

Abstract
Genome-scale interrogation of gene function using RNA interference (RNAi) holds tremendous promise for the rapid identification of chemically tractable cancer cell vulnerabilities. Limiting the potential of this technology is the inability to rapidly delineate the mechanistic basis of phenotypic outcomes and thus inform the development of molecularly targeted therapeutic strategies. We outline here methods to deconstruct cellular phenotypes induced by RNAi-mediated gene targeting using multiplexed reporter systems that allow monitoring of key cancer cell-associated processes. This high-content screening methodology is versatile and can be readily adapted for the screening of other types of large molecular libraries.
Keywords: Cancer Biology, Issue 77, Medicine, Genetics, Cellular Biology, Molecular Biology, Biochemistry, Cancer Biology, Bioengineering, Genomics, Drug Discovery, RNA Interference, Cell Biology, Neoplasms, luciferase reporters, functional genomics, chemical biology, high-throughput screening technology, signal transduction, PCR, transfection, assay
Introduction
A variety of luciferase-based reporters for monitoring a diverse array of cell biological processes are commercially available. The majority of these DNA constructs encode luciferase proteins that are induced to express by specific cell stimuli or perturbations. The most robust transcriptionally based reporters place the expression of a luciferase enzyme under the control of synthetic enhancer elements or gene promoter regions well-established for their reliability in reporting a physiologically relevant transcriptional event1,2. There are also trans-reporter luciferase systems that utilize GAL4-UAS to drive luciferase protein expression. Gal4 is a yeast transcriptional activator and the UAS is an enhancer to which Gal4 specifically binds to activate the transcription of gene sequences placed down-stream of it3. These types of luciferase systems are typically supported by two DNA plasmids - one encodes the GAL4 protein fused to the regulatory portion of a protein of interest, and the second encodes a luciferase protein placed under the control of one or more UAS sequences. Thus, the cellular luciferase signal reflects the activity level of the protein of interest. Another common use of luciferase-based reporters is for monitoring protein stability whereby a protein of interest is fused to a luciferase enzyme4. Regardless of the type of reporter, a caveat emptor is noted here as one cannot assume the specificity of any reporter to have been well interrogated. Thus, due diligence is required in the incorporation of new reporter constructs in any research strategy.
The selection of the luciferase enzyme read-out can be as important as the reliability of the DNA elements that control its expression. Whereas firefly luciferase (FL) is the most commonly used enzyme in commercially available constructs, the advent of new luciferase reporter systems that do not require ATP (as in the case of FL-based reactions) or that incorporate more stable enzymes that emit stronger signals promise to improve the efficiency and reliability of this research platform. An important note regarding the selection of luciferase reporters in chemical studies is the susceptibility of FL to chemical inhibition that could give rise to false reports. In our experience, other enzymes such as Renilla or Gaussia luciferase (RL and GL, respectively) that utilize coelenterazine as a substrate are less readily inhibited by small molecules5.
The most common format for multiplexing luciferase read-outs entails the use of FL and RL largely given the availability of easy-to-use kits with the ability to sequentially measure enzymatic activities from a single sample. In most kits, samples are first exposed to luciferin to reveal levels of FL activity followed by a quenching reagent that is simultaneously deposited with coelenterazine to yield a secondary RL signal. With the addition of other luciferases that can be secreted such as GL or that use yet another substrate such as Cypridina luciferase (CL), a greater number of possibilities for generating high content data using luciferases have emerged. An example of a genome-wide RNAi screen using this technique can be found here6. These technological advances have in turn spurred the development of specific mechanisms to quench each type of luciferase enzyme in order to limit cross-talk7. In our hands, we note a greater frequency of "edge effects" (inexplicable trends in cellular activity associated with the edge of high titer culture plates), with secreted luciferases such as GL or CL. On the other hand, such secreted luciferases are useful for kinetic assays as they enable signal sampling without adulterating cell viability.
For our study presented here, we will simultaneously monitor the activity of three cancer-relevant cellular processes: the p53, Kras, and Wnt signal transduction pathways. The reporters incorporated in our study are the pp53-TA-Luc FL reporter from Clontech (henceforth referred to as the p53-FL reporter)8, an Elk1-GAL4/UAS-CL reporter system (henceforth Elk-1 reporter; Agilent), and the 8X TCF RL reporter that incorporates multiple synthetic enhancer elements recognized by the Wnt pathway transcriptional regulators known as T-cell factors (or TCFs)9-11.
Protocol
Entire protocol takes approximately 4 days.
1. Preparation of Cells for siRNA and Reporter Transfection
We will introduce here a protocol for interrogating siRNA libraries using a small set of siRNAs (384 different siRNA pools array in 96 well format with each pool consisting of 4x siRNAs targeting a single gene) from a genome-wide siRNA library for illustrative purposes. For this particular study, we will exploit HCT116 cells that exhibit deviant Wnt and Kras signaling, and p53 activity. The appropriate cell line in studies aimed at interrogating other cellular processes of interest will have to be identified and evaluated in a similar fashion.
Wash 5 x 10 cm2 plates of 80-90% confluent HCT116 cells with PBS then harvest cells by trypsinization using 1 ml of trypsinization solution/plate followed by neutralization with 10 ml of Dulbecco's modified eagle medium (DMEM) containing 2% fetal bovine serum (FBS) and 1% penicillin/streptomycin/plate.
Transfer suspended cells to a 50 ml conical tube and centrifugate at ~130 x g for 5 min, remove supernatant, and re-suspend cell pellet in 10 ml of DMEM/ 2%FBS/ 1% penicillin/streptomycin.
Count the cell number with a cell counter, and then make a cell solution with 105 cells/ml, keep 150 ml cell suspension for the next step. Plate 104 cells (100 μl) in each well of a 96 well cell culture plate using a Multidrop automated liquid dispenser (henceforth Multidrop). The cell suspension should be sufficient in this case for plating 12 x 96 well plates.
Incubate the plates with cells at 37 °C in an incubator with 5% CO2 while preparing the transfection mixture.
2. Preparing the Reporter Construct Stock Solution
Isolate high-quality reporter construct DNA using standard midiprep kits (NucleoBond Xtra Midi produced by Clontech) and dilute the DNA to a final concentration of 1 mg/ml for each reporter.
Prepare 100 μl of reporter construct stock solution by combining 30 μl of p53-FL, 30 μl of 8X TCF-RL, 30 μl of Elk1-Gal4, 6 μl UAS-CL and 4 μl of H2O to achieve a DNA mix with a final 1:1:1:0.2 ratio of the reporters. Final DNA concentration of this DNA mix stock solution is 0.3 mg/ml.
3. Preparing the siRNA/DNA Transfection Mix
In this part of the protocol, we will prepare siRNA/DNA transfection mixes that will be sufficient for transfecting 12 x 96 well plates. For this test library of 4 x 96 well plate of siRNAs, we will transfect each siRNA pool in triplicate. The transfection reagent we will use is called Effectene (Qiagen). In our hands this is the only reagent that works for the simultaneous delivery of siRNA and DNA into cultured cells.
Dilute 80 μl of reporter construct stock solution in 8 ml of EC buffer (Effectene Kit) to achieve a DNA solution with a final concentration of 3 μg/ml. In general, prepare enough DNA solution mix for immediate use.
Plate 20 μl of this DNA solution to each well of a 96 well/plate. The number of 96 well plates will depend on how many siRNA pools will be tested. For example, in this case we will need 4 x 96 well plates to evaluate 384 different siRNA pools.
The siRNAs to be tested should be at a stock concentration of 5 μM. If not, they can be replica plated and diluted to this concentration using the recommended dilution buffer associated with each library.
Transfer 2 μl of each 5 μM siRNA stock to the corresponding well of the PCR plate containing the diluted DNA stock with a Biomek Automated Liquid Handler. Depending on the scale of the study, a multi-channel pipettor may suffice.
Now, we will add 1 μl of Enhancer solution (Effectene kit) to each well of the DNA/siRNA plate. This again can be accomplished either with an automated liquid handler or multichannel pipettor.
Allow the Enhancer reaction to occur at RT for 5 min then add 3 μl of Effectene transfection reagent to each well and incubate at RT for an additional 10 min.
To stop transfection complex formation, add 10 μl of DMEM/ 2% FBS/ 1% penicillin/streptomycin to each well of the PCR plate.
Transfer 10 μl of the transfection mix to each well of the 96 well plate containing the cells (retrieved from the cell incubator). As each transfection will be performed in triplicate, the same mix will be applied to two additional 96 well plates containing cells.
Incubate the cells with transfection mixtures added at 37 °C in a humidified environment with 5% CO2 for 36 hr.
4. Determine Luciferase Activities from Cell Samples
After 36 hr, luciferase activities are ready for measurement in transfected cells. The endpoint should be determined on a per-experiment basis. In our study, a 36 hr incubation time had previously yielded a sufficiently robust signal for meaning outcome determination. As this study incorporates multiple reporters (one secreted into the culture medium, and two others expressed in the cell cytoplasm), we will obtain measurements from both culture medium as well as a cellular lysate.
- Detection of CL activity in the culture medium:
- 20 μl of culture medium is replica-plated in a white opaque 96 well plate (either using the Biomek Liquid Handler or multichannel pipettor).
- Add 20 μl of CL assay buffer (Targeting Systems) to each well.
- Add 10 μl of Cypridina luciferin substrate (Targeting Systems) to each well and detect CL activity using a luminometer (the Pherastar plate reader produced by BMG for example).
- Detection of FL and RL activity:
- Lyse the cells by first removing the culture medium. This can be accomplished rapidly by turning plate upside down and tossing medium in a sink. Remaining medium can be removed by gently tapping the upside down plate on paper towels. Add 30 μl of 1X Passive Lysis Buffer (Promega) to each well of cells using the Multidrop and place on a platform rocker (Bellco is one company that makes these) set a medium speed for 5 min at the RT.
- Add 20 μl of the Luciferase Assay Reagent II (part of the Promega Dual Luciferase Kit) using the Multidrop and immediately measure FL activity using the luminometer. Next, add Stop & Glo Reagent (Promega) that will quench the FL activity and allow measurement of RL activity. Note: unless you are using substrate mixes that allow extended signal duration (such as Dual-Glo Luciferase Kits from Promega), the use of flash kits requires rapid measurement upon substrate addition (within 5 min).
Representative Results
Despite advances in mapping the mutational landscape of various cancers using massive genome sequencing efforts12-14, we still do not have in place a systematic approach for translating these observations into intervention strategies. In colorectal cancer (CRC), mutations that are predicted to affect components of three cellular processes - Wnt/β-catenin, p53, and Kras signaling - are found in 99% of all tumors suggesting they are key drivers of cellular transformation in the gut13. Thus, cancer genome datasets are likely enriched for genes that support common denominator cancer promoting processes, and that could be exploited for rapid identification of cancer cell vulnerabilities. To investigate this hypothesis, we have devised strategies amenable to high-throughput interrogation of gene function with respect to Wnt/β-catenin, p53, and Kras signaling. We first demonstrate the specificity of each reporter system in single reporter tests (Figure 1) and then provide evidence that these reporters could be used together for simultaneous measurements of gene activity of these three important cellular processes (Figure 2).
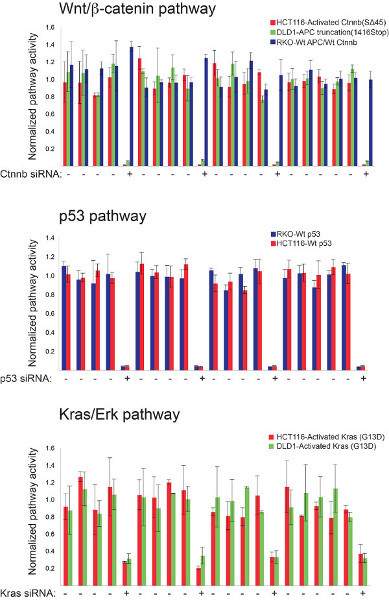 Figure 1. High-throughput luciferase-based assays for monitoring Wnt/β-catenin, p53 and Kras/Erk pathway status in mammalian cells. The robustness of three different screening platforms were individually assessed prior to multiplexing using control siRNAs targeting well-established pathway components. Indicated colorectal cancer cell lines were transfected with the 8X TCF, pp53-TA-Luc, or Elk-1 firefly luciferase-encoding reporter constructs along with pools of siRNAs targeting β-catenin, p53, or Kras, respectively. Pathway-relevant genotypes for each cell line are indicated. The strength of each screening platform was assessed by the reproducibility of these siRNAs-induced effects on the relevant signal transduction pathway. The β-catenin siRNAs have no effect on 8X TCF reporter activity in RKO cells in contrast to DLD-1 and HCT-116 cells that are respectively deficient in the APC pathway suppressor protein and express an activated form of β-catenin. Click here to view larger figure.
Figure 1. High-throughput luciferase-based assays for monitoring Wnt/β-catenin, p53 and Kras/Erk pathway status in mammalian cells. The robustness of three different screening platforms were individually assessed prior to multiplexing using control siRNAs targeting well-established pathway components. Indicated colorectal cancer cell lines were transfected with the 8X TCF, pp53-TA-Luc, or Elk-1 firefly luciferase-encoding reporter constructs along with pools of siRNAs targeting β-catenin, p53, or Kras, respectively. Pathway-relevant genotypes for each cell line are indicated. The strength of each screening platform was assessed by the reproducibility of these siRNAs-induced effects on the relevant signal transduction pathway. The β-catenin siRNAs have no effect on 8X TCF reporter activity in RKO cells in contrast to DLD-1 and HCT-116 cells that are respectively deficient in the APC pathway suppressor protein and express an activated form of β-catenin. Click here to view larger figure.
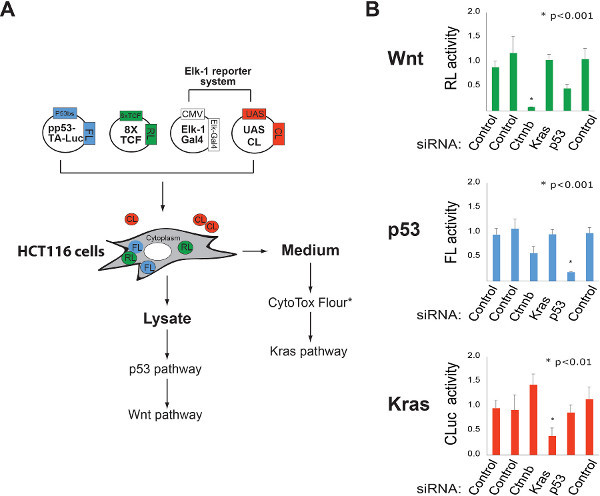 Figure 2. Multiplexing luciferase based reporters for high-content cellular analysis of gene function. A. Schematic representation of a strategy for simultaneously monitoring Wnt/β-catenin, p53, and Kras pathway activities. FL= firefly luciferase, RL= Renilla luciferase, CL= Cypridina luciferase (secreted enzyme). FL and RL activities in lysate are determined using luciferin and coelenterazine substrates. CL activity in the culture medium is measured using oxyluciferin. *Other reporter systems can be incorporated into this protocol to increase the content of each experiment. For example, the CytoTox Flour assay can be used to monitor cellular toxicity by sampling the levels of an intracellular protease released into the culture medium. The addition of the CytoTox assay reagent does not influence the activity of the CL reporter. B. Identification of pathway-specific vulnerabilities using RNAi. The ability of the multiplexed luciferase system described in "A" to quantify gene dedication to cellular function was tested using the indicated siRNA pools. Click here to view larger figure.
Figure 2. Multiplexing luciferase based reporters for high-content cellular analysis of gene function. A. Schematic representation of a strategy for simultaneously monitoring Wnt/β-catenin, p53, and Kras pathway activities. FL= firefly luciferase, RL= Renilla luciferase, CL= Cypridina luciferase (secreted enzyme). FL and RL activities in lysate are determined using luciferin and coelenterazine substrates. CL activity in the culture medium is measured using oxyluciferin. *Other reporter systems can be incorporated into this protocol to increase the content of each experiment. For example, the CytoTox Flour assay can be used to monitor cellular toxicity by sampling the levels of an intracellular protease released into the culture medium. The addition of the CytoTox assay reagent does not influence the activity of the CL reporter. B. Identification of pathway-specific vulnerabilities using RNAi. The ability of the multiplexed luciferase system described in "A" to quantify gene dedication to cellular function was tested using the indicated siRNA pools. Click here to view larger figure.
Discussion
There are several purveyors of luciferase-based reporter systems (such as Promega, Targeting Systems, New England BIolabs, and Thermo Fisher) that provide useful on-line resources for those entertaining a high-throughput screen for the first time. In addition, a number of excellent reviews regarding optimizing the use of high-throughput screens for gene discovery and how RNAi-based screening results can be integrated with other -omics-based datasets should be helpful15,16. Methods for the selection of "hits" from high-throughput screens are the subject of another discussion that is beyond the scope of this report. Several reviews on this topic are available17,18. However, an important criterion for validating selected hits from screens based on the use of transcriptionally activated reporters is the ability for the same perturbagen (RNAi- or chemically based) to induce the same effect on validated target genes as measured using RT-PCR, qPCR, or microarray analysis. Moreover, keep in mind that the magnitude of response as measured by the reporter is typically higher than the induction of an endogenous reporter given that a feature of a successfully synthetic reporter is to provide a robust signal. Finally, an advantage of this co-transfection strategy is the ease with which one can rapidly assemble different reporter cocktails for monitoring a desired set of cellular activities. Although more labor intensive, the use of cell lines stably harboring the reporters would likely increase the signal to noise ratio given the variability of response is now primarily limited to siRNA transfection efficiency alone.
Disclosures
O.K. and L.L are authors on a pending patent relating to multiplexed luciferase technology.
Acknowledgments
We acknowledge funding support from CPRIT (RP100119) and the Welch Foundation (I-1665).
References
- Bronstein I, Fortin J, Stanley PE, Stewart GS, Kricka LJ. Chemiluminescent and bioluminescent reporter gene assays. Analytical Biochemistry. 1994;219:169–181. doi: 10.1006/abio.1994.1254. [DOI] [PubMed] [Google Scholar]
- Miraglia LJ, King FJ, Damoiseaux R. Seeing the light: luminescent reporter gene assays. Combinatorial Chemistry & High Throughput Screening. 2011;14:648–657. doi: 10.2174/138620711796504389. [DOI] [PubMed] [Google Scholar]
- McGuire SE, Roman G, Davis RL. Gene expression systems in Drosophila: a synthesis of time and space. Trends in Genetics: TIG. 2004;20:384–391. doi: 10.1016/j.tig.2004.06.012. [DOI] [PubMed] [Google Scholar]
- Smirnova NA, et al. Development of Neh2-luciferase reporter and its application for high throughput screening and real-time monitoring of Nrf2 activators. Chem. Biol. 2011;18:752–765. doi: 10.1016/j.chembiol.2011.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, et al. Small molecule-mediated disruption of Wnt-dependent signaling in tissue regeneration and cancer. Nat. Chem. Biol. 2009;5:100–107. doi: 10.1038/nchembio.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W, et al. A genome-wide RNAi screen for Wnt/beta-catenin pathway components identifies unexpected roles for TCF transcription factors in cancer. Proc. Natl. Acad. Sci. U.S.A. 2008;105:9697–9702. doi: 10.1073/pnas.0804709105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lum LD, Kulak O, inventors. Multiplexed Luciferase Reporter Assay Systems. United States patent. 2012.
- Funk WD, Pak DT, Karas RH, Wright WE, Shay JW. A transcriptionally active DNA-binding site for human p53 protein complexes. Mol. Cell Biol. 1992;12:2866–2871. doi: 10.1128/mcb.12.6.2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DasGupta R, Kaykas A, Moon RT, Perrimon N. Functional genomic analysis of the Wnt-wingless signaling pathway. Science. 2005;308:826–833. doi: 10.1126/science.1109374. [DOI] [PubMed] [Google Scholar]
- Korinek V, et al. Depletion of epithelial stem-cell compartments in the small intestine of mice lacking Tcf-4. Nat. Genet. 1998;19:379–383. doi: 10.1038/1270. [DOI] [PubMed] [Google Scholar]
- Korinek V, et al. Two members of the Tcf family implicated in Wnt/beta-catenin signaling during embryogenesis in the mouse. Mol. Cell Biol. 1998;18:1248–1256. doi: 10.1128/mcb.18.3.1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koboldt DC, et al. Comprehensive molecular portraits of human breast tumours. Nature. 2012. [DOI] [PMC free article] [PubMed]
- Wood LD, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–1113. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- Berndt JD, Biechele TL, Moon RT, Major MB. Integrative analysis of genome-wide RNA interference screens. Science Signaling. 2009;2:pt4. doi: 10.1126/scisignal.270pt4. [DOI] [PubMed] [Google Scholar]
- Falschlehner C, Steinbrink S, Erdmann G, Boutros M. High-throughput RNAi screening to dissect cellular pathways: a how-to guide. Biotechnology Journal. 2010;5:368–376. doi: 10.1002/biot.200900277. [DOI] [PubMed] [Google Scholar]
- Boutros M, Bras LP, Huber W. Analysis of cell-based RNAi screens. Genome Biology. 2006;7:R66. doi: 10.1186/gb-2006-7-7-r66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert DF, et al. A novel multiplex cell viability assay for high-throughput RNAi screening. PLoS ONE. 2011;6:e28338. doi: 10.1371/journal.pone.0028338. [DOI] [PMC free article] [PubMed] [Google Scholar]