

Abstract
Retaining glycosyltransferase enzymes retain the stereochemistry of the donor glycosidic linkage after transfer to an acceptor molecule. The mechanism these enzymes utilize to achieve retention of the anomeric stereochemistry has been a matter of much debate. Re-analysis of previously released structural data from retaining and inverting glycosyltransferases allows competing mechanistic proposals to be evaluated. The binding of metal-nucleotide-sugars between inverting and retaining enzymes is conformationally unique and requires the donor substrate to occupy two different orientations in the two types of glycosyltransferases. The available structures of retaining glycosyltransferases lack appropriately positioned enzymatic dipolar residues to initiate or stabilize the intermediates of a dissociative mechanism. Further, available structures show that the acceptor nucleophile and anomeric carbon of the donor sugar are in close proximity. Structural features support orthogonal (front-side) attack from a position lying ≤90° from the C1-O phosphate bond for retaining enzymes. These structural conclusions are consistent with the geometric conclusions of recent kinetic and computational studies.
Introduction
Understanding the fundamental structure-function relationships of glycosyltransferase enzymes is an essential step in the directed development new drugable inhibitors. Glycosyltransferases synthesize biological oligo- and polysaccharides, many of which have been associated with disease processes. In addition to being the defective product of a number of human genetic disorders e.g. [1]–[3], glycosyltransferases play critical roles in many facets of infection (e.g. [4]–[10]), immunity (e.g. [11]–[13]) and cancer (e.g. [14]–[22]. Despite the potential of these targets, the underlying biochemical mechanism of the glycosyltransferases is still poorly understood and is hampering focussed drug development.
Leloir glycosyltransferases donate a monosaccharide unit from a nucleotide-sugar (“glycosyl donor”) to a “glycosyl acceptor”, typically a hydroxyl group of an oligosaccharide [23]. Two stereochemical classes are known. Retaining glycosyltransferase enzymes preserve the stereochemistry about the anomeric carbon atom of the donor sugarin the new glycosidic linkage i.e. an axial donor stereochemistry results in an axial stereochemistry in the product. Inverting glycosyltransferases invert the anomeric stereochemistry i.e. an axial donor becomes equatorial in the product. The mechanism of the inverting reaction is widely accepted and is mechanistically straightforward; the acceptor hydroxyl acts as a nucleophile and approaches the anomeric carbon from the opposite side to the donor-nucleoside linkage eventually resulting in inversion of anomeric stereochemistry as the nucleoside leaves. The mechanism for retaining glycosyl transfer stereospecificity is more problematic and remains a matter of debate. Mechanisms can be broadly classified as proceeding with primarily dissociative (SN1) or primarily associative (SN2)character. Postulate mechanisms are outlined in Figure 1 .
Figure 1. Proposed glycosyltransferase mechanisms.

(A)A double displacement mechanism utilizing two inversions with net retention of stereochemistry involving a covalent glycosyl-enzyme intermediate. The individual steps are inverting via (B) an SN2 process. Inverting Leloir glycosyltransferases promote a backside nucleophilic attack on C1 by the acceptor from an inline (usually equatorial) position, with resulting inversion of the anomeric bond stereochemistry. Alternative mechanisms for retaining glycosyltransferases include: (C) an orthogonal mechanism consisting of nucleophilic attack on C1 by the acceptor concurrent with leaving group loss from a position approximately at right angles to the C1-leaving group axis; (D) an SNi mechanism involving an intermediate with oxocarbenium character followed by rapid internal nucleophilic attach by the acceptor nucleophile; or (E) an SN1 mechanism involving a discreet oxocarbenium intermediate. All mechanisms require proton transfers of the hydroxyl hydrogen of the acceptor to an enzymatic baseor the departing leaving group.
The earliest mechanism proposed to explain retaining glycosyltransfer was the double displacement mechanism ( Fig. 1A ): an initial nucleophilic substitution provided by an enzyme nucleophile forms an inverted covalent enzyme-carbohydrate intermediate which is in turn attacked by the acceptor molecule leading to a net retention of anomeric stereochemistry [24]. Each step of this process occurs as described above for inverting glycosyltransferases via backside attack through a single transition state resulting in inversion ( Fig. 1B ). Although there is mass spectrometric evidence that migh support the existence of a covalent glycosyl-enzyme intermediate in retaining glycosyltransferases [25], [26], such an intermediate has not been detected structurally, kinetically or spectroscopically [27]–[33].
Retaining substitution with dissociative character ( Fig. 1D,E ) has been proposed as an alternative [27]–[33]. Nucleotide diphosphates (NDPs) are excellent leaving groups, and the resulting oxocarbenium cation could be stabilized by adjacent protein dipoles. Both of these factors would favour a dissociative process. However, unimolecular dissociation would result in the loss of stereochemical integrity. No partially inverted products have ever been described for a retaining glycosyltransferases. Enzymes that do involve dissociative character are hydrolases or transferases that do not transfer stereocenters (reviewed in [34]). Thus proposed dissociative pathways also require that steric hindrance is provided by the enzyme to force the generation of retained product ( Fig. 1E ).
Another dissociative variant is called SNi (Nucelophilic Substitution with internal return; Fig. 1D ). This mechanism involves partial nucleotide diphosphate dissociation and charge development within a polar active site cage prior to nucleophilic attack by the acceptor [35]. This is usually drawn as a dissociative transition state with a long distance interaction between the anomeric carbon atom and the incoming nucleophile and a shorter interaction with the departing leaving group. This leads to a short-lived intermediate ion pair which rapidly collapses in a second step. SNi has previously been invoked to explain gas-phase chemical reactions, butits acceptance as a suitable pathway for retaining glycosyltransferases has met with resistance [25], [26].
Recent kinetic investigation of trehalose-6-phosphate synthase, a metal-free retaining glycosyltransferase, concludes that the available evidence favors a “front-side SNi” intermediate having substantial dissociative character at the rate-limiting transition state [36]. The same conclusion is supported by kinetic and computational studies of the solvolysis of isotopically labeled α-D-glycopyranosyl fluorides in hexafluoro-2-propanol in which the kinetic isotope effects are most consistent with a “front-face” geometry [37]; the authors favor a stepwise SNi intermediate, although the data show a concerted transition-state with both the leaving group and the incoming nucleophile in close proximity to the anomeric carbon gives closely similar computed isotope effects. Finally, a computational study of lipopolysaccharyl-α-1,4-galactosyltransferase C, a Leloir retaining glycosyltransferase finds a front-side geometry at the transition state which is described as “SNi-like” with significant charge development in the donor sugar as the transition state is reached [38].
It has also been suggested that retaining transfer may contain both dissociative and associative elements [39], and the two are not mutually exclusive. Absolute distinction between associative and dissociative reaction pathways is not always possible; dissociative pathways progress into associative pathways as the transition state develops a less stable and shorter-lived oxocarbenium cation intermediate [40]; the mechanism illustrated as Fig. 1C reflects this continuum from the SNi mechanism of Fig. 1D , involving a discrete if short-lived intermediate, to the SN2 case of Fig. 1B involving only a single transition state without an intermediate. The precise character of the Fig. 1C mechanism depends upon the intimate details of charge development and nucleophilic attack (vide infra). We use the term “orthogonal” for this mechanism to mean a process involving the nucleophile and the leaving group on the same side (a.k.a. “front-side” attack) where the approach of the nucleophile is approximately orthogonal to the breaking bond axis, and proceeding in a single step from reactants to products without an intermediate.
Knowledge of the mechanistic details of glycosyltransferases can be derived from a number of experimental approaches of which structural studies play a central role in providing starting points for computation, and geometrical constraints on the enzymatic groups required to interpret the kinetics. The modest degree of sequence homology among glycosyltransferase families has made the prediction of tertiary structures difficult. However, structural determinations in recent years have revealed that the catalytic domains of most glycosyltransferases display one of two fold types designated GT-A or GT-B [41], [42]. With few exceptions, the donor binding Rossmann folds of glycosyltransferases contain a “DXD motif” that consists of an Asp-X-Asp amino acid triplet used to coordinate the phosphates of the donor molecule through a divalent cation with octahedral geometry. Some inverting enzymes do not require a divalent metal cofactor, though to date there is only one retaining Leloir-type enzyme that has been characterized as metal independent [43].
A neutron structure of the human retaining enzyme GTA at LANCE PCS (PDB 4DHH associated with [44]) has been reported. More detailed analysis of this structure has revealed an aprotic active site that appears to be incompatible with a dissociative mechanism. To examine the generality of this observation, we report a re-investigation of the published geometric presentation between donor and acceptor substrates in the enzymatic active sites of previously reported GT-A fold glycosyltransferases. The analysis of the structures, together with literature data from NMR, MS, kinetics, and computational studies,point to the orthogonal mechanism for retaining glycosyltransferases as both the simplest and the most consistent with the available data.
Methods
Deposited GT-A fold PDBs identified by CAZy were analyzed for geometric parameters using SetoRibbon,a continued development of SETOR [45] with adaptations for high throughput geometric analysis. Of the eleven families with deposited structures ( Table 1 ), four were found which had unambiguous densities complete for donor (nucleotide and monosaccharide) and acceptor molecules (or analogs): GT-6 retaining enzyme human blood-group A glycosyltransferase, GTA (L266M/G268A [46]); GT-7 inverting enzyme 1,4-galactosymtransferase T1, GalT1 (wt [47]); GT-8 retaining enzyme lipooligosaccaride transferase C, LgtC (C128/174S [48]); and GT-43 inverting enzyme β-1,3-glucuronosyl transferase 1, GlcAT-1 (M344H [49], [50]). Retaining enzymes GTA and LgtC were both crystallized with deoxy-acceptor analogs, which allowed confident modeling of their respective nucleophilic atoms. Inverting enzymes GlcAT-I and GalT1 required combining separate donor-bound and acceptor-bound structures for analysis, and the accuracy of the analyzed geometry is potentially reduced as the same steric constrains as the bisubstrate liganded active site are not necessarily applicable. For example unambiguous density for the carbohydrate moiety of retaining enzyme GTA mutants have been observed in 4 distinct conformations [46], [51], though all but one conformation is incompatible with catalytic turnover. Structures have been deposited of the two retaining enzymes with both donor and acceptor analogs bound simultaneously, where the deviation of the analogs from positions observed occupied by the natural acceptors is only ∼0.1 Å.
Table 1. GT-A fold glycosyltransferase families with deposited structures.
| Family | Example enzyme | Stereo-specificity | Example Complex(es) |
| GT-2 | SpsA | Inverting | UDP |
| GT-6 | GTA | Retaining | UDP-Gal+Gal-Fuc |
| GT-7 | GalT1 | Inverting | UDP-Gal, GlcNAc-GlcNAc |
| GT-8 | LgtC | Retaining | UDP-Gal+Gal-Glc |
| GT-13 | GnT1 | Inverting | UDP-GalNAc, UDP-Glc |
| GT-15 | Kre2 | Retaining | GDP+Man+GlcNAc |
| GT-27 | GNAc:Pep | Retaining | UDP+GlcNAc |
| GT-43 | GlcAT-I | Inverting | UDP-GlcUA, UDP+Gal-Gal-Xyl |
| GT-55 | MpgS | Retaining | GDP-Man |
| GT-64 | Extl2 | Retaining | UDP-GalNAc |
| GT-78 | MgS | Retaining | GDP |
| GT-81 | ManT | Retaining | GDP |
Bold underlined families were assessed to have unambiguous whole acceptor and donor molecule electron density for analysis; those in italics have donor density.
The geometry between the phosphates and acidic residues that coordinate the divalent metal cofactor (M) were determined with SetoRibbon, a continued development of SETOR [45] with adaptations for high throughput geometric analysis. The molecular geometry surrounding the metal is roughly octahedral, though when very acute bidentate Asp coordination is employed this can be skewed to nearly trigonal prismatic dimensions. For consistency we have labeled the α-phosphate O2 as a at the apex of the coordination octahedron, and place the second β-phosphate O1 as b in the clockwise position of the projection with the remaining coordinating atoms labeled c-f as illustrated in Figure 2 . The metal cation is the focal point of the alignments analyzed. Instead of optimizing RMS for the polypeptide chain, alignments were made using metal cation M, α-phosphate O1 and β-phosphate O2 as fixed positions from which to compute relative distances and angles.
Figure 2. The a-f nomenclature used to describe octahedral binding partners.

Inverting enzymes such as GalT1 (top) achieve nearly perfect octahedral geometry about the coordinated metal ion (displayed angles of 81° and 91° compared to ideal octahedral 90° bond angles) with subsequent “inline” (approaching 180°) placement of the acceptor nucleophile for classic inverting SN2 backside attack. Retaining enzymes such as GTA (bottom), however, use an arrangement of acidic residues, often with acute bidentate Asp coordination, which severely skews metal geometry (displayed angles of 54° and 115°) and allots sufficient room between phosphate oxygens for orthogonal attack from the acceptor. U is uridine, C1 is donor galactose C1.
Geometric parameters that pertain to the discussion of the mechanism include the nucleophilic distance from the acceptor nucleophilic oxygen atom Nu to the donor monosaccharide electrophilic center (C1 for all of the enzymes analyzed),the angle between the incoming nucleophile and leaving group β-phosphate oxygen O3, the distance between Nu and O3, the distance to the closest enzyme electronegative atom observed for O3 and C1, the angle between O3 and C1 nearest dipole vectors, and the angles between donor β-phosphate O1 and the adjacent coordinating acidic ligands of the metal ion. These are listed in Table 1 . Protein macrodipoles (represented in Figure 3 ) were estimated with the Protein Dipole Moments Server [52]and compared to the path between Nu and C1.
Figure 3. Reaction center dipoles.

Opposed to the placement of the acceptor nucleophile (Green spheres), the closest polar residues to leaving group β-phosphate O3 and C1 lay acutely (67° and 75°, respectively) for inverting enzymes (A,B) and lie nearly in-line (171° and 155°, respectively) for retaining enzymes GTA (C,D). This may help to stabilize the associative intermediates without hindering the opposite angle of attack from the acceptor molecule nucleophile. Also, the O3– C1vectors lay looselyperpendicular to the enzyme macrodipole vectors to stabilize the inverting transition states (green arrows) (A,B), and loosely parallel to stabilize the retaining transition states (C,D) (green ⊕, dipole oriented with the cationic end above the page and the anionic end in the page).
Results and Discussion
Analysis of Available Structural Data
It is well accepted that dissociative (SN1 or SNi) mechanisms require activation and stabilization of the ions as they form [53]–[55]. In homogenous solution this is usually achieved by a polar protic solvent such as water. Within an enzyme, solvent molecules are thought to be excluded from the proximity to the donor sugar anomeric carbon (C1) electrophile of these enzymes to prevent destructive donor hydrolysis from nucleophilic attack by water. The neutron structure of GTA shows no water in proximity to the active site; indeed, the active site is aprotic to a distance of >4.5 Å from the reaction site. In the absence of solvent it falls upon the enzyme to provide correctly positioned and oriented dipoles with which to stabilize anion pair intermediates. The closest observed enzymatic polar groups to donor sugar C1, β-phosphate nucleofuge (atom O3) and ring O5 for a number of retaining and inverting enzymes are outlined in Table 2 . These centers would share the charge of an intermediate oxocarbeniumion as would develop in a SN1 or SNi process. All lie too far away (∼4.5 Å) to initiate a dissociative mechanism, but may extend the lifetime of a dipola rtransition state.
Table 2. Active site residue identities and geometric values.
| Stereospecificity | Inverting | Retaining | |||||
| Example enzyme | GlcAT-I | GalT1 | GnT1 | LgtC | GTA | Extl2 | ManT |
| PDB(1) | 1V84 | 1TVY | 2AM3 | 1GA8 | 2RJ7 | 1OMZ | 2WVL |
| PDB(2) | 1KWS | 1TW5 | |||||
| Nu – C1 dist. | 4.4 Å | 4.2 Å | 4.0 Åa | 2.2 Å | 2.5 Å | ||
| <Nu-C1-O3 | 160° | 165° | 151°a | 90° | 74° | NA | NA |
| Nu – O3 dist. | 5.8 Å | 5.6 Å | 5.4 Åa | 2.8 Å | 2.2 Å | ||
| O3– nearest polar X | H2Ob | K279 | Y184 | H78 | K346 | H2Ob | Y268 |
| O3- X dist. | 4.4 Å | 4.4 Å | 5.4 Å | 4.7 Å | 5.6 Å | 3.8 Å | 4.4 Å |
| <X-O3-C1 | 91° | 80° | 87° | 171° | 149° | 131° | 59° |
| C1 nearest polar Y | H308 | W314 | D211 | Q189 | E303 | R293 | D167 |
| C1-Y dist | 3.6 Å | 4.5 Å | 5.2 Å | 3.5 Å | 4.8 Å | 3.7 Å | 3.5Å |
| <Y-C1-O3 | 67° | 75° | 71° | 162° | 155° | 167° | 142° |
| O5 nearest polar Z | R156 | W314 | D291 | Q189 | R352 | R293 | D168 |
| O5-Z dist. | 5.9 Å | 3.4 Å | 3.9 Å | 4.2 Å | 5.8 Å | 3.2 Å | 3.8 Å |
| <Z-O5-C1 | 113° | 123° | 96° | 82° | 83° | 97° | 68° |
| c c | H2Ob | H2Ob | H2Ob | D103 | D211 | H2Ob | NA |
| <bMc c | 89° | 82° | 87° | 105° | 116° | 101° | NA |
| f c | D196 | H347 | H2Ob | D105 | D213 | H2Ob | N313 |
| <bMf c | 114° | 104° | 95° | 92° | 90° | 88° | 82° |
With the exception of the GTA neutron diffraction studies [44] hydrogen atoms are not directly observed, so distances are given between centers of non-hydrogen atoms. Italic enzyme names indicate the model did not contain an acceptor molecule.
PDB 2AM3 has a glycerol molecule modeled as an acceptor.
It is likely that the active species are not actually water molecules, but residues in disordered regions of the polypeptide.
b, c and f are octahedral binding partners to the coordinated metal atom M as described in Figure 2 .
For retaining enzymes GTA and ManT the closest nucleophiles to C1 (Glu303 and Asp167, respectively) could be considered candidate nucleophiles for a double displacement reaction, however structurally conserved nucleophiles are absent in many reported retaining enzymes including LgtC and Extl2 which have respective Gln and Arg residues in this position. The closest polar groups to donor sugar C1, O5 and phosphate O3 vary considerably, can carry either positive or negative charges, and often their mutation does not inhibit catalysis (eg. [25]). Furthermore, glycosyl transfer still proceeds when O5 is substituted with sulfur [56], and as such an intermediate thiocarbenium ion is unlikely to be stabilized to the same extent by donation from sulfur as in a regular oxocarbenium intermediate.
It is noteworthy that the active site architecture for proximal dipoles is conserved and distinct for retaining and inverting enzymes ( Fig. 3 , Table 2 ). For the inverting enzymes the C1 bond to the leaving group O3 lay at acute angles from adjacent polar residues ( Fig. 3A: 67° to His308 N∈ of GlcAT-1; Fig. 3B: 80° to Lys279Nζ and 75° to Trp314 N∈ of GalT1). In retaining enzymes the corresponding angles are obtuse ( Fig. 3C 162 to His78 N∈ of LgtC; Fig 3D: 155° to Lys 346 Nζ and 155° to Glu303 O∈ of GTA). Significantly, this positions the polar groups and enzyme macrodipoles that stabilize the retaining and inverting transition states to lie approximately orthogonal to each other ( Fig. 3 ). The orientations of the protein macrodipoles are conserved among retaining enzymes where they lie roughly perpendicular to the nucleophile approach as expected for stabilization of developing partial cationic charge without influencing leaving group departure or nucleophilic attack. The macrodipoles of inverting enzymes are similarly conserved, but are oriented parallel to the line of nucleophile approach, oriented to assist such an attack.
Further, the proximity of nucleophile (Nu) and electrophile (C1) in the retaining enzymes places tight constraints on the extent of dissociation possible before nucleophilic approach becomes the dominant interaction. The position of acceptor nucleophiles modeled from deoxy-acceptor crystal structures are observed at distances much less than 3 Å (2.5 Å for GTA and 2.2 Å for LgtC) from donor C1, whereas they wouldbe expected to reside greater than 3 Å away to allow UDP dissociation prior to nucleophilic attack [57].The computed transition states for glucopyranosyl fluoride solvolyses place the acceptor oxygen 3.02Å from C1 in the SNi transition state and at 2.25 Å in the associative “front side” transition state [37]; the corresponding distance computed for the “SNi-like” transition state of a galactosyl transferase is 2.3 Å [38].Nu and C1 can be much greater than 3 Å in a precatalytic conformation as is observed for both analyzed models of inverting enzymes (4.4 Å for GlcAT-I and 4.2 Å for GalT1).
Comparing the biologically active GTA structures to inverting enzymes such as galactosyltrasferasesβ4GalT1 reveals that the enzymes bind distinct metal-nucleotide-sugar conformers ( Figs. 2 & 3 ), where the metal coordinating angle <b-M-c is less than <b-M-f for inverting enzymes and the opposite for retaining enzymes ( Table 2 ). Inverting enzymes position C1 for inline nucleophilic attack from the acceptor at an angle nearly 180° to the leaving group, while retaining enzymes position these groups at roughly 90° with respect to the C1-leaving group axis ( Fig. 2 165° in inverting GalT1; 74° in retaining GTA). This is accomplished by inverting and retaining enzymes orienting their metal-nucleotide-sugar binding Rossmann folds approximately perpendicular to one another ( Figure 4 ). Although geometrically distinct, in-line (inverting) and orthogonal transition states are not dissimilar; retaining enzymes apparently orient their acceptors to an apical position of a trigonal bipyramidal transition state with the leaving group occupying one of the equatorial positions. This orientation is formally accessible as a pseudo-rotation of the trigona bipyramidal geometry of the SN2 transition state, which is facilitated by structurally conserved obtusely oriented enzymatic dipoles for retaining enzymes, and is complemented by conserved acute dipoles for retaining enzymes ( Fig. 3 ). Concurrent closing of the Nu-C1 distance with leaving group loss and concurrent opening of the H-C1-Nu angle would result in associative retention of the donor’s anomeric stereochemistry ( Fig. 1C ). A similar reaction pathway has been suggested based on structural studies involving glycomimetic inhibitors [36], [58] and quantum chemical calculations [59], however the proposed mechanisms were still referred to as “SNi-like” implying the mechanism proceeds with a rate-limiting dissociative transition state and an intermediate of some finite lifetime.
Figure 4. Retaining and inverting enzymes are entirely orthogonal.
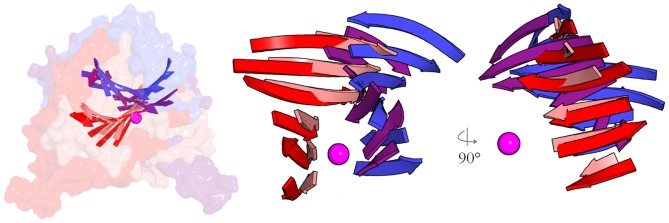
Theβ-sheets of the metal-nucleotide-sugar binding GT-A foldsof glycosyltransferase structures are superimposed by centering on the metal ion (magenta sphere) and the coordinated phosphates reveal that the general architecture of entire inverting or retaining enzymes are skewed by ∼90°. Color coding: purple, inverting GlcAT-I; blue, inverting GalT1; red, retaining GTA; pink, retaining LgtC. Left panel shows the superimposed solvent-accessible surfaces of the four structures with the folds embedded; the right panels isolate the β-sheets and show orthogonal perspectives.
Analysis of other Published Data
NMR analysis of donor hydrolysis facilitated by retaining glycosyltransferase enzymes in the absence of acceptor indicates that the cleaved monosaccharides are attacked by water from a retained position [59], [60]. This is inconsistent with a dissociative mechanism, as the steric constraints imparted by the enzyme’s fully liganded closed position would not be at play, with solvent molecules occupying both equatorial and axial positions.
Glycosyltransferases are bi-substrate enzymes and some mechanistic features can be inferred from the overall kinetic schemes observed. Double displacement should follow ping-pong kinetics as it develops a covalent intermediate, which can be identified on a Lineweaver-Burke plot as parallel lines at varied donor substrate concentrations; as is seen for trans-sialidase, for which such a mechanism has good precedent [61]. This is not observed for retaining glycosyltransferases such as MshA, assingle values for the acceptor KM have been reported even when detailed bisubstrate Michaelis–Menten kinetic data has been collected [62].This kinetic evidence clearly does not support a 2-step mechanism with a covalent intermediate for this Leloir retaining glycosyltransferase.
The double displacement mechanism has strong precedent for enzymes that do not use metallic co-factors (reviewed in [34]) such as glycoside hydrolases, in which covalent glycosyl-enzyme intermediates have been trapped in crystal structures by using fluoridated substrates (e.g. [63], [64]). Such a species should be easier to trap for a glycosyltransferase as the strong donor leaving groups would leave a covalent sugar-enzyme intermediate in an energy well with the second attack being the rate limiting step [39], and there have been intensive attempts to trap such an intermediate. The only reports of enzyme-glycosyl intermediates have come from two independent ESI-MS studies, which identified apparent covalent intermediates using postulate nucleophile mutants [25], [26].One case showed the covalent species substituted remotely from the acceptor and produced at a rate much slower than enzymatic turnover, an observation of limited relevance to the catalytic mechanism. The other case showed the enzyme-glycosyl intermediate bound to the mutated cysteine; however, such an intermediate has not been observed by means other than MS.It has been suggested that these species could be the results of charged carbocation monosaccharides introduced in the gas phase by the electrospray conditions that undergo reaction with enzyme nucleophiles to produce such glycosylated species (reviewed in [65]). Kinetic isotope effect data [36] are also strong evidence against a stable covalent intermediate.
While double-displacement should follow ping-pong bi-substrate kinetics, SNi and associative mechanisms should follow either random associative or Theorell-Chance mechanisms. The latter is followed by a retaining galactosyl transferase [66]. The distinction between these is whether or not the ternary species builds to an extent that is kinetically significant. A developed SNi intermediate must avoid water attack, so a well-structured ternary complex formed in a random associative scheme is a reasonable possibility. There is no need for a long-lived ternary complex in an orthogonal mechanism. Thus the observation of Theorell-Chance kinetics is consistent with, but does not compel an orthogonal mechanism for the group transfer transition state.
Mechanistic Proposal
The foregoing establishes that there is little direct evidence that Leloir retaining glycosyltransferases utilize a double-displacement or a fully developed SN1 mechanism ( Fig. 1A or E). The focus therefore shifts to the dissociative pathway SNi and the orthogonal pathway ( Fig. 1D or C). The distinction between an SNiand an orthogonal mechanism is found in the reaction profile and the timing of bond formation and bond breakage: if nucleophilic attack precedes or is concurrent with leaving group dissociation [59] with no enzymatic cage required to stabilize an oxocarbenium intermediate then there can be little dissociative character to the mechanism. The physical organic literature describes an associative mechanism as ANDN indicating a single transition state with association of the nucleophile fully concurrent with departure of the leaving group. An alternative in which dissociation is slightly ahead of association would be DNAN but in this case as well there is a single transition state without an intermediate. TheSNi pathway must involve an intermediate in a two-step process. It is described as DN*ANSS or DN ‡*ANSS [66]with the notations denoting differing depths of the energetic well occupied by the intermediate of the two-step process.
The available structural and kinetic data presented above are most consistent with an orthogonal mechanism of the DNAN type. The assumed geometric and energetic consequences of the various relevant transition states and intermediates are sketched in Figure 5 to visually highlight the distinctions. The geometrical changes of the alternative mechanisms are illustrated in the More O’Ferrall-Jencks diagram ( Fig. 5 left). The geometric consequences of the orthogonal mechanism are that the bond-making and bond-breaking phases are more closely coordinated than in the SNi trajectory. The energetic consequences are given in Fig.5 right with the curves offset for clarity. The orthogonal mechanism involves a single barrier without intermediate, while the SNi and the more dissociative SN1reaction profiles involve and intermediate with two transition states. The geometric location of the transition states is indicated in Fig.5 left with asterisks. The proposed orthogonal transition state likely lies close in energy to the transition state leading to a SNi intermediate. The key issue is that these pathways differ solely in the number of barriers and intermediates invoked. The DNAN process we favour is in fact identical in energetic profile with the one determined computationally [38]. These authors described their trajectory as “SNi-like”. We disagree with this description as the trajectory does not involve an intermediate, so cannot be SNi by definition [66]; our use of “orthogonal” makes this distinction clearer.
Figure 5. Geometries and energetics of mechanisms.

(left) More O'Ferrall-Jencks plot illustrating the concurrent reaction coordinate geometry changes of proposed mechanisms. (right) Comparative reaction profile diagrams for dissociative (SN1, SNi) and orthogonal pathways. The relative energetics and rate-limiting transition state locations of the three pathways are speculative and are offset for clarity, but both SN1 and SNi displacement would certainly involve an intermediate in anenergy well whereas the orthogonal mechanism does not.
Conclusions
The foregoing structural and kinetic analyses are most consistent with an orthogonal pathway for glycosyltransferases that retain anomeric stereochemistry. From the structural perspective, retaining and inverting enzymes are observed to bind and to act upon distinct conformers of the metal nucleotide sugar complex. The donor substrate trajectory architecture observed for retaining enzymes is conserved so as to present the transferring monosaccharide anomeric electrophile from an orthogonal orientation. The distances observed between the approaching nucleophile and C1 are too close to support full development of dissociation in the structures of both LgtC and GTA.
A double displacement mechanism requires an appropriately positioned and structurally conserved nucleophile in the active site. The active sites of many retaining enzymes do not contain well-positioned candidate nucleophiles, and those that have been proposed are often not sequentially or spatially conserved. In many cases, alanine mutagenesis of the proposed nucleophiles does not always abolish enzyme activity [26], [36].
Structural and kinetic evidence lies in favor of a single step orthogonal displacement. The substitution is positioned to initiate with nucleophilic attack and proceed through a trigonal bipyramidal transition state with incoming acceptor Nu axial and concurrently transferring a proton to an equatorial leaving β phosphate O3. This makes C1 the focal point for a pseudorotation that pivots O3 towards axial and Nu towards equatorial for retention ( Fig. 1C ). This mechanism provides the shortest physical route to glycosyltransfer ( Fig. 5 ), avoiding energy wells and intermediates that have been elusive to detection. An orthogonal process avoids generation of even a short-lived oxocarbenium intermediate; it is therefore the simplest of the alternatives and the only one consistent with all available evidence.
Funding Statement
This work was supported by funding from the Canadian Institutes of Health Research (www.cihr.gc.ca) MOP-77655 to SVE, who is a Michael Smith Foundation for Health Research (msfhr.org) Senior Scholar. Ongoing support to TMF was from the Natural Science and Engineering Research Council of Canada (www.nserc-csnrg.gc.ca). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Pastores GM, Elstein D, Hrebicek M, Zimran A (2007) Effect of miglustat on bone disease in adults with type 1 Gaucher disease: a pooled analysis of three multinational, open-label studies. Clin Ther 29: 1645–1654. [DOI] [PubMed] [Google Scholar]
- 2. Yoshida A, Kobayashi K, Manya H, Taniguchi K, Kano H, et al. (2001) Muscular dystrophy and neuronal migration disorder caused by mutations in a glycosyltransferase, POMGnT1. Dev Cell 1: 717–724. [DOI] [PubMed] [Google Scholar]
- 3. Wennekes T, van den Berg RJ, Donker W, van der Marel GA, Strijland A, et al. (2007) Development of adamantan-1-yl-methoxy-functionalized 1-deoxynojirimycin derivatives as selective inhibitors of glucosylceramide metabolism in man. J Org Chem 72: 1088–1097. [DOI] [PubMed] [Google Scholar]
- 4. Ma B, Simala-Grant JL, Taylor DE (2006) Fucosylation in prokaryotes and eukaryotes. Glycobiology 16: 158R–184R. [DOI] [PubMed] [Google Scholar]
- 5. Umesiri FE, Sanki AK, Boucau J, Ronning DR, Sucheck SJ (2010) Recent advances toward the inhibition of mAG and LAM synthesis in Mycobacterium tuberculosis. Med Res Rev 30: 290–326. [DOI] [PubMed] [Google Scholar]
- 6. Mas E, Pasqualini E, Caillol N, El Battari A, Crotte C, et al. (1998) Fucosyltransferase activities in human pancreatic tissue: comparative study between cancer tissues and established tumoral cell lines. Glycobiology 8: 605–613. [DOI] [PubMed] [Google Scholar]
- 7. Mulichak AM, Losey HC, Walsh CT, Garavito RM (2001) Structure of the UDP-Glucosyltransferase GtfB that modifies the heptapeptide aglycone in the biosynthesis of vancomycin group antibiotics. Structure 9: 547–557. [DOI] [PubMed] [Google Scholar]
- 8. Raetz CRH, Whitfield C (2002) Lipopolysaccharide endotoxins. Annual Review of Biochemistry 71: 635–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Harrington PR, Lindesmith L, Yount B, Moe CL, Baric RS (2002) Binding of Norwalk virus-like particles to ABH histo-blood group antigens is blocked by antisera from infected human volunteers or experimentally vaccinated mice. J Virol 76: 12335–12343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wong SM, St Michael F, Cox A, Ram S, Akerley BJ (2011) ArcA-Regulated Glycosyltransferase Lic2B Promotes Complement Evasion and Pathogenesis of Nontypeable Haemophilus influenzae. Infect Immun 79: 1971–1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Freiberger F, Claus H, Günzel A, Oltmann-Norden I, Vionnet J, et al. (2007) Biochemical characterization of a Neisseria meningitidis polysialyltransferase reveals novel functional motifs in bacterial sialyltransferases. Molecular Microbiology 65: 1258–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Byrne GW, Stalboerger PG, Du Z, Davis TR, McGregor CG (2011) Identification of new carbohydrate and membrane protein antigens in cardiac xenotransplantation. Transplantation 91: 287–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Weil R, 3rd, Nozawa M, Chernack W, McIntosh R, Reemtsma K (1973) Effect of Concanavalin A on rat heart allografts. Surg Forum 24: 194–196. [PubMed] [Google Scholar]
- 14. Hakomori S (1996) Tumor malignancy defined by aberrant glycosylation and sphingo(glyco)lipid metabolism. Cancer Res 56: 5309–5318. [PubMed] [Google Scholar]
- 15. Gschaidmeier H, Seidel A, Burchell B, Bock KW (1995) Formation of mono- and diglucuronides and other glycosides of benzo(a)pyrene-3,6-quinol by V79 cell-expressed human phenol UDP-glucuronosyltransferases of the UGT1 gene complex. Biochem Pharmacol 49: 1601–1606. [DOI] [PubMed] [Google Scholar]
- 16. Yamamoto H, Kaneko Y, Rebbaa A, Bremer EG, Moskal JR (1997) alpha 2,6-sialyltransferase gene transfection into a human glioma cell line (U373 MG) results in decreased invasivity. Journal of Neurochemistry 68: 2566–2576. [DOI] [PubMed] [Google Scholar]
- 17. Werther JL, Rivera-MacMurray S, Bruckner H, Tatematsu M, Itzkowitz SH (1994) Mucin-associated sialosyl-Tn antigen expression in gastric cancer correlates with an adverse outcome. Br J Cancer 69: 613–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ravindranath MH, Tsuchida T, Morton DL, Irie RF (1991) Ganglioside Gm3-Gd3 Ratio as an Index for the Management of Melanoma. Cancer 67: 3029–3035. [DOI] [PubMed] [Google Scholar]
- 19. Mathieu S, Gerolami R, Luis J, Carmona S, Kol O, et al. (2007) Introducing alpha(1,2)-linked fucose into hepatocarcinoma cells inhibits vasculogenesis and tumor growth. International Journal of Cancer 121: 1680–1689. [DOI] [PubMed] [Google Scholar]
- 20. Marionneau S, Le Moullac-Vaidye B, Le Pendu J (2002) Expression of histo-blood group A antigen increases resistance to apoptosis and facilitates escape from immune control of rat colon carcinoma cells. Glycobiology 12: 851–856. [DOI] [PubMed] [Google Scholar]
- 21. Itzkowitz SH, Dahiya R, Byrd JC, Kim YS (1990) Blood group antigen synthesis and degradation in normal and cancerous colonic tissues. Gastroenterology 99: 431–442. [DOI] [PubMed] [Google Scholar]
- 22. Buzzi S, Buzzi L (1974) Cancer immunity after treatment of Ehrlich tumor with diphtheria toxin. Cancer Res 34: 3481–3486. [PubMed] [Google Scholar]
- 23. Kapitonov D, Yu RK (1999) Conserved domains of glycosyltransferases. Glycobiology 9: 961–978. [DOI] [PubMed] [Google Scholar]
- 24. Chelsky D, Parsons SM (1975) Stereochemical course of the adenosine triphosphate phosphoribosyltransferase reaction in histidine biosynthesis. J Biol Chem 250: 5669–5673. [PubMed] [Google Scholar]
- 25. Soya N, Fang Y, Palcic MM, Klassen J (2011) Trapping and characterization of covalent intermediates of mutant retaining glycosyltransferases. Glycobiology 21: 547–552. [DOI] [PubMed] [Google Scholar]
- 26. Lairson LL, Chiu CPC, Ly HD, He SM, Wakarchuk WW, et al. (2004) Intermediate trapping on a mutant retaining alpha-galactosyltransferase identifies an unexpected aspartate residue. Journal of Biological Chemistry 279: 28339–28344. [DOI] [PubMed] [Google Scholar]
- 27. Martinez-Fleites C, Proctor M, Roberts S, Bolam DN, Gilbert HJ, et al. (2006) Insights into the synthesis of lipopolysaccharide and antibiotics through the structures of two retaining glycosyltransferases from family GT4. Chemistry & Biology 13: 1143–1152. [DOI] [PubMed] [Google Scholar]
- 28. Gibson RP, Turkenburg JP, Charnock SJ, Lloyd R, Davies GJ (2002) Insights into trehalose synthesis provided by the structure of the retaining glucosyltransferase OtsA. Chem Biol 9: 1337–1346. [DOI] [PubMed] [Google Scholar]
- 29. Pedersen LC, Dong J, Taniguchi F, Kitagawa H, Krahn JM, et al. (2003) Crystal structure of an alpha 1,4-N-acetylhexosaminyltransferase (EXTL2), a member of the exostosin gene family involved in heparan sulfate biosynthesis. Journal of Biological Chemistry 278: 14420–14428. [DOI] [PubMed] [Google Scholar]
- 30. Lobsanov YD, Romero PA, Sleno B, Yu BM, Yip P, et al. (2004) Structure of Kre2p/Mnt1p - A yeast alpha 1,2-mannosyltransferase involved in mannoprotein biosynthesis. Journal of Biological Chemistry 279: 17921–17931. [DOI] [PubMed] [Google Scholar]
- 31. Sommer N, Depping R, Piotrowski M, Ruger W (2004) Bacteriophage T4 alpha-glucosyltransferase: a novel interaction with gp45 and aspects of the catalytic mechanism. Biochemical and Biophysical Research Communications 323: 809–815. [DOI] [PubMed] [Google Scholar]
- 32. Reinert DJ, Jank T, Aktories K, Schulz GE (2005) Structural basis for the function of Clostridium difficile toxin B. Journal of Molecular Biology. 351: 973–981. [DOI] [PubMed] [Google Scholar]
- 33. Flint J, Taylor E, Yang M, Bolam DN, Tailford LE, et al. (2005) Structural dissection and high-throughput screening of mannosylglycerate synthase. Nat Struct Mol Biol 12: 608–614. [DOI] [PubMed] [Google Scholar]
- 34. Nagano N, Noguchi T, Akiyama Y (2007) Systematic comparison of catalytic mechanisms of hydrolysis and transfer reactions classified in the EzCatDB database. Proteins: Structure, Function, and Bioinformatics 66: 147–159. [DOI] [PubMed] [Google Scholar]
- 35. Sinnott ML, Jencks WP (1980) Solvolysis of D-Glucopyranosyl Derivatives in Mixtures of Ethanol and 2,2,2-Trifluoroethanol. Journal of the American Chemical Society 102: 2026–2032. [Google Scholar]
- 36. Lee SS, Hong SY, Errey JC, Izumi A, Davies GJ, et al. (2011) Mechanistic evidence for a front-side, S(N)i-type reaction in a retaining glycosyltransferase. Nat Chem Biol 7: 631–638. [DOI] [PubMed] [Google Scholar]
- 37. Chan J, Tang A, Bennet AJ (2011) A Stepwise Solvent-Promoted SNi Reaction of α-d-Glucopyranosyl Fluoride: Mechanistic Implications for Retaining Glycosyltransferases. Journal of the American Chemical Society 134: 1212–1220. [DOI] [PubMed] [Google Scholar]
- 38. Gomez H, Polyak I, Thiel W, Lluch JM, Masgrau L (2012) Retaining Glycosyltransferase Mechanism Studied by QM/MM Methods: Lipopolysaccharyl-alpha-1,4-galactosyltransferase C Transfers alpha-Galactose via an Oxocarbenium Ion-like Transition State. Journal of the American Chemical Society 134: 4743–4752. [DOI] [PubMed] [Google Scholar]
- 39. Lairson LL, Henrissat B, Davies GJ, Withers SG (2008) Glycosyltransferases: Structures, functions, and mechanisms. Annual Review of Biochemistry 77: 521–555. [DOI] [PubMed] [Google Scholar]
- 40. Katritzky AR, Brycki BE (1990) The mechanisms of nucleophilic substitution in aliphatic compounds. Chemical Society Reviews 19: 83–105. [Google Scholar]
- 41. Bourne Y, Henrissat B (2001) Glycoside hydrolases and glycosyltransferases: families and functional modules. Curr Opin Struct Biol 11: 593–600. [DOI] [PubMed] [Google Scholar]
- 42. Coutinho PM, Deleury E, Davies GJ, Henrissat B (2003) An evolving hierarchical family classification for glycosyltransferases. J Mol Biol 328: 307–317. [DOI] [PubMed] [Google Scholar]
- 43. Tumbale P, Brew K (2009) Characterization of a metal-independent CAZy family 6 glycosyltransferase from Bacteroides ovatus. J Biol Chem 284: 25126–25134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schuman B, Fisher SZ, Kovalevsky A, Borisova SN, Palcic MM, et al. (2011) Preliminary joint neutron time-of-flight and X-ray crystallographic study of human ABO(H) blood group A glycosyltransferase. Acta Crystallographica Section F 67: 258–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Evans SV (1993) SETOR: hardware-lighted three-dimensional solid model representations of macromolecules. J Mol Graph 11: 134–138, 127–138. [DOI] [PubMed]
- 46. Alfaro JA, Zheng RB, Persson M, Letts JA, Polakowski R, et al. (2008) ABO(H) blood group A and B glycosyltransferases recognize substrate via specific conformational changes. J Biol Chem 283: 10097–10108. [DOI] [PubMed] [Google Scholar]
- 47. Ramakrishnan B, Boeggeman E, Qasba PK (2004) Effect of the Met344His mutation on the conformational dynamics of bovine beta-1,4-galactosyltransferase: crystal structure of the Met344His mutant in complex with chitobiose. Biochemistry 43: 12513–12522. [DOI] [PubMed] [Google Scholar]
- 48. Persson K, Ly HD, Dieckelmann M, Wakarchuk WW, Withers SG, et al. (2001) Crystal structure of the retaining galactosyltransferase LgtC from Neisseria meningitidis in complex with donor and acceptor sugar analogs. Nat Struct Biol 8: 166–175. [DOI] [PubMed] [Google Scholar]
- 49. Kakuda S, Shiba T, Ishiguro M, Tagawa H, Oka S, et al. (2004) Structural basis for acceptor substrate recognition of a human glucuronyltransferase, GlcAT-P, an enzyme critical in the biosynthesis of the carbohydrate epitope HNK-1. J Biol Chem 279: 22693–22703. [DOI] [PubMed] [Google Scholar]
- 50. Pedersen LC, Darden TA, Negishi M (2002) Crystal structure of beta 1,3-glucuronyltransferase I in complex with active donor substrate UDP-GlcUA. J Biol Chem 277: 21869–21873. [DOI] [PubMed] [Google Scholar]
- 51. Schuman B, Persson M, Landry RC, Polakowski R, Weadge JT, et al. (2010) Cysteine-to-serine mutants dramatically reorder the active site of human ABO(H) blood group B glycosyltransferase without affecting activity: structural insights into cooperative substrate binding. J Mol Biol 402: 399–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Felder CE, Prilusky J, Silman I, Sussman JL (2007) A server and database for dipole moments of proteins. Nucleic Acids Research 35: W512–W521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Grote RF, Hynes JT (1980) The stable states picture of chemical reactions. II. Rate constants for condensed and gas phase reaction models. The Journal of Chemical Physics 73: 2715–2732. [Google Scholar]
- 54. Gertner BJ, Wilson KR, Zichi DA, Lee S, Hynes JT (1988) Non-equilibrium solvation in SN1 and SN2 reactions in polar solvents. Faraday Discussions of the Chemical Society 85: 297–308. [Google Scholar]
- 55. Kim HJ, Hynes JT (1990) Role of solvent electronic polarization in electron-transfer processes. The Journal of Physical Chemistry 94: 2736–2740. [Google Scholar]
- 56. Adlercreutz D, Yoshimura Y, Mannerstedt K, Wakarchuk WW, Bennett EP, et al. (2012) Thiogalactopyranosides are Resistant to Hydrolysis by α-Galactosidases. ChemBioChem 13: 1673–1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Schramm VL, Shi W (2001) Atomic motion in enzymatic reaction coordinates. Current Opinion in Structural Biology 11: 657–665. [DOI] [PubMed] [Google Scholar]
- 58. Errey JC, Lee SS, Gibson RP, Martinez Fleites C, Barry CS, et al. (2010) Mechanistic insight into enzymatic glycosyl transfer with retention of configuration through analysis of glycomimetic inhibitors. Angew Chem Int Ed Engl 49: 1234–1237. [DOI] [PubMed] [Google Scholar]
- 59. André I, Tvaroska I, Carver JP (2003) On the reaction pathways and determination of transition-state structures for retaining [alpha]-galactosyltransferases. Carbohydrate Research 338: 865–877. [DOI] [PubMed] [Google Scholar]
- 60. Sindhuwinata N, Munoz E, Munoz FJ, Palcic MM, Peters H, et al. (2010) Binding of an acceptor substrate analog enhances the enzymatic activity of human blood group B galactosyltransferase. Glycobiology 20: 718–723. [DOI] [PubMed] [Google Scholar]
- 61. Cheng J, Huang S, Yu H, Li Y, Lau K, et al. (2010) Trans-sialidase activity of Photobacterium damsela alpha2,6-sialyltransferase and its application in the synthesis of sialosides. Glycobiology 20: 260–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Vetting MW, Frantom PA, Blanchard JS (2008) Structural and enzymatic analysis of MshA from Corynebacterium glutamicum: substrate-assisted catalysis. J Biol Chem 283: 15834–15844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Howard S, He S, Withers SG (1998) Identification of the active site nucleophile in jack bean alpha-mannosidase using 5-fluoro-beta-L-gulosyl fluoride. J Biol Chem 273: 2067–2072. [DOI] [PubMed] [Google Scholar]
- 64. Numao S, Kuntz DA, Withers SG, Rose DR (2003) Insights into the mechanism of Drosophila melanogaster Golgi alpha-mannosidase II through the structural analysis of covalent reaction intermediates. Journal of Biological Chemistry 278: 48074–48083. [DOI] [PubMed] [Google Scholar]
- 65. Di Marco VB, Bombi GG (2006) Electrospray mass spectrometry (ESI-MS) in the study of metal-ligand solution equilibria. Mass Spectrom Rev 25: 347–379. [DOI] [PubMed] [Google Scholar]
- 66. Guthrie RD, Jencks WP (1989) IUPAC recommendations for the representation of reaction mechanisms. Accounts of Chemical Research 22: 343–349. [Google Scholar]