

Abstract
Advanced chronic kidney disease (CKD) is associated with impaired exercise capacity, skeletal muscle dysfunction, and oxidative stress. Mitochondria are the primary source for energy production and generation of reactive oxygen species (ROS). Mitochondrial state 3 respiration, mitochondrial complex I enzyme activity, and tissue porin/actin ratio were determined in the gastrocnemius muscle of male SD rats 14 weeks after 5/6 nephrectomy (CKD) or sham-operation (control). The CKD group exhibited azotemia, hypertension, significant reduction (-39%) of state 3 mitochondrial respiration, and a significant increase in the mitochondrial complex I enzyme activity. The latter is the first step in oxidative phosphorylation, a process linked to production of ROS. These abnormalities were associated with a significant reduction in muscle porin/β actin ratio denoting substantial reduction of mitochondrial mass in skeletal muscle of animals with CKD. CKD results in impaired mitochondrial respiration, reduced muscle mitochondrial mass, depressed energy production and increased ROS generation in the skeletal muscle. These events can simultaneously contribute to the reduction of exercise capacity and oxidative stress in CKD.
Keywords: End stage renal disease, oxidative stress, muscle weakness, exercise capacity, uremia
Introduction
Chronic kidney disease (CKD) has a profound negative impact on skeletal muscle function and structure leading to muscle atrophy and diminished exercise capacity [1-5]. The mechanisms by which CKD adversely affects skeletal muscle are complex and multifaceted and include: I- reduced substrate availability/delivery (uremic anorexia, Amino Acid removal by dialysis, insulin resistance, lipoprotein lipase and VLDL receptor deficiencies), impaired perfusion (peripheral vascular disease, CHF) and decreased oxygen carrying capacity (anemia)) II- catabolic state (inflammation, metabolic acidosis, growth hormone (GH) resistance, gonadal dysfunction, uremic neuropathy and physical inactivity) and III- impaired energy production (mitochondrial dysfunction/depletion) [1-5]. As the main engine of ATP generation, mitochondria represent the power plant of the aerobic cells and are among the most abundant organelles in myocytes. Consequently their potential deficiency or dysfunction can contribute to the impaired exercise capacity in CKD. In addition, mitochondrial dysfunction can lead to increased production of reactive oxygen species (ROS) and oxidative stress and promote cellular apoptosis, thereby contributing to the loss of muscle mass in this condition [1,6,7].
The available data on the effect of CKD on mitochondrial function, structure and abundance in skeletal muscle are limited and inconclusive. Earlier studies have revealed significant reduction in citrate synthase expression in CKD rats and a trend for reduced mitochondrial complex III and IV activity and altered skeletal muscle energy metabolism (increase dependence on non-oxidative ATP production) in patients with CKD [8-10]. The present study was designed to examine the effect of CKD on mitochondrial ATP generation capacity, the mitochondrial respiratory chain complex enzyme activities, the expression of the respiratory chain complex proteins, and the abundance of mitochondria in the skeletal muscle.
Methods
Animals
Male Sprague–Dawley rats with an average body weight of 225-250 g (Harlan Sprague-Dawley, Inc., Indianapolis, IL) were used in this study. Animals were housed in a climate-controlled vivarium with 12-h day and night cycles and were fed a standard laboratory diet (Purina Mills, Brentwood, MO) and water ad libitum. The animals were randomly assigned to the CKD and sham-operated control groups. The CKD group underwent 5/6 nephrectomy (5/6 Nx) by surgical resection of the upper and lower thirds of the left kidney, followed by right nephrectomy 7 days later. The control group underwent sham operation. The procedures were carried out under general anesthesia (sodium pentobarbital, 50 mg/kg i.p.) using strict hemostasis and aseptic techniques. Six animals were included in each group.
Timed urine collections were carried out at baseline and at week 12, using metabolic cages. Urine protein concentration (Chondrex, Inc., Redmond, WA) was determined in the 24-h urine samples. Blood pressure was determined by tail cuff plethysmography (CODA2, Kent Scientific Corporation, Torrington, CT). Briefly, conscious rats were placed in a restrainer on a warming pad and allowed to rest inside the cage for 15 min before blood pressure measurements. Rat tails were placed inside a tail cuff, and the cuff was inflated and released several times to allow the animal to be conditioned to the procedure. At the conclusion of the observation period (14 weeks) animals were anesthetized (sodium pentobarbital, 50 mg/kg i.p.) and euthanized by exsanguinations using cardiac puncture. Gastrocnemius muscle was immediately removed and a piece was excised for polarography as described below. The remaining tissue was cleaned with phosphate-buffered saline, snap-frozen in liquid nitrogen, and stored at -70°C until processed. Plasma total cholesterol (Stanbio Laboratory, Boerne, TX), triglyceride (Stanbio Laboratory, Boerne, TX), urea (Bioassay Systems, Hayward, CA), and creatinine (Bioassay Systems, Hayward, CA) were analyzed using the specified products. Creatinine clearance was calculated using standard equation and the 24 hour urine creatinine concentration (Bioassay systems, Hayward, CA). The experimental protocol was approved by the Institutional Animal Care and Use Committee of the University of California (Irvine, CA).
Mitochondria preparation
The skeletal muscles were homogenized in a mitochondria isolation buffer (225 mM mannitol, 75 mM sucrose, 10 mM MOPS (pH 7.2), 1 mM EGTA, 0.5% BSA, 3 μg/ml aprotinin, 3 μg/ml leupeptin, 2 mM phenylmethyl-sulfonyl fluoride (PMSF), 20 mM NaF, 10 mM NaPP, and 2 mM Na3VO4) on ice and homogenized with Dounce homogenizer. The homogenized mixture was centrifuged at 1000 g, 4°C for 15 min to remove cell debris, and the supernatants were spun down at 16,100 g, 4°C for 15 min to obtain the isolated mitochondria pellet. The pellets were re-suspended in mitochondria isolation buffer and used to assay oxygen consumption by poloragrophy, enzyme complex activity, and protein abundance by western blot.
Mitochondrial oxygen consumption
Respiration rates were measured at 25°C using an oxygraph system (Oxytherm; Hansatech Instruments, Norfolk, UK) with the mixture containing 20 mM pyruvate + 1 mM malate to test the mitochondrial respiratory chain activity. State 2 was measured in the presence of respiratory substrates without adenosine 5’-diphosphate (ADP). State 3 was measured after addition of 1 mM ADP. Respiratory control ratio (RCR) was calculated by dividing State 3 by State 2 respiration rate.
Oxidative phosphorylation complex activity assays
Mitochondria membranes were ruptured by freeze-thaw cycles, and OXPHO complex activities were measured as previously described [11]. In brief, equal amounts of mitochondria proteins were assayed and normalized to porin levels as outlined below.
Complex I + III
One ml reaction buffer consisting of 10 mM Tris-HCl (pH 8.0), 1 mg/ml BSA, 8 μM oxidized cytochrome c, and 40.8 μM KCN was added to the cuvette, and incubated at 37°C for 3 min with mitochondria proteins. The reaction was started by adding 0.8 mM NADH. Cytochrome c reduction at 550-540 nm was recorded for 3 min. 4 μM rotenone was added and absorbance at 550-540 nm was recorded for 3 min to quantify the rotenone-sensitive activity.
Complex II
One ml reaction buffer consisting of 10 mM KH2PO4 (pH 7.8), EDTA 2 mM, 1 mg/ml BSA, 80 μM DCPIP, 240 μM KCN, 4 μM rotenone and 200 μM ATP was placed in a cuvette and equal amount of mitochondria proteins was added to each reaction sample. The reaction was started by adding 80 μM decylubiquinone. The activities were measured by changes of absorbance at 600 nm for 3 min.
Complex IV
Equal amounts of mitochondria proteins were mixed with 1 ml reaction buffer containing 10 mM KH2PO4 (pH 6.5), 1 mg/ml BSA, 0.25 M sucrose and placed in the cuvette. The reaction was started by adding 10 μM reduced cytochrome c. The activity was recorded by the absorbance at 550-540 nm for 3 min.
Complex V
Fresh mitochondria proteins were added to 800 μl pre-warmed distilled water and 200 μl pre-warmed reaction buffer containing 50 mM Tris-HCl (pH 8.0), 1 mM NADH, 5 mg/ml BSA, 20 mM MgCl2, 50 mM KCl, 2.5 mM ATP, 15 μM carbonyl cyanide m-chlorophenylhydrazone, 10 mM phosphoenol pyruvate, 5 μM antimycin and 4 U of lactate dehydrogenase and pyruvate kinase at 37°C. The activity was measured by the absorbance at 340 nm for 3 min. 12 μM oligomycin was added to the reaction mixture to determine the oligomycin-sensitive complex V activity.
Western blot analyses
The skeletal muscles were homogenized in a mitochondria isolation buffer (225 mM mannitol, 75 mM sucrose, 10 mM MOPS (pH 7.2), 1 mM EGTA, 0.5% BSA, 3 μg/ml aprotinin, 3 μg/ml leupeptin, 2 mM phenylmethyl-sulfonyl fluoride (PMSF), 20 mM NaF, 10 mM NaPP, and 2 mM Na3VO4) on ice with Dounce homogenizer. The homogenized mixture was centrifuged at 1000 g, 4°C for 15 min to remove cell debris, and the supernatants were spun down at 16,100 g, 4°C for 15 min to obtain the isolated mitochondria pellet. The pellets were re-suspended in mitochondria isolation buffer. Protein concentration was measured prior to each Western blot analysis using a BCA Protein Assay Kit purchased from Pierce Biotechnology (Rockford, Ill., USA) following the manufacturer’s protocol. Aliquots containing 5-30 μg of protein were fractionated on 4-20% Bis-Tris gels (Invitrogen, Calif., USA) at 120 V for 2 h. After electrophoresis, proteins were transferred to Hybond enhanced chemiluminescence (ECL) membrane (Amersham Life Science, Arlington Heights, Ill., USA). The membrane was incubated for 1 h in blocking buffer (1× Tris-buffered saline, TBS, 0.1% Tween 20, 5% nonfat dry milk) and then overnight in the same buffer containing the primary antibody. All antibodies against Complex IV subunit IV, Complex IV subunit I, ATP Synthase Alpha, ATP Synthase Beta, and Porin, were purchased from Mitosciences. Beta actin antibody was purchased through Santa Cruz Biotechnology. Membrane was then washed three times for 7 min in 1× TBS, 0.1% Tween 20 before a 2-hour incubation in blocking buffer (1× TBS, 0.1% Tween 20, 5% nonfat dry milk) plus diluted horseradish peroxidase-linked anti-mouse IgG (Amersham Life Science). The washing procedures were repeated before the membranes were developed with chemiluminescent agents (ECL; Amersham Life Science) and subjected to autoluminography for 1 s to 5 min.
For measurement of porin/actin ratios, whole tissue lysates were prepared and used for Western blotting. Briefly, Frozen tissue was homogenized in 1 ml of 20 mM Tris-HCl (pH 7.5) buffer containing 2 mM MgCl2, 0.2 M sucrose and protease inhibitor cocktail (Sigma, St. Louis). The crude extract was centrifuged at 2,000 g at 4°C for 15 min to remove tissue debris. The supernatant was used for Western blot analyses.
Data analysis
Student’s t-test was used in statistical evaluation of the data which are shown as mean ± SEM. P-values <0.05 were considered significant.
Results
General data
As expected, compared with the sham-operated control rats, the CKD group showed a significant increase in arterial pressure and serum concentrations of creatinine, urea nitrogen, triglycerides, and cholesterol as well as urinary protein excretion (Table 1).
Table 1.
Plasma concentrations of creatinine (Cr), urea, triglycerides, and cholesterol, Creatinine clearance (Ccr), urine protein excretion and systolic and diastolic blood pressure in the CKD and normal control rats
| Control | CKD | |
|---|---|---|
| Cr, mg/dl | 0.50 ± 0.06 | 1.56 ± 0.23* |
| Urea, mg/dl | 54.3 ± 1.8 | 114.7 ± 4.8* |
| Ccr, ml/min-1.kg-1 | 5.62 ± 0.53 | 1.74 ± 0.19* |
| Urine protein, mg/24 h | 6.7 ± 0.9 | 80.3 ± 3.7* |
| Triglycerides, mg/dl | 45.8 ± 8.2 | 99.7 ± 2.1* |
| Total cholesterol, mg/dl | 71.2 ± 4.0 | 221.2 ± 10.3* |
| SBP, mmHg | 123.5 ± 6.0 | 161.4 ± 2.0* |
| DBP, mmHg | 87.5 ± 3.3 | 117.0 ± 3.2* |
Values are means ± SEM (n=5-6 in each group), Cr, creatinine; Ccr, creatinine clearance; SBP and DBP, systolic and diastolic blood pressure.
P<0.05.
Mitochondrial oxygen consumption by polarography
Data are summarized in Figure 1A. There was a 39% reduction in state 3 mitochondrial respiration in isolated skeletal muscle from CKD animals when compared with the control group (*p<0.05).
Figure 1.

A: State 3 oxygen consumption rates in purified mitochondria from gastrocnemius muscle were measured by the polarography method in the 5/6 nephrectomized (CKD) and sham-operated control rats. Data represent mean ± SEM (n=6). (*p<0.05). B: OXPHO complex activities in the gastrocnemius muscle of the 5/6 nephrectomized (CKD) and sham-operated control rats. OXPHO complex activities were normalized with the porin contents in each mitochondria sample. Data are expressed as the mean ± SEM (n=6 per group). (*p<0.05).
Mitochondrial enzyme complex activities
Data are summarized in Figure 1B. Mitochondrial enzyme complex I plus III activity was significantly increased (+18%) in the isolated skeletal muscle mitochondria from the CKD group when compared with sham-operated controls. This reaction constitutes the first step in oxidative phosphorylation by catalyzing oxidation of reduced nicotinamide adenine dinucleotide (NADH NAD+), a reaction which is coupled with production of ROS. In contrast, activity of the complex IV was reduced (-13%) while the activities of the remaining enzyme complexes were unchanged in the CKD animals when compared with the control rats.
Mitochondrial protein abundance
While there was a reduction in Complex IV subunit IV protein abundance in the isolated skeletal muscle mitochondria from CKD animals when compared with sham-operated controls, the difference did not reach statistical significance. The protein abundance of Complex IV subunit I, ATP Synthase Alpha, and ATP Synthase Beta was unchanged in skeletal muscle mitochondria from CKD animals when compared with controls (Figure 2). It should be noted that the lack of significant difference in the abundance of the given proteins does not imply absence of significant difference in their abundance in the whole tissue. This is because the measurements were obtained in isolated mitochondria and normalized against porin, mitochondrial housekeeping protein. Consequently normal values do not exclude potential depletion of cellular mitochondria and their protein constituents. To address this issue we determined relative abundance of mitochondrial housekeeping protein, porin, against cellular housekeeping protein β actin in the whole tissue protein extract to determine possible effect of CKD on the skeletal muscle mitochondrial mass.
Figure 2.
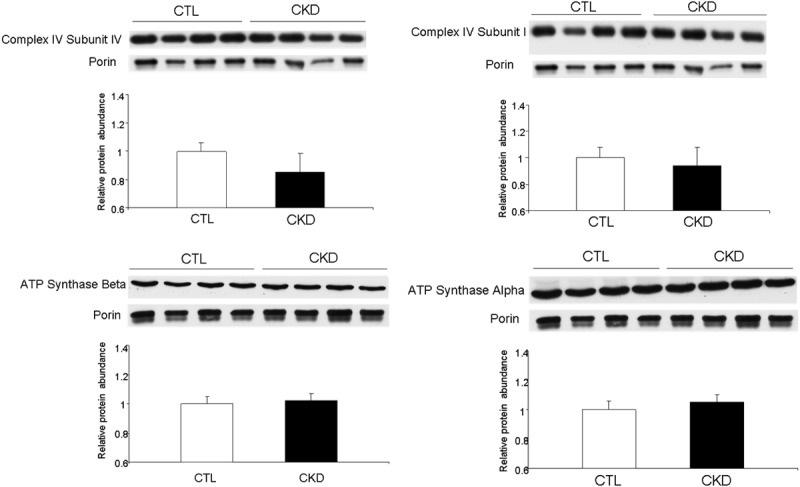
Representative Western blots and group data depicting gastrocnemius muscle Complex IV subunit IV, Complex IV subunit I, ATP Synthase Alpha, and ATP Synthase Beta protein abundance in the isolated skeletal mitochondria of the 5/6 nephrectomized (CKD) and sham-operated control rats. Data are expressed as the mean ± SEM (n=6 per group). (*p<0.05).
Whole tissue porin abundance
Porin/β actin ratio determined in the whole muscle tissue homogenate was markedly reduced in the CKD group as compared to the normal control rats. This observation indicates a significant reduction of mitochondrial mass in the skeletal muscle of the CKD animals. Thus mitochondrial dysfunction in CKD is compounded by mitochondrial depletion in skeletal muscle (Figure 3).
Figure 3.
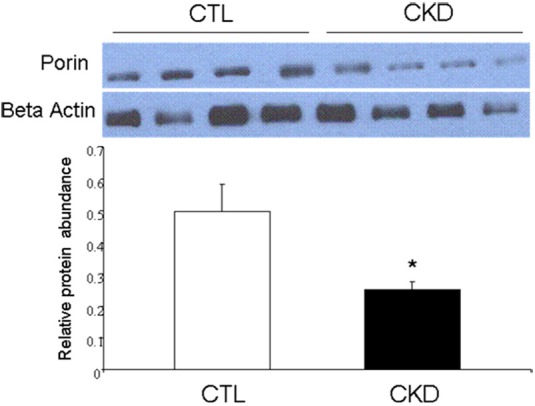
Representative Western blots and group data depicting porin abundance in gastrocnemius muscle homogenates from the 5/6 nephrectomized (CKD) and sham-operated control rats. Data are expressed as the mean ± SEM (n=6 per group). (*p<0.05).
Discussion
While the skeletal muscle is significantly and adversely impacted by CKD, the etiology of muscle dysfunction in CKD has not been definitively elucidated. The potential mechanisms by which CKD may negatively impact skeletal muscle are multifaceted and include alterations in muscle perfusion, substrate delivery, and catabolic state mediated by various factors such as metabolic acidosis, corticosteroids, pro-inflammatory cytokines, and decreased physical activity, among others [12,13]. While the mitochondria play a key role in energy metabolism of all organs especially the skeletal muscle, the available data on the effect of CKD on mitochondrial function, structure and abundance in skeletal muscle remains limited.
Several studies have been conducted to determine the potential role of mitochondrial dysfunction in CKD-induced skeletal muscle abnormalities. While skeletal muscle mitochondrial function did not appear to be significantly impaired in CKD patients when compared with controls, there were trends toward decreased citrate synthase (CS) as well as depressed mitochondrial complex III and IV activity [10]. The activity of CS is often used as an index of cellular oxidative capacity [14]. Earlier studies have shown significant abnormalities in energy metabolism in the muscles of CKD patients including dependence on non-oxidative ATP synthesis and decreased mitochondrial capacity during recovery [9]. We have reported a significant (31%) decrease in CS activity in the skeletal muscles of CKD (5/6 nephrectomized) rats [8]. Davis et al. [15] had previously reported that exercise training (swim training) could prevent the decrements in CS activity in 3/4 nephrectomized rats. Similarly, we found that voluntary wheel-running exercise blunted the de-conditioning effect of CKD in rats by preserving the skeletal muscle oxidative capacity [8]. Therefore, although the results of human studies are inconclusive, the results of animal studies suggest that oxidative capacity of the muscle is impaired in CKD animals and can be ameliorated by exercise.
In the current study, we found that CKD animals studied 12 wks after 5/6 nephrectomy exhibited a significant reduction in mitochondrial respiratory capacity. This further confirms the impaired oxidative capacity observed in previous studies and indicates that mitochondrial dysfunction is partially responsible for this finding. In order to determine the underlying mechanism for impaired oxidative capacity observed in animals with CKD we measured the activity of the five complexes of the electron transport chain. We found a marked reduction in mitochondrial Complex IV activity and Complex IV subunit IV protein abundance in the skeletal muscles of CKD animals when compared with the sham-operated controls. Although the differences did not reach statistical significance, there was a trend towards reduction of activity and protein abundance which is consistent with reduced mitochondrial respiratory capacity. The reduction in Complex IV activity and protein abundance may be, in part, responsible for impaired oxidative capacity in CKD.
In addition, we found a significant increase in Complex I and III activity in CKD animals when compared with the sham-operated controls. This can lead to increased ROS production and oxidative stress in skeletal muscle. Oxidative stress has been proposed as a principal mediator of skeletal muscle atrophy and sarcopenia. Increased oxidative burden can promote expression of genes encoding catabolic factors and activate apoptotic pathways thereby contributing to skeletal muscle atrophy [16-20]. Furthermore, prolonged exposure to relatively high levels of oxidants reduces mitochondrial membrane integrity, and causes mitochondrial dysfunction in skeletal muscle leading to decreased mitochondrial oxidative capacity as shown in animals with CKD here and in earlier studies [21,22]. The functional consequences of increased oxidative stress in skeletal muscle of CKD animals include decreased specific force, altered myofilament function, and muscle fatigue [23-25].
Porin is the principal protein constituent of the mitochondrial outer membrane and as such its abundance in the whole tissue can serve as a marker of mitochondrial mass. In the current study we found a significant reduction in the skeletal muscle porin/actin ratio in the CKD animals compared with the normal control rats. This finding points to the CKD-induced mitochondrial depletion in skeletal muscle. This phenomenon can, in part, accounts for the previous studies which revealed reduction in CS activity in skeletal muscle in CKD animals and contribute to the associated reductions in energy production and oxidative capacity [8]. The reduction in skeletal muscle mitochondrial mass in CKD may be due to reduced replication and increased destruction of mitochondria driven by diminished physical activity, oxidative stress and as-yet other unidentified factors. Our results seem to point to increased ROS as a driving force behind this decreased mitochondrial content.
In conclusion, the CKD animals exhibited: 1- Marked reduction (-39%) of state 3 mitochondrial respiration; 2- Significant increase in the mitochondrial complex I plus III enzyme activity (18%) which is a major source of ROS generation; 3- Reduction of the mitochondrial complex IV activity and Complex IV subunit IV protein; and 4- Significant reduction of tissue Porin/β actin ratio which points to diminished skeletal muscle mitochondrial mass. Together these results illustrate the adverse effect of uremia on the mitochondrial function, structure, and abundance in the skeletal muscle and their contribution to diminished exercise capacity in CKD.
Acknowledgements
We gratefully acknowledge the support of staff at UC Irvine animal care facilities.
This work was supported in part by a grant from the NIH DK082130 (H.M.) and National Kidney Foundation of Southern California (P.Y.).
Disclosure of conflict of interest
The authors have no conflicts of interest to disclose.
References
- 1.Adams GR, Vaziri ND. Skeletal muscle dysfunction in chronic renal failure: effects of exercise. Am J Physiol Renal Physiol. 2006;290:F753–61. doi: 10.1152/ajprenal.00296.2005. [DOI] [PubMed] [Google Scholar]
- 2.Workeneh BT, Mitch WE. Review of muscle wasting associated with chronic kidney disease. Am J Clin Nutr. 2010;91:1128S–1132S. doi: 10.3945/ajcn.2010.28608B. [DOI] [PubMed] [Google Scholar]
- 3.Conjard A, Ferrier B, Martin M, Caillette A, Carrier H, Baverel G. Effects of chronic renal failure on enzymes of energy metabolism in individual human muscle fibers. J Am Soc Nephrol. 1995;6:68–74. doi: 10.1681/ASN.V6168. [DOI] [PubMed] [Google Scholar]
- 4.Adey D, Kumar R, McCarthy JT, Nair KS. Reduced synthesis of muscle proteins in chronic renal failure. Am J Physiol Endocrinol Metab. 2000;278:E219–E225. doi: 10.1152/ajpendo.2000.278.2.E219. [DOI] [PubMed] [Google Scholar]
- 5.Bailey JL, Zheng B, Hu Z, Price SR, Mitch WE. Chronic kidney disease causes defects in signaling through the insulin receptor substrate/phosphatidylinositol 3-kinase/Akt pathway: implications for muscle atrophy. J Am Soc Nephrol. 2006;17:1388–1394. doi: 10.1681/ASN.2004100842. [DOI] [PubMed] [Google Scholar]
- 6.Naik E, Dixit VM. Mitochondrial reactive oxygen species drive proinflammatory cytokine production. J Exp Med. 2011;208:417–20. doi: 10.1084/jem.20110367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vendelbo MH, Nair KS. Mitochondrial longevity pathways. Biochim Biophys Acta. 2011;1813:634–44. doi: 10.1016/j.bbamcr.2011.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Adams GR, Zhan CD, Haddad FA, Vaziri ND. Voluntary exercise during chronic renal failure in rats. Med Sci Sports Exerc. 2005;37:557–562. doi: 10.1249/01.mss.0000159006.87769.67. [DOI] [PubMed] [Google Scholar]
- 9.Kemp GJ, Thompson CH, Taylor DJ, Radda GK. ATP production and mechanical work in exercising skeletal muscle: a theoretical analysis applied to 31P magnetic resonance spectroscopic studies of dialyzed uremic patients. Magn Reson Med. 1995;33:601–609. doi: 10.1002/mrm.1910330504. [DOI] [PubMed] [Google Scholar]
- 10.Miró O, Marrades RM, Roca J, Sala E, Masanés F, Campistol JM, Torregrosa JV, Casademont J, Wagner PD, Cardellach F. Skeletal muscle mitochondrial function is preserved in young patients with chronic renal failure. Am J Kidney Dis. 2002;39:1025–1031. doi: 10.1053/ajkd.2002.32776. [DOI] [PubMed] [Google Scholar]
- 11.Barrientos A. In vivo and in organello assessment of OXPHO activities. Methods. 2002;26:307–316. doi: 10.1016/S1046-2023(02)00036-1. [DOI] [PubMed] [Google Scholar]
- 12.Rajan VR, Mitch WE. Muscle wasting in chronic kidney disease: the role of the ubiquitin proteasome system and its clinical impact. Pediatr Nephrol. 2008;23:527–35. doi: 10.1007/s00467-007-0594-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Han HQ, Mitch WE. Targeting the myostatin signaling pathway to treat muscle wasting diseases. Curr Opin Support Palliat Care. 2011;5:334–41. doi: 10.1097/SPC.0b013e32834bddf9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Holloszy JO, Oscai LB, Don IJ, Mole PA. Mitochondrial citric acid cycle and related enzymes: adaptive response to exercise. Biochem Biophys Res Commun. 1970;40:1368–1373. doi: 10.1016/0006-291x(70)90017-3. [DOI] [PubMed] [Google Scholar]
- 15.Davis TA, Karl IE, Goldberg AP, Harter HR. Effects of exercise training on muscle protein catabolism in uremia. Kidney Int Suppl. 1983;16:S52–7. [PubMed] [Google Scholar]
- 16.DeMartino GN, Ordway GA. Ubiquitin-proteasome pathway of intracellular protein degradation: implications for muscle atrophy during unloading. Exerc Sport Sci Rev. 1998;26:219–252. [PubMed] [Google Scholar]
- 17.Li YP, Chen Y, Li AS, Reid MB. Hydrogen peroxide stimulates ubiquitin-conjugating activity and expression of genes for specific E2 and E3 proteins in skeletal muscle myotubes. Am J Physiol Cell Physiol. 2003;285:C806–C812. doi: 10.1152/ajpcell.00129.2003. [DOI] [PubMed] [Google Scholar]
- 18.Mantovani G, Madeddu C, Macciò A, Gramignano G, Lusso MR, Massa E, Astara G, Serpe R. Cancer-related anorexia/cachexia syndrome and oxidative stress: an innovative approach beyond current treatment. Cancer Epidemiol Biomarkers Prev. 2004;13:1651–1659. [PubMed] [Google Scholar]
- 19.Kagan VE, Tyurina YY, Bayir H, Chu CT, Kapralov AA, Vlasova II, Belikova NA, Tyurin VA, Amoscato A, Epperly M, Greenberger J, Dekosky S, Shvedova AA, Jiang J. The “pro-apoptotic genies” get out of mitochondria: oxidative lipidomics and redox activity of cytochrome c/cardiolipin complexes. Chem Biol Interact. 2006;163:15–28. doi: 10.1016/j.cbi.2006.04.019. [DOI] [PubMed] [Google Scholar]
- 20.Siu PM, Always SE. Mitochondria-associated apoptotic signaling in denervated rat skeletal muscle. J Physiol. 2005;565:309–323. doi: 10.1113/jphysiol.2004.081083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bejma J, Ji LL. Aging and acute exercise enhance free radical generation in rat skeletal muscle. J Appl Physiol. 1999;87:465–70. doi: 10.1152/jappl.1999.87.1.465. [DOI] [PubMed] [Google Scholar]
- 22.Yokota T, Kinugawa S, Hirabayashi K, Matsushima S, Inoue N, Ohta Y, Hamaguchi S, Sobirin MA, Ono T, Suga T, Kuroda S, Tanaka S, Terasaki F, Okita K, Tsutsui H. Oxidative stress in skeletal muscle impairs mitochondrial respiration and limits exercise capacity in type 2 diabetic mice. Am J Physiol Heart Circ Physiol. 2009;297:H1069–77. doi: 10.1152/ajpheart.00267.2009. [DOI] [PubMed] [Google Scholar]
- 23.Clanton TL, Zuo L, Klawitter P. Oxidants and skeletal muscle function: physiologic and pathophysiologic implications. Proc Soc Exp Biol Med. 1999;222:253–262. doi: 10.1046/j.1525-1373.1999.d01-142.x. [DOI] [PubMed] [Google Scholar]
- 24.Andrade FH, Reid MB, Allen DG, Westerblad H. Effect of hydrogen peroxide and dithiothreitol on contractile function of single skeletal muscle fibres from the mouse. J Physiol. 1998;509:565–575. doi: 10.1111/j.1469-7793.1998.565bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reid MB. Free radicals and muscle fatigue: of ROS, canaries, and the IOC. Free Radic Biol Med. 2008;44:169–179. doi: 10.1016/j.freeradbiomed.2007.03.002. [DOI] [PubMed] [Google Scholar]