

Abstract.
Collagen fibers can be imaged with second harmonic generation (SHG) and are associated with efficient tumor cell locomotion. Preferential locomotion along these fibers correlates with a more aggressively metastatic phenotype, and changes in SHG emission properties accompany changes in metastatic outcome. We therefore attempted to elucidate the cellular and molecular machinery that influences SHG in order to understand how the microstructure of tumor collagen fibers is regulated. By quantifying SHG and immunofluorescence (IF) from tumors grown in mice with and without stromal tumor necrosis factor (TNF)- and in the presence or absence of tumor-associated macrophages (TAMs), we determined that depletion of TAMs alters tumor collagen fibrillar microstructure as quantified by SHG and IF. Furthermore, we determined that abrogation of TNF- expression by tumor stromal cells also alters fibrillar microstructure and that subsequent depletion of TAMs has no further effect. In each case, metastatic burden correlated with optical readouts of collagen microstructure. Our results implicate TAMs and stromal TNF- as regulators of breast tumor collagen microstructure and suggest that this regulation plays a role in tumor metastasis. Furthermore, these results indicate that quantification of SHG represents a useful strategy for evaluating the cells and molecular pathways responsible for manipulating fibrillar collagen in breast tumor models.
Keywords: second harmonic generation, cancer, extracellular matrix, collagen, metastasis, multiphoton microscopy
1. Introduction
Fibrillar collagen is an extracellular matrix protein providing significant structural support to tumors and is characterized in part by its strong signal when imaged with a light-scattering process called second harmonic generation, or SHG. Collagen fibers that produce significant detectable SHG have been noted in breast tumor models as pathways of improved tumor cell locomotion.1,2 This efficient, biased movement along SHG-producing fibers contrasts strongly with the random walk exhibited by cells moving independently of the fibers and also positively correlates with increased metastatic behavior.3,4 This prometastatic role is enhanced by a tendency of SHG-producing fibers to orient themselves radially within tumors, extending through the tumor-host interface.3 The areas where these fibers cross the interface are associated with tumor cells intravasating into healthy tissue, one step in metastasis to distant organs.3 In human breast cancer samples, the presence of oriented SHG-producing fibers is an independent prognostic factor for disease-free survival, independent of grade, size, and receptor status.5 In addition to these morphological properties of SHG-producing collagen fibers (direction, tortuosity, etc.), the SHG emission properties of the fibers themselves are of interest, as SHG scattering directionality has been shown to change in the transition from healthy ovarian tissue to ovarian cancer6 and to evolve in concert with metastatic outcome in breast cancer patient samples.7 Finally, in addition to being used to study metastasis, SHG has been used to study drug transport, where therapeutic alteration of the tumor collagenous matrix led to an alteration in molecular transport within a tumor model.8 These connections between SHG properties and metastasis/transport suggest that the next logical step is to understand the molecular machinery by which tumors control their collagenous matrix assembly and hence define their SHG properties. Consequently, in this work, we begin dissecting the cells and molecular signals responsible for manipulating tumor collagen fiber properties to which SHG is sensitive. This may, in turn, lead to novel therapeutic targets to manipulate the tumor matrix and hence alters metastatic output and/or drug delivery.
SHG is defined as the nonabsorptive combination of two excitation photons into one emission photon of exactly half the wavelength and twice the energy of the individual incoming photons.9–11 SHG is a coherent phenomenon (the scatterers produce emission waves exhibiting a constant phase relationship), and as such depends on the ordering of the individual scatterers.12 In the tumor, SHG is produced primarily by fibrillar collagen8 and the scatterers are the individual collagen triple helices. These helices are bundled together end-to-end and side-by-side into fibrils, which are in turn bundled into generally regularly spaced arrays producing the collagen fiber. As a result of the coherent nature of SHG emission, the SHG epidetected from a given fiber is influenced by the amount of collagen triple helices in the focal volume as well as the diameter of the fibrils, their spacing, and the degree of order versus disorder in their packing.11–15 In this work, we will define a change in collagen “microstructure” as a change in these last three parameters (fibril diameter, spacing, and packing order). To help distinguish changes in fiber microstructure from simple alterations in local fibril density, we will compare SHG emission signal to signal from collagen I immunofluorescence (IF) in the manner of Diop-Frimpong et al.16 and Ingman et al.17 We chose to quantify collagen content with collagen I IF because collagen I is the primary fibrillar collagen that generates SHG in vivo, and while other fibrillar collagens such as III and V can contribute to SHG emission,18,19 in vivo they exist in molar ratios to collagen I of significantly less than one.20,21 The SHG signal from a given focal point in a multiphoton laser-scanning microscope is sensitive to collagen fiber microstructure (as defined above) as well as fiber content, whereas IF yields information as to the total number of accessible epitopes present in the focal volume and is therefore sensitive to fiber content. We will therefore define the ratio of SHG to collagen I IF as an “ordering index,” or OI. Changes in the OI can then be interpreted as changes primarily in collagen fiber microstructure, distinct from changes in total collagen fiber content which will tend to cancel out in the numerator and denominator. The OI can be determined readily on a pixel-by-pixel basis using simultaneous image capture in two color channels or can be averaged over entire image fields.
To use this SHG-based tool to determine if a given candidate cell type or signal plays a role in defining tumor collagen microstructure, we must now identify some candidate cell types and/or signals to test. Breast tumors are not only composed solely of tumor cells but also host cell types including hematopoietic and stromal cells of which up to approximately 50% may be macrophages.22,23 Leukocyte infiltration in the breast tumor, originally thought to simply indicate an antitumor response, has been recognized as far more complicated. Clinical data indicate that high densities of leukocyte infiltration, particularly tumor-associated macrophage (TAM) invasion, correlate with poor clinical prognosis and increased instance of metastatic disease.24,25 TAMs are uniquely suited to tumorigenic promotion—they produce a wide variety of growth factors, cytokines, matrix-altering enzymes, and chemokines that coordinate to assist in matrix remodeling and angiogenesis.22 Hence, we have identified the TAM as a promising candidate for a cell that manipulates collagen microstructure.
The identification of the TAM as a candidate cell that may manipulate collagen microstructure in breast tumor models is supported by one key recent study, in which mice deficient in colony-stimulating factor 1 (a protein essential to macrophage proliferation, survival, and chemotactic recruitment) showed marked alterations in terminal end bud formation in the mammary duct.17 The abortive development of the end bud was noted as well as a steep decrease in the SHG around the mammary duct. Curiously enough, the total amount of collagen present as detected by IF was unaffected. This represents a decrease in OI accompanying the loss of macrophages in the breast bud area and identifies macrophages as a cell type capable of affecting OI in at least one model system. A previous study showed that upon treatment with the hormone relaxin, soft tissue sarcomas respond by altering collagen I structure with SHG intensity as a readout.8 Relaxin binds specifically to macrophage glucocorticoid receptors, which results in alterations in macrophage cytokine expression, including negative modulation of TNF- expression by macrophages in vitro.26,27 Hence, we have identified TAMs as candidate cells, and TNF- signaling as a candidate signaling molecule to manipulate the OI. Interestingly, TNF- is also shown to be essential to primary growth and metastatic progression in Lewis lung carcinoma.28
We, therefore, hypothesize that TAMs can affect collagen fiber microstructure in tumors, as measured by the OI, and that this is accomplished through TAM expression of TNF-. To test this hypothesis, we grew mammary fat pad (MFP) tumors using a breast tumor cell line (E0771, a mammary adenocarcinoma derived from C57Bl/6 mice) in wild-type mice and mice genetically incapable of expressing TNF-. Additionally, in both types of mice, TAMs were depleted by periodic injections of clodronate-containing liposomes (ClodL).29 OI and metastatic burden were evaluated, and we show for the first time that the expression of TNF- by the host stromal cells of the tumor affects collagen microstructure (as measured by OI), that TAMs affect collagen microstructure in tumors, and that the influence of TAMs and stromal TNF- expression is not additive. We also show that these effects on collagen microstructure each correlate with a significant decrease in metastatic events. Consequently, these results may provide a platform for therapies that manipulate collagen microstructure and thereby impact the metastatic output of the tumor. This work further serves to demonstrate the utility of SHG as an additional tool to reveal cells and signals which play a role in defining tumor collagen properties.
2. Materials and Methods
2.1. Cells and Reagents
A murine medullary mammary adenocarcinoma syngeneic with C57Bl/6 mice (E0771, Roswell Park Cancer Institute, Buffalo, NY) was maintained in RPMI 1640 medium (Gibco/Invitrogen, Carlsbad, CA) supplemented with 10% gamma-irradiated fetal calf serum (HyClone/Thermo-Fisher, Waltham, MA) and Primocin (InvivoGen, San Diego, CA). Cells were passaged no more than five times before being replaced from frozen stocks. After harvesting with 25% trypsin/ethylenediaminetetraacetic acid (EDTA), cells were centrifuged and resuspended in sterile phosphate buffered saline (PBS) and kept on ice until implantation into a mammary fat pad. T47D and MCF-7 human breast cancer cell lines [American Type Culture Collection (ATCC), Manassas, VA] were also cultured in RPMI 1640 medium for use in in vitro proliferation assays. RAW264.7 transformed murine macrophages (ATCC) were used as a positive control for TNF- production and were cultured in Dulbecco’s modified Eagle's medium (DMEM) supplemented with glucose, 10% fetal calf serum (FCS), and Primocin. To activate RAW264.7 cells, lipopolysaccharide (LPS, Escherichia coli serotype 026:B6, Sigma-Aldrich, St. Louis, MO) was added to media at for 24 h prior to assaying for TNF-. All lines were tested for mycoplasma contamination bi-monthly using MycoFluor detection kit (Invitrogen, Carlsbad, CA) and only certified mycoplasma-free cultures were used for implantation.
2.2. Purification of Tumor CD11b+ Cells by Magnetic Separation
E0771 tumors grown in mammary fat pads were finely minced, washed with RPMI 1640 and then shaken in medium supplemented with of collagenase type D (Sigma-Aldrich, St. Louis, MO) at 37°C for 180 min. The cell dispersion was passed through metal mesh (100-μm pore size) and washed three times in RPMI 1640, then resuspended in MACS buffer (Miltenyi Biotec, Auburn, CA) containing 2% EDTA and 5% bovine serum albumin (BSA) in PBS. To harvest cells, the single-cell suspension was incubated with anti-CD11b antibody conjugated to magnetic beads (Miltenyi Biotec, Auburn, CA). The cell suspension was then applied to a type LS positive selection column with MidiMACS (Miltenyi Biotec) according to manufacturer instructions. Selected cells were cultured to confluence in DMEM supplemented with 10% FCS and Primocin.
2.3. Animals and Husbandry
C57Bl/6 female mice (Jackson Laboratories, Bar Harbor, ME) were used between 15 and 19 weeks of age. Mice were housed in two-way static (nonventilated) conditions in groups of five, and were allowed ad libitum access to standard food and water. To determine the effects of global deletion of TNF-, female B6.129S- (Jackson Labs) mice were used between 15 and 19 weeks of age and housed as above. C57Bl/6 animals represent a valid control genotype for this knockout. All animal work was done in accordance with University Committee for Animal Resources regulations.
2.4. Tumor Implantation and Liposome Administration
Animals were anesthetized with a ketamine/xylazine mixture ( body weight) delivered intraperitoneally (i.p.). The ventral surface of the animal was depilated and E0771 cells were implanted in the right inguinal mammary fat pad using a 27-gauge needle. Four hours following this procedure, mice were administered either PBS- or clodronate-containing liposomes (Encapsula NanoSciences, Nashville, TN, stock solution) i.p. at body weight on day 0.29 After the first injection, liposomes were injected every third day at body weight. The long and short axes of each tumor were measured on days 3, 9, 15, 21, and 27 with digital calipers. Tumor volume was calculated using the formula for a prolate spheroid and standardized to volume at day 3 (multiplicity). On day 27 postimplantation, animals were sacrificed by sodium pentobarbital overdose and subsequent cervical dislocation. Half the tumor was frozen in dry ice for sectioning and immunohistochemistry. The other half was placed in cell culture media and prepared for flow cytometric analysis. Lungs were placed in 10% neutral-buffered formalin for hematoxylin and eosin (H&E) staining.
2.5. Flow Cytometry
To prepare a single cell suspension, tumors were finely minced, washed with RPMI 1640, and strained through a 70-μm mesh filter to remove debris. After centrifuging two times, the cells were counted by cytometer with trypan blue exclusion. Viable cells () were incubated at 4°C for 15 min in ACK buffer to lyse red blood cells, centrifuged, and resuspended in flow wash buffer consisting of 1% BSA and 25% sodium azide in sterile PBS. Cells were incubated for 30 min with 25-μL anti-mouse CD16/CD32 antibody at dilution (BD Pharmingen, Franklin Lakes, NJ) to block receptors and then were resuspended in 100 μL buffer containing either: (1) buffer only (autofluorescence); (2) peridinin chlorophyll-conjugated rat [BD Pharmingen (isotype control)], or (3) fluorescein isothiocyanate (FITC)-conjugated rat () anti-mouse F4/80 (Abcam, Cambridge, MA). Cells were incubated in the dark for 30 min at 4°C. Cells were then centrifuged in flow wash buffer twice and resuspended in 4% paraformaldehyde. Samples were stored in the dark at 4°C until cytometric analysis using a FACSAria flow cytometer (BD Biosciences, Franklin Lakes, NJ) equipped with FlowDiVa software.
2.6. TNF- Determination
A Quantikine enzyme-linked immunosorbent assay (ELISA) kit specific for mouse TNF- (R&D Systems, Minneapolis) with a sensitivity of was employed to measure TNF- in cell culture supernatants and tumor lysates. To prepare cell culture supernatants, cells were grown to 70% confluence in a T-75 tissue culture flask and then media was removed and replaced with reduced serum media for 48 h. This media was removed, centrifuged, and immediately assayed for TNF-. Tumor lysates were prepared by homogenization of excised tumor tissue in RIPA buffer containing sodium deoxycholate and HALT protease/phosphatase inhibitor (Pierce Protein Research, Rockford, IL). Lysates were centrifuged at for 10 min and supernatants were removed for assay. Total protein concentration in tumor lysates was determined with a bicinchoninic acid assay (Pierce Protein Research, Rockford, IL) per manufacturer specifications. For the ELISA, absorption was measured at 450 nm using a mutliwell plate reader (Synergy HT, BioTek Instruments Inc, Winooski, VT). Curve fitting and sample concentration calculations were conducted with Gen5 software (Biotek). Results are presented as TNF- concentration standardized to sample protein concentration.
2.7. Proliferation Assays
In vitro proliferation was determined with the use of a fluorescent CyQuant Cell Proliferation Assay (Molecular Probes/Invitrogen, Carlsbad, CA) following manufacturer’s instructions. To detect DNA fluorescence, a plate reader (BioTek) exciting bound dye at 480 nm and detecting at 520 nm was used. This assay effectively measures viable cell number, and we have determined that fluorescent intensity correlates directly with actual cell number (data not shown).
2.8. Immunohistochemistry
Snap-frozen tumor halves were sectioned at 7 μm on a cryostat (Reichert-Jung, Depew, NY) at and static-mounted on positively charged slides (VWR, Radnor, Pennsylvania). Each slide contained at least four sections from each experimental group to minimize possible staining artifacts between samples. Sections were fixed in a mixture of acetone/methanol for 20 min at . Slides were rehydrated twice in sterile PBS for 5 min and then placed in peroxidase blocking solution (5% BSA, 2% Triton X-100) for 1 h, followed by two 5 min PBS washes. Sections were incubated at room temperature for 2 h in a humidified chamber with PBS containing 5% BSA and combinations of the following antibodies: (1) rabbit anti-mouse collagen I (Abcam, Cambridge, MA), dilution; (2) FITC-conjugated rat anti-mouse CD31 (BD Pharmingen, Franklin Lakes, NJ), 1:500 dilution; or (3) FITC-conjugated rat anti-mouse F4/80, dilution. To detect rabbit anti-mouse collagen I, sections were then washed and incubated for 2 h at room temperature in AlexaFluor 594-conjugated goat anti-rabbit IgG (Invitrogen, Carlsbad, CA), dilution. Optimal antibody dilutions were predetermined. Slides were then washed and coverslipped in Prolong Gold AntiFade without DAPI (Invitrogen, Carlsbad, CA) and allowed to dry 24 h before imaging.
2.9. Evaluation of Metastatic Burden
Automated H&E staining (Dako, Carpinteria, CA) was performed on 3 μm rotary microtome sections of paraffin-embedded lungs. H&E-stained lung sections were obtained at 50 μm intervals through both lobes of the lung and evaluated by a blinded observer using brightfield microscopy (Olympus BX-51, Center Valley, PA). Malignant cells lose their differentiation and undergo increased proliferation, frequently exhibiting enlarged and variable sized nuclei as well as evidence of accelerated mitosis. Consequently, accepted criteria for recognizing metastatic foci in H&E-stained sections include a high ratio of hematoxylin relative to eosin, surrounding abnormalities in lung structure, abnormal shape/size of nuclei and/or presence of abnormal mitotic spindles, and differences in cell shape and size.30 We used these criteria to identify lung metastases, with results presented as the number of metastases in 10 sections per animal.
2.10. Imaging and Image Analysis
Slides were imaged by a blinded observer using a custom-built two-photon microscope (Olympus). Two-photon (2P) excitation was achieved by a MaiTai Ti:sapphire laser providing 100 fs pulses at 80 MHz and 810 nm. Beam scanning and image acquisition were performed with a Fluoview FV300 scanning system interfaced with a BX61WI upright microscope (Olympus, Center Valley, PA). The focusing objective was a XLUMPLFL20xW water immersion lens (, 0.95 N.A., Olympus). The objective focused the 810 nm excitation beam on the sample and at the same time collected both backscattered SHG signal and the two-photon excited IF from the antibody of interest. The backscattered SHG and IF signals were separated from the excitation beam by a short pass dichroic mirror (670 DCSX, Chroma, Rockingham, VT), with the SHG and IF emissions then separated into two channels with a long pass dichroic mirror (475 DCSX, Chroma, Rockingham, VT). SHG was collected with a bandpass filter centered at 405 nm (HQ405/30m-2P, Chroma) and detected by a photomultiplier tube (PMT) (HC125-02 Hamamatsu Corporation, Hamamatsu, Japan). The IF signal was collected with a bandpass filter centered at 635 nm (HQ635/30m-2P, Chroma). For capturing this red IF channel, a red-sensitive PMT was used (Hamamatsu HC125-01), and the emission signal was additionally filtered (Chroma E700SP-2P) to block any stray near-infrared infra-red (IR) laser light. Resulting images are 680 μm across. Laser power was monitored and kept constant throughout the experiment and across experimental repetitions, as were PMT voltage, gain, and offset. Ten tissue slices were chosen by a blinded observer, spaced throughout each tumor half (the other half is used to verify macrophage depletion) and five fields of view were generated from each slice. The five imaged fields of view formed an “x” pattern; one in the tumor center, then four adjacent diagonal fields of view (i.e., one to the upper left of the first central image, with the corners of the images not quite touching, one to the upper right of the first central image, etc.). Hence, 50 image pairs were analyzed as described below to produce a single average SHG/IF value for each tumor.
Image analysis was performed as follows. In IMAGE J (Rasband, W. S., IMAGE J, U. S. National Institutes of Health, Bethesda, Maryland, USA, http://imagej.nih.gov/ij/, 1997–2011), the background in each channel (SHG and IF) was defined by the average pixel counts of an image with no excitation laser and subtracted from the raw SHG and IF images, respectively. A single common threshold was then chosen by a blinded observer which passed the most collagen pixels while rejecting the most background pixels (typically individual pixels not clearly laying in a fibrillar pattern). The common threshold was then applied to all SHG images, setting collagen fiber pixels to 1 and any remaining dim background pixels to zero, producing an “SHG mask.” The same thresholding procedure was followed with IF images, producing an “IF mask.” The SHG image was multiplied by the SHG mask, producing the “masked SHG image,” and the IF image was multiplied by the IF mask to produce the “masked IF image.” The average pixel count of the masked SHG image, divided by the average pixel count of the SHG mask, is then the average pixel count of those pixels above threshold, i.e., within collagen fibers, likewise with the masked IF image. The average SHG pixel count, average IF pixel count, and their ratio, of those pixels within collagen fibers, are then reported as the images’ SHG, IF, and OI, respectively. Note that due to fluctuations in pixel counts, there are occasional pixels which are above threshold in the SHG image but below threshold (and hence set to zero) in the IF image. Consequently, the SHG/IF ratio is infinity for those pixels. This makes it problematic to calculate an average SHG/IF for the imaged region by first producing an SHG/IF ratio image and then calculating the average pixel count of that image. We avoid this problem by first separately calculating the average SHG and IF pixel counts of the whole imaged region and then dividing these two (nonzero) numbers to produce the SHG/IF for the whole imaged region.
2.11. Statistical Analysis
Statistical analysis was performed using Prism 5 software (GraphPad, La Jolla, CA). Student’s (unpaired) -tests were employed to make pairwise comparisons where appropriate. To analyze the effects of macrophage depletion and TNF- knockout, two-way analysis of variance (ANOVA) was used. Significant main effects or interactions were analyzed with Bonferroni posttests to adjust for multiple comparisons. Tumor growth curves were assessed by two-way ANOVA with repeated measures, followed by multiple group comparisons using Bonferroni posthoc analysis. Probability values () less than or equal to 0.05 were considered significant differences between groups. When is reported for animal experiments in the figures or text, it represents the number of animals studied.
3. Results
3.1. E0771 does not Produce TNF- nor Proliferate in Response to TNF- In Vitro or In Vivo
Pilot experiments were conducted to determine: (1) if E0771 cells produce TNF- and (2) if E0771 responds to TNF-. E0771 cells cultured with 10% FCS to 75% confluence did not produce measurable TNF- [, Fig. 1(a)]. By comparison, RAW 264.7 macrophages were also tested at 75% confluence in both normal and LPS-activated conditions and produced significant levels of TNF- with or without activation by LPS [Fig. 1(a)]. To confirm that TAMs from E0771 tumors produce TNF-, macrophages were isolated from E0771 tumors using magnetic antibody separation targeted to CD11b.31 These TAMs also produced TNF- at detectable levels in culture [Fig. 1(a)]. Furthermore, E0771 tumors were grown in both wild-type and TNF- animals. TNF- was readily detectable in wild-type-derived E0771 tumor lysates at , , and dilutions, but not in those grown in TNF- mice, where levels were again undetectable [, Fig. 1(b)]. Taken together, these results suggest that the E0771 cell line is not a source of TNF- production in vitro or in vivo and that E0771-associated macrophages, as expected, are a primary source of this signal in vivo.
Fig. 1.
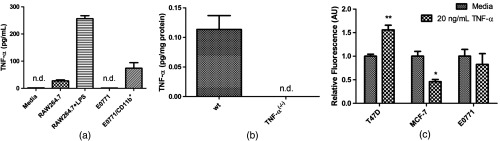
E0771 breast cancer cells do not produce nor respond to TNF-. (a): In vitro TNF- production: Supernatants from E0771 tumor cells showed no detectable TNF- in vitro (E0771 bar). Supernatants from TAMs isolated from E0771 tumors, in contrast, did produce detectable TNF- ( bar). To confirm sensitivity of the assay, RAW264.7-transformed murine macrophages produced detectable TNF- both when unstimulated (RAW264.7 bar) and upon activation with LPS for 24 h (RAW264.7+LPS bar). TNF- ELISA sensitivity was , samples for all but , where . Both the media alone control (media bar) and E0771 supernatants (E0771 bar) registered below sensitivity (not detectable). (b): In vivo TNF- production: also by this same ELISA assay, TNF- was detectable in E0771 tumors grown in C57Bl/6 mice (wt bar) but not in mice lacking TNF- [TNF- bar]. (c): Proliferation responses to TNF-: three breast tumor cell lines (T47D, MCF-7, and E0771) were treated with media control or TNF-. Pairwise comparisons indicate that T47D proliferation was significantly elevated by TNF- at 48 h (), MCF-7 proliferation was significantly decreased (), whereas only E0771 proliferation was unchanged in response to TNF-. Proliferation was assessed by fluorescent intensity of CyQuant DNA-binding dye standardized to that of cells in untreated media, per group.
The proliferative response of E0771 to TNF- was determined using a fluorescent DNA-binding dye kit (CyQuant). In another model of breast cancer (T47D), TNF- was shown to directly promote mitogenic signaling by binding to TNFR1 and activating p42/p44 MAPK, JNK, PI3-K/Akt pathways, and NF- transcriptional activation.32 Furthermore, in that model, application of TNF- supported tumor growth. Conversely, TNF- induced apoptosis in MCF-7 breast cancer cells in vitro.33 To determine the response of E0771 cells to TNF-, we incubated cells for 48 h in either media or media containing TNF- [Fig. 1(c)]. TNF- did not significantly alter proliferation of E0771, in direct contrast to the human breast tumor cell line T47D, which showed a marked increase in proliferation in response to TNF-, as reported.32 Furthermore, MCF-7 cells exposed to this level of TNF- exhibited significant reduction in proliferation, also as expected.33 Therefore, TNF- does not significantly stimulate E0771 proliferation nor significantly inhibit its growth.
3.2. ClodL Selectively Impairs Macrophage Viability In Vitro and Depletes TAMs In Vivo
ClodL are commonly used to selectively deplete macrophages in tumors.29 Macrophages that phagocytose ClodL are destroyed by release of clodronate into the cytoplasm following degradation of the liposome shell and subsequent formation of a lethal metabolite.34 To determine if TAM susceptibility to ClodL is specific, we tested the viability of E0771 cells, fibroblasts (HFF-1, transformed human foreskin fibroblasts), and RAW264.7, a transformed murine macrophage cell line in the presence of PBS-containing liposomes (PBSL) or ClodL. ClodL did not alter viability of E0771 or HFF-1 cells relative to PBSL-treated controls. By contrast, RAW264.7 viability was significantly reduced by ClodL [Fig. 2(a)]. Depletion of TAMs in vivo was quantified by flow cytometric analysis of F4/80, a marker for mature macrophages.35 ClodL treatment in E0771-bearing mice dramatically reduced the percentage of F4/80-positive cells in E0771 tumors in both wild-type and TNF- mice [Fig. 2(b)]. Together, these results demonstrate that ClodL treatment is selective for TAMs in E0771 tumors. Figure 2(b) also demonstrates that the percentage of E0771-associated F4/80-positive macrophages is not statistically different between wild-type and TNF- mice ().
Fig. 2.
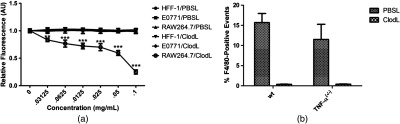
ClodL selectively reduces macrophage viability in vitro and in vivo. (a) Graded dose–response treatments with Clodronate (0 to ) or PBS control liposomes demonstrated that at all Clodronate concentrations tested, only macrophage RAW264.7 cells, but not nonmacrophage E0771 breast tumor cells or HFF-1 fibroblasts, were sensitive to Clodronate depletion ( each data point, for RAW264.7 PBSL versus ClodL at all doses). For each cell type, fluorescent intensities were standardized to the PBS control liposome group (no ClodL), to account for differences in proliferative rate inherent to cell type. Liposomes containing PBS (vehicle) only also did not affect proliferation of RAW264.7 macrophages at any liposome concentration (RAW264.7/PBSL trace). (b) Flow cytometric analysis indicated that endogenous TAMs were depleted by in vivo Clodronate treatment in both wild-type (wt) and TNF- mice [; ; TNF-; TNF-]. Moreover, these data also indicate that under baseline (PBSL, i.e., no depletion) conditions, macrophage populations were not significantly different in wt versus TNF-) animals.
3.3. Effects of TAM Depletion and TNF- Knockout on Intensity of SHG-Producing Fibers
To determine if TAM depletion or TNF- knockout affects the intensity of SHG-producing fibers in the E0771 tumor, tumors were grown orthotopically in either wild-type or TNF- mice treated with liposomes containing clodronate to deplete TAMs. Control mice were treated with liposomes containing PBS. SHG imaging of excised E0771 tumors reveals a typical fibrillar structure (Fig. 3). Figure 4(a) shows that either TAM depletion or TNF- knockout reduced the intensity of SHG-producing fibers in E0771 tumors. By two-way ANOVA, a strong main effect of TAM depletion () and a strong interaction between TAM depletion and TNF- knockout () was revealed. By Bonferroni’s posttest for multiple comparisons, the intensity of SHG-producing fibers in wild-type/PBSL mice was significantly elevated relative to all other groups (). Intensity of SHG-producing fibers in tumors from TNF- mice was also greater than that in tumors from wild-type/ClodL mice () but not relative to TNF- mice (). Intensity of SHG-producing fibers did not differ between the two ClodL-treated groups. These results demonstrate that TAM depletion or attenuation of stromal TNF- reduces intensity of SHG-producing fibers in the E0771 tumor. Furthermore, in the absence of TAMs, the inability to produce TNF- did not produce a greater effect than either treatment alone. This suggests that nonmacrophage sources of TNF- do not significantly influence SHG intensity.
Fig. 3.

SHG image of E0771 murine mammary adenocarcinoma excised from the mammary fat pad of a C57Bl/6 mouse. The tumor sample was excised, sectioned, and fixed. With 810-nm excitation, typical collagen fibrils are imaged via SHG at 405-nm emission.
Fig. 4.

Effects of macrophage depletion and TNF- knockout on SHG and IF signals in the E0771 tumor. (a) SHG-producing fibers in E0771 tumors in wild-type/PBSL mice were significantly brighter compared to all other groups ( for all comparisons). In addition, SHG-producing fibers in E0771 tumors in wild-type/ClodL mice were significantly dimmer compared to TNF- tumors (). No other group was found significantly different from another (). , , TNF-, and TNF-). (b) Collagen I immunofluorescence (IF) was significantly lower in E0771 tumors in wild-type/PBSL mice compared to all other groups ( for all comparisons). In addition, collagen I IF in E0771 tumors in TNF- mice was significantly lower than in E0771 tumors in TNF- mice. No other group was found significantly different from another ().
3.4. Effects of TAM Depletion and TNF- Knockout on Collagen Fiber IF Intensity
SHG is dependent upon collagen content as well as fiber microstructure. Therefore, in order to produce a more direct measure of fiber microstructure, we normalize the SHG intensity to the total amount of collagen present. As collagen type I is the primary fibrillar collagen in vivo,20,21 we quantify collagen fibril content using fluorescently labeled anticollagen type I antibodies.16,17 TAM depletion and stromal TNF- knockout both alter detected collagen I fiber IF in the E0771 tumor [Fig. 4(b)]. By two-way ANOVA, we see a strong main effect of both TAM depletion and TNF- knockout (both ) and a significant degree of interaction () was detected. By multiple comparison analysis, anticollagen type I IF was significantly lower in wild-type/PBSL mice compared to the three other groups (). Furthermore, although IF signal in tumors grown in wild-type/ClodL mice is statistically equivalent to that of both TNF- knockout groups (), tumors grown in TNF- mice exhibit less IF than do tumors grown in TNF- mice (). We therefore conclude that tumor growth in the presence of TAM depletion results in a significant increase in anticollagen type I IF in E0771 tumors. Growth during attenuation of stromal TNF- accomplishes this to the same extent. Furthermore, in the absence of TAMs, depletion of stromal TNF- has no further effect on IF. However, in the absence of stromal TNF-, depletion of TAMs further increases IF. The combined abrogation of TAMs and TNF- did not produce a greater increase in IF signal intensity than TAM depletion alone.
3.5. Effects of TAM Depletion and TNF- Knockout on OI
Division of the SHG signal by the IF signal produces the order index (OI), which is primarily sensitive to changes in fibrillar microstructure, such as fibril diameter, spacing, and order versus disorder in fibrillar packing.11–15 In Fig. 5, OI was reduced by TAM depletion with ClodL or stromal knockout of TNF-, with a main effect of and , respectively, with a significant interaction (). Bonferroni post-ho analysis shows that wild-type/PBSL mice have significantly elevated OI relative to the three remaining groups (). However, none of the three remaining groups exhibit significant differences between their mean OI values (). We therefore conclude that tumor growth in the presence of TAM depletion results in a significant decrease in OI in E0771 tumors, as does growth during attenuation of stromal TNF-, to the same extent. Furthermore, combined abrogation of TAMs and TNF- did not produce a greater decrease in OI than either depletion alone.
Fig. 5.

Effects of macrophage depletion and TNF- knockout on OI. E0771 tumors grown in wild-type/PBSL mice had a significantly higher order index (OI, defined as SHG intensity/IF intensity) relative to all other treatment groups (). No other group was found significantly different from another ().
3.6. Metastatic Burden Correlates Negatively with Both TAM Presence and TNF- Expression
To evaluate the possibility of a relationship between modulations in OI and modulations in metastatic burden, lungs were resected, sectioned, and stained with H&E and then imaged to determine the extent of metastasis (Fig. 6). E0771 readily metastasizes to the lung from the primary site,36 and two-way ANOVA shows a strong main effect of both TAM depletion and abrogation of TNF- on metastatic burden ( and , respectively). The degree of interaction is again significant (). Bonferroni posthoc analysis shows that while wild-type/PBSL mice exhibit significantly () higher metastatic burden than any of the other groups, the other three groups do not show distinguishably different metastatic burdens (). We conclude that both TAM depletion and TNF- knockout result in significant decreases in metastatic events and that these decreases correlate well with OI values in the E0771 tumor.
Fig. 6.

Lung metastatic burden decreases sharply with both macrophage depletion and TNF- knockout. E0771 tumors grown in wild-type/PBSL mice generated significantly higher numbers of lung metastases, compared to E0771 tumors in TAM depleted and TNF- knockout mice (). Metastatic output was assessed across 10 sections of lung per group. No other group was found significantly different from another (). , , TNF-, and TNF-.
3.7. ClodL and TNF- Knockout Reduce E0771 Tumor Growth In Vivo
To determine if TAM depletion or TNF- knockout affect tumor growth, E0771 was implanted in PBSL- or ClodL-treated wild-type or TNF- mice and tumor volume was measured over the course of 27 days. The data presented in Fig. 7 were normalized at each time point relative to the volume at day 3 posttumor inoculation. By repeated measure of two-way ANOVA, a treatment by time interaction was revealed (). Wild-type/PBSL mice bore significantly larger tumors at day 27 (, Bonferroni posttest) compared to any other group. Tumor growth did not differ among the other three groups.
Fig. 7.

E0771 tumor volume multiplicity decreases with TAM depletion or TNF- knockout, but the effects are not synergistic. After 27 days, E0771 tumors grown in wild-type mice on PBSL therapy () were larger than those grown in wild-type mice on ClodL therapy (), as well as those grown in TNF- knockout mice exposed to either PBSL or ClodL (, for both). No other group was found significantly different from another at 27 days, and no significant differences in tumor volume multiplicity were found at earlier timepoints (). Tumor growth was assessed by normalizing measurements to tumor size at day 3 (i.e., tumor volume multiplicity) to minimize the possibility that slightly different numbers of cells may have been injected despite constant injection volumes and cell densities.
4. Discussion
In this study, our goal was to demonstrate the use of SHG as a tool to help detect cells and signals involved in establishing tumor collagen microstructure. We did this by elucidating a novel mechanistic role for TAMs and stromal TNF- in the evolution of collagen microstructure in a murine model of breast cancer. In doing so, we made use of the E0771 murine mammary adenocarcinoma, a metastatic cell line syngeneic with the C57Bl/6 mouse. The choice of this cell line was predicated on the availability of the knockout strain B6.129S-, which is incapable of expressing TNF-. We first established that the E0771 tumor model does not produce TNF-, is nonresponsive to TNF-, and that it also does not have an intrinsic cellular response to liposome-encapsulated clodronate therapies. Next, we established that these therapies are able to alter the microstructure of collagen I fibers (as measured by the OI) and metastatic burden.
Taken together, our OI data suggest that in the E0771 model of metastatic breast cancer, TAMs influence collagen microstructure (Fig. 5, columns 1 and 2 are different) and that stromal TNF- influences collagen microstructure (Fig. 5, columns 1 and 3 are different). This suggests three main models: (A) TAMs and TNF- influence OI independently, (B) stromal TNF- influences OI via its action on TAMs, and (C) TAMs influence OI via expression of TNF-. Model A is unlikely because in the absence of TAMs, modulation of stromal TNF- has no further effect on OI (Fig. 5, columns 2 and 4 are the same) and in the absence of stromal TNF- modulation of TAMs has no further effect on OI (Fig. 5, columns 3 and 4 are the same). However, models B and C are consistent with our observations and can be further refined. If stromal TNF- influences OI via action on TAMs (model B), our data further suggest that stromal TNF- acts primarily via TAMs. This is because in the absence of TAMs, modulation of stromal TNF- has no further effect on OI (Fig. 5, columns 2 and 4 are the same). Furthermore, in that model, our data also suggest that no other molecule plays a significant role in stimulating TAMs to influence OI. This is because in the absence of stromal TNF- modulation of TAMs has no further effect on OI (Fig. 5, columns 3 and 4 are the same). Lastly, if TAMs influence OI via their expression of TNF- (model C), our data suggest that TAMs influence collagen structure primarily through their TNF- expression and not via other mechanisms. This is because in the absence of stromal TNF-, modulation of TAMs has no further effect on OI (Fig. 5, columns 3 and 4 are the same). Furthermore, in that model, our data also suggest that there are no other significant host cell types utilizing TNF- to influence collagen microstructure. This is because in the absence of TAMs modulation of stromal TNF- has no further effect on OI (Fig. 5, columns 2 and 4 are the same).
While we used OI to detect a role for TAMs and stromal TNF- in manipulating collagen microstructure, the information that OI provides about the actual changes in matrix microstructure caused by modulating TAMs or stromal TNF- is limited. Consequently, results must be interpreted carefully. This is primarily due to the fact that epidetected SHG signal depends on several different physical parameters (e.g., fibril diameter, spacing, packing order), and changing any of these parameters alone or in combination will likely result in a change in OI. Furthermore, the relationship between epidetected SHG (and hence OI) and a specific fiber physical parameters can be complex, with one illustrative case being the dependence of epidetected SHG on the diameter of fibrils within a collagen fiber. An increase in fibril diameter could result in either increased or decreased epidetected SHG signal due to the predicted sinusoidal dependence of emitted SHG directionality on fibril diameter.13 Hence, it is the presence of a modulation of OI, regardless of sign, due to modulation of a cell/signal which indicates that the cell/signal in question influences collagen microstructure. However, the sign of the OI change (i.e., increasing or decreasing) does not tell us how the relevant parameters (e.g., diameter, spacing, packing order) are changing, or what combination of them are changing.
In addition to our primary observations detecting a role for TAMs and stromal TNF- in modulating OI, there are two secondary observations: TAM presence was able to modulate IF in the absence of stromal TNF- [Fig. 4(b), columns 3 and 4 are different] and TAM depletion produced a greater SHG reduction than attenuation of stromal TNF- [Fig. 4(a), columns 2 and 3 are different). In model B (stromal TNF- operates via TAMs), these two observations imply that there may be other less significant mechanisms, in parallel with stromal TNF-, which induce TAMs to affect tumor collagen. In model C (TAMs operate via TNF-), these two observations imply that there may be other less significant mechanisms by which TAMs affect tumor collagen in addition to their expression of TNF-. However, the observation that attenuation of TAM presence produced a greater SHG reduction than the attenuation of stromal TNF- is based upon quantitative comparison of the results of two very different experimental manipulations (TAM depletion versus TNF- knockout) and may simply be a result of different efficiencies in the two methods.
When macrophage recruitment is specifically disabled in developing mouse mammary tissue, the microstructure of collagen fibers but not their total amount, as measured by SHG and IF, is altered with negative consequence to normal maturation.17 Interestingly, in contrast to our observations, this same study noted a similar decrease in SHG but without any elevation in IF. This may be due to myriad differences between normal and diseased mammary fat pad, in particular, the well-documented ability of cancer cells to produce stimuli inducing fibroblasts to transition to a more active myofibroblast phenotype, with one consequence being elevated levels of collagen production.37 Additionally, this may simply be due to differences in antibody–epitope binding between two different model systems.
Our observations that TAMs and stromal TNF- play a role in altering tumor collagen microstructure are consistent with established roles for both in modulating tumor stroma. Autocrine TNF- signaling has been noted as necessary to induce high levels of expression of monocyte matrix metalloproteinase (MMP)-9, a gelatinase that is active on basement membrane, via the transcription factor Egr-1.38–41 TNF- has also been shown to sharply increase MMP-14 levels in concert with CCL4, a C-C chemokine, in a monocytic human cell line.42 MMP-14 is able to subsequently activate MMP-2, another gelatinase capable of degrading basement membrane. However, note that the nonfibrillar collagens in basement membrane do not generate significant SHG and were not studied here. Importantly, both direct application of TNF- and indirect production of it through LPS stimulation of macrophages have been shown to upregulate MMP-1a, -1b, -3, and -13 in rat synovia, and these MMPs can directly affect the fibrillar collagen responsible for SHG signal.43 On a genetic level, TNF- is able to induce COX-2 promoter activation via upregulation of p300 binding and p50 acetylation in human foreskin fibroblasts, which is an important step in inflammation, angiogenesis, and tumor promotion.44–46
Our demonstration of a novel role for TAMs and TNF- in manipulating collagen microstructure in the tumor extracellular matrix naturally leads us to ask: what are the possible consequences of this manipulation? As discussed above, there is evidence in the literature that collagen microstructure, as quantified with SHG, influences tumor metastasis.1–5,7 Consistent with this, our data demonstrate that alterations in collagen microstructure as quantified by the OI correlated with alterations in metastatic output (Figs. 5 and 6). In each case, wild-type mice treated with PBSL had significantly greater OI and metastatic output than all other treatment groups, and all other treatment groups were not statistically significantly different from each other.
The link between TAMs, stromal TNF-, and metastatic output in this tumor model is consistent with observations that myeloid cells and their TNF- expression are necessary for the formation of Lewis lung carcinoma metastasis.28 However, while our observations establish an interesting link between TAMs, stromal TNF-, and metastatic output in this tumor model, it does not prove that the metastatic effects are transduced via collagen fiber microstructure itself. There may be other effects of TAMs and stromal TNF- expression that assist in the induction of alterations in metastatic output. For example, our data also demonstrate that tumor growth is correlated with OI (albeit only at the last time point studied), and in turn with metastatic burden (Figs. 5–7). Hence, TAMs and stromal TNF- may instead influence tumor growth via alterations in collagen matrix microstructure (note that TNF- does not directly affect E0771 proliferation in vitro), and the altered metastatic output may be a result of the different tumor burden or all three effects (OI, tumor growth, and metastatic output) may share a common cause but operate via independent mechanisms.
In the future, it may be productive to explore how collagen microstructure in different subregions of the tumor (lobular, ductal, perilobular, periductal, distant, etc.) is influenced by these manipulations. While the orthotopic cell injection model used here does not typically preserve the ductal and lobular structure that can be found in and around examples of the human disease, the MMTV mouse model of breast cancer does.47 By crossbreeding MMTV mice with different transgenic strains, one could explore how collagen microstructure in different breast tumor subregions is regulated. However, using a different measure of collagen microstructure (the SHG forward-scattering to backwards-scattering ratio), we found that in samples of human breast cancer, there is no significant difference in collagen microstructure between different breast tumor subregions.7
5. Conclusions
In summary, this study serves to demonstrate the ability of SHG to help identify key cells and signals that play a role in establishing tumor collagen microstructure. We show that TAMs as well as stromal TNF- expression are responsible for significant alterations in the microstructure of collagen fibers within a metastatic breast tumor model (as indicated by the OI). We further demonstrate that this effect is implemented either via TAM expression of TNF- or stromal TNF- action on TAMs and that parallel pathways via other cell types or other cytokines are considerably less significant. Lastly, we show that the effects of TAMs and stromal TNF- on tumor collagen microstructure are correlated with effects on metastatic output, consistent with previous literature connecting SHG and metastatic output.
Discovering a key role for tumor-associated macrophages and stromal TNF- in influencing the microstructural properties of tumor collagen, as measured by SHG, is interesting because of the previously observed effects of these SHG-producing fibers on metastasis of the primary breast tumor, as well as the previously observed relationship between SHG signal and drug transport in tumors.1–5,7,8 These findings suggest that manipulation of SHG measures of collagen microstructure may, in turn, manipulate the transport of drugs through tumor tissue as well as manipulate metastatic output of tumors.
Acknowledgments
The authors wish to acknowledge Tracy Bubel and Khawarl-Jamma M. Liverpool for their indispensable technical assistance, Diana Scott for assistance with H&E automation, Michele Au for assistance with flow cytometry, and Dr. Peter Salzman for his assistance with statistical analysis. This work was funded by Department of Defense Breast Cancer Research Program (BCRP) Era of Hope Scholar Research Award W81XWH-09-1-0405 and NIH Director’s New Innovator Award 1DP2OD006501 (to EBB), BCRP Idea Award W81XWH-10-01-0087 and NIH R21CA152777-01 (to KSM), and NIH R21DA030256 (to SWP).
References
- 1.Wang W., et al. , “Single cell behavior in metastatic primary mammary tumors correlated with gene expression patterns revealed by molecular profiling,” Cancer Res. 62(21), 6278–6288 (2002). [PubMed] [Google Scholar]
- 2.Sidani M., et al. , “Probing the microenvironment of mammary tumors using multiphoton microscopy,” J. Mammary Gland Biol. Neoplasia 11(2), 151–163 (2006). 10.1007/s10911-006-9021-5 [DOI] [PubMed] [Google Scholar]
- 3.Provenzano P. P., et al. , “Collagen reorganization at the tumor-stromal interface facilitates local invasion,” BMC Med. 4(1), 38 (2006). 10.1186/1741-7015-4-38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wyckoff J. B., et al. , “Direct visualization of macrophage-assisted tumor cell intravasation in mammary tumors,” Cancer Res. 67(6), 2649–2656 (2007). 10.1158/0008-5472.CAN-06-1823 [DOI] [PubMed] [Google Scholar]
- 5.Conklin M. W., et al. , “Aligned collagen is a prognostic signature for survival in human breast carcinoma,” Am. J. Pathol. 178(3), 1221–1232 (2011). 10.1016/j.ajpath.2010.11.076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nadiarnykh O., et al. , “Alterations of the extracellular matrix in ovarian cancer studied by Second Harmonic Generation imaging microscopy,” BMC Cancer 10, 94 (2010). 10.1186/1471-2407-10-94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burke K., Tang P., Brown E., “Second harmonic generation reveals matrix alterations during breast tumor progression,” J. Biomed. Opt. 18(3), 31106 (2013). 10.1117/1.JBO.18.3.031106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brown E., et al. , “Dynamic imaging of collagen and its modulation in tumors in vivo using second-harmonic generation,” Nat. Med. 9(6), 796–800 (2003). 10.1038/nm879 [DOI] [PubMed] [Google Scholar]
- 9.Zipfel W. R., et al. , “Live tissue intrinsic emission microscopy using multiphoton-excited native fluorescence and second harmonic generation,” Proc. Natl. Acad. Sci. U.S.A. 100(12), 7075–7080 (2003). 10.1073/pnas.0832308100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Raub C. B., et al. , “Noninvasive assessment of collagen gel microstructure and mechanics using multiphoton microscopy,” Biophys. J. 92(6), 2212–2222 (2007). 10.1529/biophysj.106.097998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Han X., et al. , “Second harmonic properties of tumor collagen: determining the structural relationship between reactive stroma and healthy stroma,” Opt. Express 16(3), 1846–1859 (2008). 10.1364/OE.16.001846 [DOI] [PubMed] [Google Scholar]
- 12.Moreaux L., et al. , “Coherent scattering in multi-harmonic light microscopy,” Biophys. J. 80(3), 1568–1574 (2001). 10.1016/S0006-3495(01)76129-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Williams R. M., Zipfel W. R., Webb W. W., “Interpreting second-harmonic generation images of collagen I fibrils,” Biophys. J. 88(2), 1377–1386 (2005). 10.1529/biophysj.104.047308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lacomb R., Nadiarnykh O., Campagnola P. J., “Quantitative second harmonic generation imaging of the diseased state osteogenesis imperfecta: experiment and simulation,” Biophys. J. 94(11), 4504–4514 (2008). 10.1529/biophysj.107.114405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lacomb R., et al. , “Phase matching considerations in second harmonic generation from tissues: effects on emission directionality, conversion efficiency and observed morphology,” Opt. Commun. 281(7), 1823–1832 (2008). 10.1016/j.optcom.2007.10.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Diop-Frimpong B., et al. , “Losartan inhibits collagen I synthesis and improves the distribution and efficacy of nanotherapeutics in tumors,” Proc. Natl. Acad. Sci. U.S.A. 108(7), 2909–2914 (2011). 10.1073/pnas.1018892108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ingman W. V., et al. , “Macrophages promote collagen fibrillogenesis around terminal end buds of the developing mammary gland,” Dev. Dyn. 235(12), 3222–3229 (2006). 10.1002/dvdy.v235:12 [DOI] [PubMed] [Google Scholar]
- 18.Ajeti V., et al. , “Structural changes in mixed Col I/Col V collagen gels probed by SHG microscopy: implications for probing stromal alterations in human breast cancer,” Biomed. Opt. Express 2(8), 2307–2316 (2011). 10.1364/BOE.2.002307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miri A. K., et al. , “Nonlinear laser scanning microscopy of human vocal folds,” Laryngoscope 122(2), 356–363 (2012). 10.1002/lary.22460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Minafra I. P., et al. , “Quantitative determination of collagen types present in the ductal infiltrating carcinoma of human mammary gland,” Cell Biol. Int. Rep. 9(3), 291–296 (1985). 10.1016/0309-1651(85)90047-5 [DOI] [PubMed] [Google Scholar]
- 21.Barsky S. H., et al. , “Increased content of Type V Collagen in desmoplasia of human breast carcinoma,” Am. J. Pathol. 108(3), 276–283 (1982). [PMC free article] [PubMed] [Google Scholar]
- 22.Pollard J. W., “Tumour-educated macrophages promote tumour progression and metastasis,” Nat. Rev. Cancer 4(1), 71–78 (2004). 10.1038/nrc1256 [DOI] [PubMed] [Google Scholar]
- 23.Direkze N. C., et al. , “Bone marrow contribution to tumor-associated myofibroblasts and fibroblasts,” Cancer Res. 64(23), 8492–8495 (2004). 10.1158/0008-5472.CAN-04-1708 [DOI] [PubMed] [Google Scholar]
- 24.Chen J. J., et al. , “Tumor-associated macrophages: the double-edged sword in cancer progression,” J. Clin. Oncol. 23(5), 953–964 (2005). 10.1200/JCO.2005.12.172 [DOI] [PubMed] [Google Scholar]
- 25.Bingle L., Brown N. J., Lewis C. E., “The role of tumour-associated macrophages in tumour progression: implications for new anticancer therapies,” J. Pathol. 196(3), 254–265 (2002). 10.1002/(ISSN)1096-9896 [DOI] [PubMed] [Google Scholar]
- 26.Dschietzig T., et al. , “The pregnancy hormone relaxin binds to and activates the human glucocorticoid receptor,” Ann. N. Y. Acad. Sci. 1041, 256–271 (2005). 10.1196/annals.1282.039 [DOI] [PubMed] [Google Scholar]
- 27.Parsell D. A., et al. , “Relaxin binds to and elicits a response from cells of the human monocytic cell line, THP-1,” J. Biol. Chem. 271(44), 27936–27941 (1996). 10.1074/jbc.271.44.27936 [DOI] [PubMed] [Google Scholar]
- 28.Kim S., et al. , “Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis,” Nature 457(7225), 102–106 (2009). 10.1038/nature07623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zeisberger S. M., et al. , “Clodronate-liposome-mediated depletion of tumour-associated macrophages: a new and highly effective antiangiogenic therapy approach,” Br. J. Cancer 95(3), 272–281 (2006). 10.1038/sj.bjc.6603240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barber M. D., Thomas J. St J., Dixon J., Breast Cancer: An Atlas of Investigation and Management, p. 118, Clinical Publishing, Ashland, OH: (2008). [Google Scholar]
- 31.Zumsteg A., et al. , “Myeloid cells contribute to tumor lymphangiogenesis,” PLoS One 4(9), e7067 (2009). 10.1371/journal.pone.0007067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rivas M. A., et al. , “TNF alpha acting on TNFR1 promotes breast cancer growth via p42/P44 MAPK, JNK, Akt and NF-kappa B-dependent pathways,” Exp. Cell Res. 314(3), 509–529 (2008). 10.1016/j.yexcr.2007.10.005 [DOI] [PubMed] [Google Scholar]
- 33.Burow M. E., et al. , “Differences in susceptibility to tumor necrosis factor alpha-induced apoptosis among MCF-7 breast cancer cell variants,” Cancer Res. 58(21), 4940–4946 (1998). [PubMed] [Google Scholar]
- 34.Frith J. C., et al. , “The molecular mechanism of action of the antiresorptive and antiinflammatory drug clodronate: evidence for the formation in vivo of a metabolite that inhibits bone resorption and causes osteoclast and macrophage apoptosis,” Arthritis Rheum. 44(9), 2201–2210 (2001). 10.1002/(ISSN)1529-0131 [DOI] [PubMed] [Google Scholar]
- 35.Nonaka K., et al. , “Skewing the Th cell phenotype toward Th1 alters the maturation of tumor-infiltrating mononuclear phagocytes,” J. Leukocytes Biol. 84(3), 679–688 (2008). 10.1189/jlb.1107729 [DOI] [PubMed] [Google Scholar]
- 36.Ewens A., Mihich E., Ehrke M. J., “Distant metastasis from subcutaneously grown E0771 medullary breast adenocarcinoma,” Anticancer Res. 25(6B), 3905–3915 (2005). [PubMed] [Google Scholar]
- 37.Kalluri R., Zeisberg M., “Fibroblasts in cancer,” Nat. Rev. Cancer 6(5), 392–401 (2006). 10.1038/nrc1877 [DOI] [PubMed] [Google Scholar]
- 38.Robinson S. C., Scott K. A., Balkwill F. R., “Chemokine stimulation of monocyte matrix metalloproteinase-9 requires endogenous TNF-alpha,” Eur. J. Immunol. 32(2), 404–412 (2002). 10.1002/(ISSN)1521-4141 [DOI] [PubMed] [Google Scholar]
- 39.Ismair M. G., et al. , “Autocrine regulation of matrix metalloproteinase-9 gene expression and secretion by tumor necrosis factor-alpha (TNF-alpha) in NB4 leukemic cells: specific involvement of TNF receptor type 1,” Leukemia 12(7), 1136–1143 (1998). 10.1038/sj.leu.2401042 [DOI] [PubMed] [Google Scholar]
- 40.Leber T. M., Balkwill F. R., “Regulation of monocyte MMP-9 production by TNF-alpha and a tumour-derived soluble factor (MMPSF),” Br. J. Cancer 78(6), 724–732 (1998). 10.1038/bjc.1998.568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shin S. Y., et al. , “Transcription factor Egr-1 is essential for maximal matrix metalloproteinase-9 transcription by tumor necrosis factor alpha,” Mol. Cancer Res. 8(4), 507–519 (2010). 10.1158/1541-7786.MCR-09-0454 [DOI] [PubMed] [Google Scholar]
- 42.Richardson V. J., “Divergent and synergistic regulation of matrix metalloprotease production by cytokines in combination with C-C chemokines,” Int. J. Immunopathol. Pharmacol. 23(3), 715–726 (2010). [DOI] [PubMed] [Google Scholar]
- 43.Hyc A., et al. , “Influence of LPS, TNF, TGF-ss1 and IL-4 on the expression of MMPs, TIMPs and selected cytokines in rat synovial membranes incubated in vitro,” Int. J. Mol. Med. 27(1), 127–137 (2011). [DOI] [PubMed] [Google Scholar]
- 44.Deng W. G., Zhu Y., Wu K. K., “Up-regulation of p300 binding and p50 acetylation in tumor necrosis factor-alpha-induced cyclooxygenase-2 promoter activation,” J. Biol. Chem. 278(7), 4770–4777 (2003). 10.1074/jbc.M209286200 [DOI] [PubMed] [Google Scholar]
- 45.Vane J. R., et al. , “Inducible isoforms of cyclooxygenase and nitric-oxide synthase in inflammation,” Proc. Natl. Acad. Sci. U.S.A. 91(6), 2046–2050 (1994). 10.1073/pnas.91.6.2046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tsujii M., DuBois R. N., “Alterations in cellular adhesion and apoptosis in epithelial cells overexpressing prostaglandin endoperoxide synthase 2,” Cell 83(3), 493–501 (1995). 10.1016/0092-8674(95)90127-2 [DOI] [PubMed] [Google Scholar]
- 47.Lin E. Y., et al. , “Progression to malignancy in the polyoma middle T oncoprotein mouse breast cancer model provides a reliable model for human diseases,” Am. J. Pathol. 163(5), 2113–2126 (2003). 10.1016/S0002-9440(10)63568-7 [DOI] [PMC free article] [PubMed] [Google Scholar]