

Abstract
In most bacteria, the nonmevalonate pathway is used to synthesize isoprene units. Dxr, the second step in the pathway, catalyzes the NADPH-dependent reductive isomerization of 1-deoxy-D-xylulose-5-phosphate (DXP) to 2-C-methyl-D-erythritol-4-phosphate (MEP). Dxr is inhibited by natural products fosmidomycin and FR900098, which bind in the DXP binding site. These compounds, while potent inhibitors of Dxr, lack whole cell activity against Mycobacterium tuberculosis (Mtb) due to their polarity. Our goal was to use the Mtb Dxr-fosmidomycin co-crystal structure to design bisubstrate ligands to bind to both the DXP and NADPH sites. Such compounds would be expected to demonstrate improved whole cell activity due to increased lipophilicity. Two series of compounds were designed and synthesized. Compounds from both series inhibited Mtb Dxr. The most potent compound (8) has an IC50 of 17.8 µM. Analysis shows 8 binds to Mtb Dxr via a novel, non-bisubstrate mechanism. Further, the diethyl ester of 8 inhibits Mtb growth making this class of compounds interesting lead molecules in the search for new antitubercular agents.
Tuberculosis (TB) remains a significant threat to global public health.1 TB is caused by the bacillus Mycobacterium tuberculosis (Mtb). According to the World Health Organization (WHO)2, one-third of the world's population is currently infected with Mtb, which is responsible for nearly 2 million deaths each year. Current treatment of uncomplicated TB requires a combination of four drugs (isoniazid, rifampin, pyrazinamide, ethambutol) over a 6–9 month period.3 Emergence of drug-resistant Mtb strains is a serious threat to TB control and treatment.4–6 Further, co-infection of TB and HIV has fueled the epidemic. Therefore, there remains an urgent need for the discovery and development of novel antitubercular agents.
As in most bacteria, Mtb synthesizes five-carbon isoprenoids via the nonmevalonate pathway (NMP). These metabolites are needed for bacterial cell wall biosynthesis and other essential processes. Humans use the alternate (mevalonate) pathway to biosynthesize the same isoprenoid units. Thus, there are no human homologs for the enzymes of the NMP. Targeting the bacterial enzymes for antibiotic development should not interfere with human isoprene biosynthesis,7 making the NMP an attractive pathway in the search for novel drugs.
The second step of the NMP is mediated by the enzyme 1-deoxy-D-xylulose 5-phosphate reductoisomerase (Dxr, Figure 1).1, 8 Dxr is essential for Mtb survival9 and catalyzes the isomerization and reduction of 1-deoxy-D-xylulose-5-phosphate (DXP) to 2-C-methyl-D-erythritol-4-phosphate (MEP). In the reaction, NADPH is a hydride donor and a metal cation (Mg2+ or Mn2+) is a required enzyme cofactor. Current anti-TB drugs do not target the NMP, thus, Dxr inhibition would be a new mechanism of action, and Dxr inhibitors would be expected to be effective against drug-resistant strains of Mtb.
Fig. 1.

Dxr catalyzes the reduction and isomerization of DXP to form MEP.
Fosmidomycin (1) and its acetyl derivative FR900098 (2) are natural products isolated from Streptomyces lavendulae (Figure 2). 10 These secondary metabolites are both known inhibitors of Dxr,11 and fosmidomycin is currently under clinical investigation due to its activity against a variety of Gram-negative and Gram-positive bacteria, as well as malaria parasites.12–14 In these pathogens, fosmidomycin is actively transported into cells via a glycerol-3-phosphate transporter, GlpT.15 Mtb, however, does not have GlpT. This, combined with both the hydrophilic nature of fosmidomycin and the highly hydrophobic Mtb cell wall, renders fosmidomycin inactive against Mtb.9, 16 However, we have shown that lipophilic prodrugs of FR900098 demonstrate effective antitubercular activity17 and act in a GlpT-independent manner.18 Additionally, a range of synthetic fosmidomycin and FR900098 analogs have been described, designed to compete with DXP at the substrate-binding site.19–27 While demonstrating potent inhibition of the purified enzyme, none of these analogs, to our knowledge, is active against intact mycobacteria. Our goal was to expand on this work designing Dxr inhibitors with a novel binding mechanism and sufficient lipophilicity to gain whole cell antimycobacterial activity.
Fig. 2.

Structures of fosmidomycin (1), FR900098 (2) and amide- or O-linked analogs (3–9).
Several crystal structures of Dxr have been reported, facilitating the rational design of novel inhibitors.28, 29 The structure of Mtb Dxr in complex with the competitive inhibitor fosmidomycin has led to the identification of binding sites in the enzyme and is an excellent template for protein-ligand docking.28 In particular, the crystal structure has revealed chelation between the retrohydroxamate moiety of fosmidomycin and a metal cation, the phosphonate binding site, and the close binding proximity of fosmidomycin and NADPH. The nicotinamide ring of NADPH binds approximately 3.5 Å from the formyl carbon atom of fosmidomycin (Figure 3A). Hence, our approach toward Dxr inhibitor development was based on the design of fosmidomycin/FR900098 analogs that target the two major binding sites in Dxr: the fosmidomycin/DXP site and the NADPH site. Our goal was to bridge these adjacent binding sites to yield a high affinity, bisubstrate ligand, while considering the need for increased lipophilicity compared with fosmidomycin/FR900098. We report here the design, synthesis, and evaluation of two series of compounds with either amide- or O-linked substituents appended to the retrohydroxamate moiety of FR900098 (Figure 2) and evidence of a novel, non-bisubstrate mode of binding.
Fig. 3.

Mtb Dxr active site and docking results. (A) Active site of Mtb Dxr with fosmidomycin (left ligand) and NADPH (right ligand) bound (pdb 2JCZ).18 Ligands are separated by ~3.5 Å. (B) Docked O-linked ligand 4. (C) Docked amide ligand 8. Protein chain A is shown as cartoon (blue), Mn2+ as a sphere (pink), ligands are shown as sticks colored by atom type, protein residues as lines colored by atom type. Hydrogen atoms have been hidden for clarity.
Results
Modeling of amide- and O-linked ligands
Several studies describe the in silico evaluation of inhibitors against Dxr.20, 30–35 These reports highlight the challenges of modeling a protein that undergoes significant conformational change upon cofactor binding. We sought to use docking to discern whether bisubstrate inhibition of Mtb Dxr with amide or O-linked compounds was feasible.
200 Compounds with the general structures shown in Figure 2 were docked into the Mtb Dxr structure.28 NADPH and fosmidomycin were removed from the active site, while Mn2+ was kept in place. The analogs retained the phosphonate and backbone of the parent compounds, designed to ensure affinity to the DXP binding site in the enzyme. The analogs were appended with (un)substituted aromatic, alkylaryl, or (cyclo)alkyl substituents to the N-hydroxy oxygen atom or the amide carbonyl group of the retrohydroxamate. These structural features were designed to increase affinity to the NADPH binding site. Specifically, we were interested in testing aromatic substituents as mimics of the nicotinamide ring of NADPH, aiming to increase affinity to that pocket. Cycloalkyl groups were used to examine the importance of an aryl substituent.
The docking results predicted that our ligands would adopt a bisubstrate binding mode and bridge the two adjacent binding sites. Representative docking images are shown in Figures 3B and 3C. In general, the amide-linked compounds were expected to bind with greater affinity compared with the O-linked compounds. This was anticipated due to differences between the two series in binding the divalent cation. Prior work has shown that the unsubstituted hydroxyl oxygen of the retrohydroxamate is required for tight coordination of the metal.36 The aryl groups from each series docked in the NADPH binding site (Figures 3B and 3C). Collectively, the docking results indicated that the two series might work well as bisubstrate inhibitors of Mtb Dxr and, as such, bind in a manner distinct from fosmidomycin and FR900098.
Synthesis
The preparation of these compounds is shown in Schemes 1 and 2 and in Supplementary Information. A new synthetic route was developed for the synthesis of the O-linked compounds 3–6, using straightforward reactions allowing fast access to small, structurally modified molecules in good yield (Scheme 1).
Scheme 1.

Synthesis of O-linked compounds 3–6. Conditions: (a) BnONHAc, NaH, NaI, THF, 70°C, 75 %; (b) H2, Pd/C, MeOH, 76 %; (c) R(CH2)nBr, NaH, THF, 70°C, 27–81 %; (d) i: TMSBr, CH2Cl2, ii: H2O, iii: NaOHaq, 61 %-quant.
Scheme 2.

Synthesis of amide ligands 7 and 8. Conditions: (a) 2N HCl, 84 %; (b) BnONH2, MeOH, 40°C, then NaBH3CN, HCl, MeOH, 79 %; (c) R(CH2)nCOCl, Et3N, CH2Cl2, 50–75 %; (d) H2, Pd/C, MeOH, 56–59 %; (e) i: TMSBr, CH2Cl2, ii: H2O, iii: NaOHaq, quant.
3-Bromopropylphosphonate 10 was prepared from triethyl phosphite and 1,3-dibromopropane, using the Arbuzov reaction under microwave irradiation.37 Compound 10 and N-(benzyloxy)acetamide were then combined under basic conditions to give 11, which yielded hydroxylamine 12 after hydrogenation. O-linked compounds 13a-d were obtained by alkylation of intermediate 12 using sodium hydride and the corresponding aryl or alkyl bromide. The four desired monosodium salts 3–6 were obtained after removal of the diethyl ester using bromotrimethylsilane, hydrolysis of the resulting silylester with water, and treatment with 1 equivalent of sodium hydroxide.
Amide ligands 7 and 8 were prepared using the synthetic route shown in Scheme 2. Aldehyde 15 was synthesized from commercially available acetal 14 in acidic conditions. Compound 15 was combined with O-benzylhydroxylamine. Reduction with sodium cyanoborohydride and hydrochloric acid gave diethyl ester 16.38 Alkylation of 16 using triethylamine and the corresponding acyl chloride gave amide intermediates 17. Monosodium salts 7 and 8 were afforded after deprotection of both the retrohydroxamate and the phosphonate ester. Amide 9 was prepared using a similar path shown in the Supplementary Information.
As has been seen with related compounds, most of the monosodium salts were isolated and evaluated as a mixture of two conformers.39 Indeed, Zinglé et al. showed that N-substituted or N- and O-substituted hydroxamic acids were usually present as a mixture of Z and E conformers because of the restricted rotation around the C-N bond.39 Moreover, the ratio was dependent on the substituent and the nature of the solvent.
Biochemical and antitubercular evaluation
To evaluate the inhibitory activity of compounds 3–9, enzyme assays were performed with purified Mtb Dxr using a reported spectrophotometric assay monitoring NADPH consumption.28, 40, 41 The half maximal inhibitory activity (IC50) of the compounds is shown in Table 1 (see also Supplementary Information, Figure 1). Among the O-linked ligands (compounds 3–6), compound 5 demonstrated the greatest inhibition, with an IC50 of 48.4 µM. The activity of the O-linked ligands is surprising and challenges the notion that chelation of the metal cation by the retrohydroxamate is required for binding to the enzyme. For the O-linked compounds, binding of the aryl substituent could compensate for diminished coordination of the cation.
Table 1.
Mtb Dxr IC50 values and GScores of amide and O-linked ligands
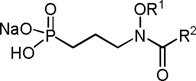 | |||
|---|---|---|---|
| compd | R1 | R2 | IC50 (µM) |
| 1 | H | H | 0.3116 |
| 2 | H | CH3 | 2.39 |
| 3 | (CH2)2Ph | CH3 | (81.5)* |
| 4 | (CH2)3Ph | CH3 | (77.5)* |
| 5 | 4-ipr-benzyl | CH3 | 48.4 |
| 6 | CH2cyclohexyl | CH3 | (83.5)* |
| 7 | H | CH2Ph | 26.9 |
| 8 | H | (CH2)3Ph | 17.8 |
| 9 | H | cyclohexyl | (80.0)* |
Values in parentheses are percent remaining enzyme activity at 100 µM.
Table 1 shows that amide ligands 7 and 8 are more potent inhibitors of the enzyme with values of 26.9 and 17.8 µM, respectively. This result may arise from more efficient coordination of the metal cation by the amide versus O-linked analogs. In the amide series, a slightly longer chain length between the retrohydroxamate and the aryl group may be preferable, as compound 8 gave slightly better inhibition than compound 7. Weaker inhibition by compounds 6 and 9, both with a nonaromatic ring, highlight the importance of having an aromatic group in the inhibitor.
Since we designed our inhibitors to occupy both the DXP and NADPH binding sites, compound 8 was further examined to discern its mechanism of inhibition. Catalysis by Mtb Dxr undergoes an ordered bi bi reaction mechanism, wherein NADPH must bind to the enzyme before DXP can bind.42 This process is reflective of a protein conformational change that occurs upon NADPH binding, resulting in the formation of the DXP binding site. Fosmidomycin and FR900098 are competitive inhibitors with respect to DXP and uncompetitive relative to NADPH42 (see also Supplementary Figure 1). Hence, NADPH must bind to Dxr before either of these inhibitors can occupy the Dxr binding site. With this mechanism in mind, we used classical competition experiments to assess the mode of binding of ligand 8.
As illustrated in Figure 4, the double reciprocal plots indicate that ligand 8 is competitive with respect to DXP, but noncompetitive relative to NADPH. Thus, in stark contrast to fosmidomycin and FR900098, the binding mechanism of compound 8 does not require the initial binding of NADPH. Since inhibitor 8 does not directly compete with the binding of NADPH, the phenylpropyl substituent of 8 is likely binding in an alternate location. Interestingly, this site is likely distinct from the NADPH site, but may promote the same structural change that gives rise to DXP binding. Studies are underway to further elucidate the nature and kinetics of this novel mode of inhibitor binding to Dxr.
Fig. 4.

Mode of inhibition by ligand 8. The Lineweaver-Burk plots indicate that 8 is competitive with respect to DXP (A), but noncompetitive with respect to NADPH (B).
To complement the characterization of compound 8 against purified Mtb Dxr, we assayed the efficacy of 8 and its lipophilic diethyl phosphonate ester (diethyl-8, 18b) against whole cell Mtb (H37Rv, Table 2). While the hydrophilic fosmidomycin, FR900098, and compound 8 do not inhibit Mtb, the diethyl phosphonate ester of fosmidomycin17 and 18b display measurable antitubercular activity (Table 2). Collectively, the enzyme and growth inhibition assays highlight the novel antitubercular activity of the amide-linked series of Dxr inhibitors, identifying compound 8 as a prototypical lead molecule for the further development of a new class of novel antimycobacterial agents.
Table 2.
Antitubercular activity of ligands and their diethyl esters
| Compd | H37Rv Mtb MIC (µg/mL) |
|---|---|
| Fosmidomycin (1) | >500 |
| FR900098 (2) | >500 |
| Diethyl-1 | 250 |
| 8 | >200 |
| Diethyl-8 (18b) | 200 |
Conclusions
There is an urgent need for the development of new, highly active, and less toxic anti-TB drugs. Dxr is an attractive target since humans do not have the enzyme or a homolog. Dxr inhibitors should be effective against drug-resistant Mtb strains, and, due to NMP’s importance in early points of bacterial metabolism, may be effective against both active and latent Mtb. Aided by an available co-crystal structure, our docking studies suggested that analogs of FR900098 with amide- or O-linked aryl substituents on the retrohydroxamate could be novel and potent inhibitors of Mtb Dxr. The inhibition data from our initial set of ligands (3–9), while not more active than the parent compounds, show promising results, with our best compound (8) having an IC50 of 17.8 µM. Indeed, our inhibition results align with prior reports of similar N-acyl analogs against E. coli and P. falciparum Dxr.26, 33, 35
Compound 8 displays a new mode of binding to Mtb Dxr not previously seen with parent compounds fosmidomycin and FR900098 or other known Dxr inhibitors. Prior modeling studies on similar N-acyl analogs indicated possible binding in a non-NADPH site.26 Ligand 8 could be using this alternate binding site and current studies are aimed at elucidating this. While this binding mode was not part of our original bisubstrate design per se, demonstration of a novel binding mode is both interesting and significant.
The MIC of the simple diethyl ester of 8 is comparable to the corresponding fosmidomycin ester and represents a new class of antitubercular compounds. As the MIC reflects both the lipophilicity and enzyme inhibition of 8, we expect it to improve as inhibitors with optimized potencies against Dxr are developed. Work is currently underway to explore an expanded set of analogs, reveal a comprehensive SAR, prove an on-target mechanism, and discern details of this new binding mode. Taken together, our data suggests new possibilities in the design and development of novel, antimycobacterial Dxr inhibitors.
Supplementary Material
Acknowledgement
This work was supported by the Intramural Research Program of NIAID/NIH, the George Washington University Department of Chemistry, the GWU University Facilitating Fund, and NIH (AI086453 to CSD). RDC was supported by George Mason University’s Department of Chemistry and Biochemistry and the U.S. Army Medical Research and Materiel Command W23RYX1291N601.
Footnotes
Electronic Supplementary Information (ESI) available: Full experimental details and spectral information for intermediates N-(benzyloxy)acetamide, 10–13a–d, 17–18a,b, and ligand 9, as well as biochemical and Mtb assay methods and full results. See DOI: 10.1039/b000000x/
Contributor Information
Kylene Kehn-Hall, Email: kkhall@gmu.edu.
Helena I. Boshoff, Email: hboshoff@niaid.nih.gov.
Robin D. Couch, Email: rcouch@gmu.edu.
Cynthia S. Dowd, Email: cdowd@gwu.edu.
Notes and references
- 1.Dye C, Glaziou P, Floyd K, Raviglione M. Annu Rev Public Health. 2013;34:271–286. doi: 10.1146/annurev-publhealth-031912-114431. [DOI] [PubMed] [Google Scholar]
- 2.Global Tuberculosis Control 2009: Epidemiology, Strategy, Financing. Geneva, Switzerland: WHO Press; 2009. [Google Scholar]
- 3.WHO. Treatment of Tuberculosis: Guideline for National Programmes. Geneva, Switzerland: 2003. [Google Scholar]
- 4.Wright A. MMWR Morb Mortal Wkly Rep. 2006;55:301–305. [PubMed] [Google Scholar]
- 5.Emergency Update, 2008: Guidelines for the programmatic management of drug-resistant tuberculosis. Geneva, Switzerland: WHO Press; 2008. [Google Scholar]
- 6.Udwadia ZF, Amale RA, Ajbani KK, Rodrigues C. Clin Infect Dis. 2012;54:579–581. doi: 10.1093/cid/cir889. [DOI] [PubMed] [Google Scholar]
- 7.Testa CA, Brown MJ. Curr Pharm Biotechnol. 2003;4:248–259. doi: 10.2174/1389201033489784. [DOI] [PubMed] [Google Scholar]
- 8.Proteau PJ. Bioorg Chem. 2004;32:483–493. doi: 10.1016/j.bioorg.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 9.Brown AC, Parish T. BMC Microbiol. 2008;8:78. doi: 10.1186/1471-2180-8-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Okuhara M, Kuroda Y, Goto T, Okamoto M, Terano H, Kohsaka M, Aoki H, Imanaka H. J Antibiot (Tokyo) 1980;33:13–17. doi: 10.7164/antibiotics.33.13. [DOI] [PubMed] [Google Scholar]
- 11.Kuzuyama T, Shimizu T, Takahashi S, Seto H. Tet. Lett. 1998;39:7913–7916. [Google Scholar]
- 12.Jomaa H, Wiesner J, Sanderbrand S, Altincicek B, Weidemeyer C, Hintz M, Turbachova I, Eberl M, Zeidler J, Lichtenthaler HK, Soldati D, Beck E. Science. 1999;285:1573–1576. doi: 10.1126/science.285.5433.1573. [DOI] [PubMed] [Google Scholar]
- 13.Singh N, Cheve G, Avery MA, McCurdy CR. Current Pharmaceutical design. 2007;13:1161–1177. doi: 10.2174/138161207780618939. [DOI] [PubMed] [Google Scholar]
- 14.Nolan Y, Martin D, Campbell VA, Lynch MA. J Neuroimmunol. 2004;151:12–23. doi: 10.1016/j.jneuroim.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 15.Sakamoto Y, Furukawa S, Ogihara H, Yamasaki M. Biosci Biotechnol Biochem. 2003;67:2030–2033. doi: 10.1271/bbb.67.2030. [DOI] [PubMed] [Google Scholar]
- 16.Dhiman RK, Schaeffer ML, Bailey AM, Testa CA, Scherman H, Crick DC. J Bacteriol. 2005;187:8395–8402. doi: 10.1128/JB.187.24.8395-8402.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Uh E, Jackson ER, San Jose G, Maddox M, Lee RE, Lee RE, Boshoff HI, Dowd CS. Bioorganic & medicinal chemistry letters. 2011;21:6973–6976. doi: 10.1016/j.bmcl.2011.09.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McKenney ES, Sargent M, Khan H, Uh E, Jackson ER, Jose GS, Couch RD, Dowd CS, van Hoek ML. PLoS One. 2012;7:e38167. doi: 10.1371/journal.pone.0038167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Andaloussi M, Henriksson LM, Wieckowska A, Lindh M, Bjorkelid C, Larsson AM, Suresh S, Iyer H, Srinivasa BR, Bergfors T, Unge T, Mowbray SL, Larhed M, Jones TA, Karlen A. Journal of medicinal chemistry. 2011;54:4964–4976. doi: 10.1021/jm2000085. [DOI] [PubMed] [Google Scholar]
- 20.Deng L, Diao J, Chen P, Pujari V, Yao Y, Cheng G, Crick DC, Prasad BV, Song Y. Journal of medicinal chemistry. 2011;54:4721–4734. doi: 10.1021/jm200363d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Devreux V, Wiesner J, Jomaa H, Rozenski J, Van der Eycken J, Van Calenbergh S. J Org Chem. 2007;72:3783–3789. doi: 10.1021/jo0700981. [DOI] [PubMed] [Google Scholar]
- 22.Devreux V, Wiesner J, Jomaa H, Van der Eycken J, Van Calenbergh S. Bioorg Med Chem Lett. 2007;17:4920–4923. doi: 10.1016/j.bmcl.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 23.Haemers T, Wiesner J, Van Poecke S, Goeman J, Henschker D, Beck E, Jomaa H, Van Calenbergh S. Bioorg Med Chem Lett. 2006;16:1888–1891. doi: 10.1016/j.bmcl.2005.12.082. [DOI] [PubMed] [Google Scholar]
- 24.Kurz T, Behrendt C, Pein M, Kaula U, Bergmann B, Walter RD. Archiv der Pharmazie. 2007;340:661–666. doi: 10.1002/ardp.200700107. [DOI] [PubMed] [Google Scholar]
- 25.Kurz T, Schluter K, Pein M, Behrendt C, Bergmann B, Walter RD. Archiv der Pharmazie. 2007;340:339–344. doi: 10.1002/ardp.200700013. [DOI] [PubMed] [Google Scholar]
- 26.Ortmann R, Wiesner J, Silber K, Klebe G, Jomaa H, Schlitzer M. Archiv der Pharmazie. 2007;340:483–490. doi: 10.1002/ardp.200700149. [DOI] [PubMed] [Google Scholar]
- 27.Verbrugghen T, Cos P, Maes L, Van Calenbergh S. Journal of medicinal chemistry. 2010;53:5342–5346. doi: 10.1021/jm100211e. [DOI] [PubMed] [Google Scholar]
- 28.Henriksson LM, Unge T, Carlsson J, Aqvist J, Mowbray SL, Jones TA. J Biol Chem. 2007;282:19905–19916. doi: 10.1074/jbc.M701935200. [DOI] [PubMed] [Google Scholar]
- 29.Mac Sweeney A, Lange R, Fernandes RP, Schulz H, Dale GE, Douangamath A, Proteau PJ, Oefner C. J Mol Biol. 2005;345:115–127. doi: 10.1016/j.jmb.2004.10.030. [DOI] [PubMed] [Google Scholar]
- 30.Andaloussi M, Lindh M, Bjorkelid C, Suresh S, Wieckowska A, Iyer H, Karlen A, Larhed M. Bioorganic & medicinal chemistry letters. 2011;21:5403–5407. doi: 10.1016/j.bmcl.2011.07.005. [DOI] [PubMed] [Google Scholar]
- 31.Bodill T, Conibear AC, Blatch GL, Lobb KA, Kaye PT. Bioorganic & medicinal chemistry. 2011;19:1321–1327. doi: 10.1016/j.bmc.2010.11.062. [DOI] [PubMed] [Google Scholar]
- 32.Deng L, Sundriyal S, Rubio V, Shi ZZ, Song Y. Journal of medicinal chemistry. 2009;52:6539–6542. doi: 10.1021/jm9012592. [DOI] [PubMed] [Google Scholar]
- 33.Giessmann D, Heidler P, Haemers T, Van Calenbergh S, Reichenberg A, Jomaa H, Weidemeyer C, Sanderbrand S, Wiesner J, Link A. Chem Biodivers. 2008;5:643–656. doi: 10.1002/cbdv.200890060. [DOI] [PubMed] [Google Scholar]
- 34.Patel AK. Int. J. Pharm. Bio. Sci. 2012;3:72–91. [Google Scholar]
- 35.Silber K, Heidler P, Kurz T, Klebe G. Journal of medicinal chemistry. 2005;48:3547–3563. doi: 10.1021/jm0491501. [DOI] [PubMed] [Google Scholar]
- 36.Marmion CJ, Griffith D, Nolan KB. European Journal of Inorganic Chemistry. 2004;15:3003–3016. [Google Scholar]
- 37.Villemin D, Simeon F, Decreus H, Jaffres P-A. Phosphorus, Sulfur and Silicon. 1998;133:209–213. [Google Scholar]
- 38.Puyau PDM, Perie JJ. Phosphorus, Sulfur and Silicon. 1996;112:71–90. [Google Scholar]
- 39.Zingle C, Kuntz L, Tritsch D, Grosdemange-Billiard C, Rohmer M. The Journal of organic chemistry. 2010;75:3203–3207. doi: 10.1021/jo9024732. [DOI] [PubMed] [Google Scholar]
- 40.Jawaid S, Seidle H, Zhou W, Abdirahman H, Abadeer M, Hix JH, van Hoek ML, Couch RD. PLoS One. 2009;4:e8288. doi: 10.1371/journal.pone.0008288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tsang A, Seidle H, Jawaid S, Zhou W, Smith C, Couch RD. PLoS One. 2011;6:e20884. doi: 10.1371/journal.pone.0020884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koppisch AT, Fox DT, Blagg BS, Poulter CD. Biochemistry. 2002;41:236–243. doi: 10.1021/bi0118207. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.