. 2007;152:1901–1910. doi: 10.1007/s00705-007-0994-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mortazavi A, Williams BA, McCue K, et al. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 83.Werner T. Next generation sequencing in functional genomics. Brief Bioinform. 2010;11:499–511. doi: 10.1093/bib/bbq018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.