

Abstract
Microglia are resident immune cells of the central nervous system. Their persistent activation in neurodegenerative diseases, traditionally attributed to neuronal dysfunction, may be due to a microglial failure to modulate the release of cytotoxic mediators such as nitric oxide (NO). The persistent activation of microglia with the subsequent release of NO vis-á-vis the accumulation of redox transition metals such as copper (Cu) in neurodegenerative diseases, prompted the hypothesis that copper would alter NO signaling by changing the redox environment of the cell and that, by altering the fate of NO, microglia would adopt a different phenotype. We have used the microglial cell model, BV2, to examine the effects of Cu(I) on NO production and activation as they have been shown to be phenotypically plastic. Our results show that cell viability is not affected by Cu(I) in BV2 microglia and that it has no effect on iNOS mRNA, protein expression and nitrite release. However, when LPS is added to Cu(I)-treated medium, nitrite release is abrogated while iNOS expression is not significantly altered. This effect is Cu(I)-specific and it is not observed with other non-redox metals, suggesting that Cu(I) modulates NO reactivity. Immunofluorescence analysis shows that the M1 (inflammatory) phenotype of BV2 microglia observed in response to LPS, is shifted to an M2 (adaptive) phenotype when Cu(I) is administered in combination with LPS. This same shift is not observed when iNOS function is inhibited by 1400W. In the present study we show that Cu(I) modulates the release of NO to the media, without altering iNOS expression, and produces a phenotypic changes in BV2 microglia.
Keywords: microglia, nitric oxide, copper, macrophage phenotype, neurodegeneration
1.Introduction
Microglia are supportive cells of the central nervous system (CNS). They play a critical role for the normal development of the CNS [1], actively monitor the surrounding microenvironment [2] and, when deviations are detected, engage in responses to restore the normal milieu. Microglia represent one effecter arm of the CNS innate immunity as they are involved in pathogen recognition [3] and are the first to respond to help protect the CNS from invading pathogens and from the damaging consequences of neural trauma and disease. Microglia are morphologically plastic, and can take on one of three forms: ramified, activated and amoeboid [4]. Ramified (or resting) microglia display small cell bodies, fine highly branched processes, and low surface antigen expression. Although attached when ramified, the motility of their processes allows microglia to survey circumscribed regions [2, 5]. Activation is marked by the shortening and thickening of cellular processes and by an increased production of immune-related proteins [6, 7]. Migration of activated cells is prompted by the release of the β-integrin marker CD11a [8], and the release of ATP and ADP from injured neurons [9], and, when necessary, activated microglia also enter the cell cycle and proliferate [10]. In addition to playing an important role in innate immunity, microglia also play a role in adaptive immunity role when, subsequent to an increase in the expression of MHC class II antigens they become antigen-presenting cells [11-13].
Copper (Cu) is an essential metal and a cofactor for many enzymes. However, exposure to high levels of Cu can be toxic. Cu can induce cellular toxicity through catalysis of the formation of reactive oxygen species (ROS), particularly in its reduced form Cu(I) [14]. Cu(I) has a great affinity for thiols (SH groups) and, thus, it is an effective catalyst for the formation and degradation of S-nitrosothiols (SNOs) [15]. Within the brain, Cu is sequestered inside astroglia where it is distributed to storage sites, enzymes and organelles for normal cellular functioning. Alterations of brain Cu levels have been observed in the pathogenesis of several neurodegenerative diseases including Alzheimer's disease [16] and familial amyotrophic lateral sclerosis [17]. Signaling for the activation of inflammatory pathways (possibly through the NF-κB pathway [18]) has been shown to be regulated by SNO formation and thus Cu(I) may significantly alter NO-mediated signaling in microglia.
In this study we examined whether microglial plasticity in response to LPS is altered by Cu; and whether such alteration depends on the fate of NO. To that end, BV2 microglia were treated with LPS in the presence of Cu(I) to determine the relative expression of markers selected for their resting or activated profile characteristics. Our study shows that BV2 microglia alter their phenotype in response to LPS when Cu(I) is present, but are unaffected by the presence of Cu(I) alone. These results suggest that Cu(I) alters the microglial response to a toxin and this alteration may contribute to neurodegeneration.
2. Materials and Methods
2.1 Cell Cultures
The immortalized murine microglia cell line BV2 was kindly provided by Dr. Nancy Lee (California Pacific Research Center, San Francisco. CA). The cells were maintained in Dulbecco's modified Eagle's Medium (DMEM; ATCC, Manassas, VA) supplemented with 10% heat-inactivated fetal bovine serum (Invitrogen, Carlsbad, CA), 100U/mL penicillin, 100μg/mL streptomycin (Gibco), and 1% L-glutamine (Lonza, Walkersville, MD) and maintained at 37°C in a humidified atmosphere of 95% O2 and 5% CO2.
2.2 Cell Stimulation
Microglia cells were plated onto 24-well culture plates at a density of 100,000 cells/well/2mL DMEM. Twenty-four hrs later, culture medium was replaced and cells were stimulated with 0, 1, or 100 μM Copper(I) chloride (Sigma-Aldrich, St. Louis, MO) dissolved in de-ionized, de-oxygenated water. Eight hrs after copper stimulation, 0, 100, or 1000 ng/mL LPS (from Escherichia coli 0128:B12; Sigma, St. Louis, MO) was added to the medium. Twenty μM 1400w Dihydrochloride (Tocris Bioscience, Ellisville, MO) was added immediately before LPS challenge.
2.3 Nitrite measurement
The nitrite release to the medium was used as a measure of NO production. Nitrite determination was performed using a potassium iodide and acetic acid mixture at room temperature and analyzed with the Ionics/Sievers Nitric Oxide Analyser 280 (NOA 280). The concentration of nitrite was calculated from a standard curve generated using NaNO2 (Sigma Chemical Company, St. Louis, MO).
2.4 Immunoblotting
After each treatment period, cells were detached with Accutase (eBioscience.com). Cells were lysed in ice-cold buffer (Lysis buffer: Hepes 20 mM, NaCl 150 mM, glycerol 10%, triton × 100 1%, EGTA 1 mM, MgCl2 1.5 mM, pH 7.4 containing protease inhibitors PMSF 1 mM, Na Pyrophosphate 10 mM, NaF 50 mM, Na Orthovanadate 2 mM, Lactacystin 1 μM, AEBSF 1 mM, EDTA 0.5 mM, Bestatin 65 μM, E-64 0.7 μM, Leupeptin 0.5 μM and Aprotinin 0.15 μM). Protein concentration in cell lysates was determined by Bradford Assay (BioRad, Hercules, CA). An equal amount of protein for each sample was separated by 10% SDS-polyacrylamide gel electrophoresis (NuPAGE; Invitrogen, Carlsbad, CA) and transferred to a nitrocellulose membrane. After blocking, the membrane was incubated with polyclonal rabbit anti-mouse i-NOS antibody (1:1000; Santa Cruz Biotechnologies, Santa Cruz, CA) and subsequently incubated in goat anti-rabbit IgG HRP conjugated antibody (1:5000; Invitrogen, Carlsbad, CA). ECL reagents (EG Healthcare, Piscataway, NJ) were used as detection system according to the manufacturer's instruction. Membranes were subsequently immersed in stripping buffer (Thermo Sceintific, Rockford, IL) and, after extensive rinsing, blocked in 5% BSA in PBS. GAPDH was utilized as a loading control and blots were reprobed with GAPDH monoclonal antibody (1:5000; Millipore, Billerica, MA) overnight, followed by the secondary antibody (1:2000; Invitrogen, Carlsbad, CA) for 2 hrs. Protein was visualized with ECL. For Arginase I (1:1000; Santa Cruz Biotechnology, Santa Cruz, CA) followed by the appropriate secondary antibody, the protocol was the same as for iNOS.
2.5 Quantitative RT/PCR
Total RNA from the cell suspension was extracted with TRIzol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. Briefly, RNA was precipitated using isopropyl alcohol. After centrifugation (10000g, 10 min, 4°C), the pellet was washed in 70% (vol/vol) ethanol and re-suspended in water. RNA was measured by spectrophotometry, and 1 μg of RNA was used for first strand cDNA synthesis with the SuperScript III First Strand Synthesis System for RT/PCR (Invitrogen Life Technologies, Carlsbad, CA). Quantitative real-time PCR was performed on a 7500 Real-time PCR System (Applied Biosystems, Foster City, CA). TaqMan PCR was performed using a 20-μl final reaction volume, containing 10 μl TaqMan Universal PCR Master Mix, 1 μl 20× Assays-on-Demand Gene Expression Assay Mix, and 9 μl cDNA diluted in RNAse-free water. The assay was performed in duplicate. The sequence of temperature cycles for all reactions was 2 min at 50°C, 10 min at 95°C with 40 two-step cycles (95°C for 15 s and 60°C for 60 s). Assays-on-Demand Gene expression primers to NOS2 (iNOS), IL-1β and rRNA (used as endogen) were purchased from Applied Biosystems (Foster City, CA). The PCR signal of the target transcript was related to that of the control by relative quantification. The 2−ΔΔCT method was used to analyze the relative changes in gene expression. The housekeeping gene Actin (Applied Biosystems, Foster City, CA) was used as internal control to normalize the PCR for the amount of RNA added to the RT reaction and the target gene expression was normalized to the control.
2.6 Immunofluorescence
Microglia were plated at a density of 50,000 cells on previously poly-L-lysinated 8-well chambers (Nalgene Nunc International, Rochester, NY) in DMEM. Twenty-four hrs after treatment, the medium was aspirated and the cells were fixed in 3% paraformaldehyde (pH 7.4), rinsed in PBS, 200mM glycine in PBS and followed by an additional rinse in PBS. Cells were permeabilized with 0.3% Triton-X, in 0.1% BSA PBS, which was omitted for surface markers, and rinsed in PBS. Cells were blocked in 5% BSA (Sigma, St. Louis, MO) in PBS for 45 min. After blocking, the cells were incubated overnight at 4°C in primary antibodies against NFκB, Cox2 (Abcam, Cambridge, MA) Dectin-1, CD11b (AbD Serotec, Raleigh, SC) or Ym1 (Stemcell Technologies, Vancouver, Canada) in a 1:100 dilution in 0.1% BSA PBS. After primary antibody incubation, cells incubated in Dectin-1 were treated with 0.3% H2O2 (as per manufacturer's instructions) for 10 min after 2 rinses in PBS. All cells were rinsed extensively in PBS with 0.1% BSA and then incubated for 2 hrs at room temperature in the appropriate FITC-conjugated IgG secondary antibody (dilution 1:200). After extensive rinses, slides were incubated for 5 min in DAPI (1ng/μl; Invitrogen, Carlsbad, CA), followed by additional rinses in PBS. Slides were subsequently coverslipped using Prolong Gold Antifade reagent (Invitrogen, Carlsbad, CA). All steps starting with the secondary antibody were performed under dimmed light conditions.
2.7 NF-κB p50 Transcription Factor Assay
Nuclear proteins from cell suspensions of BV2 microglia were extracted using the Sigma Nuclear Extract Kit (Sigma St. Louis, MO) according to the manufacturer's instructions. Activation of the NFκB p50 subunit in 5 μg nuclear extract was determined using the NFκB enzyme-linked immunosorbent assay (ELISA)-based transcription factor kit (TransAm assay) according to the manufacturer's protocol.
2.8 DAF-2 DA
To measure intracellular nitrosation, BV2 microglia were plated at a density of 300,000 cells/well/2mL, and, 24 hrs later, challenged with 100 μM Cu(I), followed 8 hrs later by 1000 ng/mL LPS. Six hrs after LPS treatment the cells were loaded with DAF-2 DA (Cell Technology, Mountain View, CA) for 20 min and washed before photomicrographs were taken, as per manufacturer's instructions. Data were analyzed with Bioquant (Image Analysis Corporation, Nashville, TN) and expressed as pixel intensity. The means represent readings taken from 4 areas in 5 different replicates.
2.9 Statistical Analysis
All experiments were run in replicate and the data are presented as means±SE. Student's T-tests and ANOVAs were used to determine statistical difference among groups with a P-value <0.05 considered statistically significant.
3. Results
3.1 LPS-induced release of nitrite is abrogated by copper
To determine the dose of LPS that would result in the maximal release of nitrite, BV2 cells were exposed to a single administration of LPS in doses ranging from 0 to 1000 ng/mL. Figure 1A shows that BV2 cells respond in a dose and time dependent manner to LPS as measured by nitrite release to the medium. All doses of LPS used resulted in significant nitrite release at 24 hrs; maximal release for the doses tested was observed with 1000 ng/mL. Although detectable, levels of nitrite are unaffected by any of the LPS doses tested 3 hrs after challenge. The subsequent gradual increase of nitrite release observed at 6 and 12 hrs after treatment, spikes at 24 hrs. This time point marks the maximal release of nitrite after which its concentration in the medium declines (data not shown).
Figure 1. Effects of LPS on nitrite release in BV2 microglia.

BV2 microglia were challenged with different doses of LPS ranging from 0 to 1000 ng/mL. Nitrite content was measured in the medium which was sampled 3, 6, 12 and 24 hrs after LPS challenge (A); * Control vs. LPS 10; + Control vs. LPS 100; Control vs LPS 1000 indicate p < 0.05. The effects of two different doses of Cu(I) on nitrite release into the medium in BV2 microglia stimulated with 1000 ng/mL LPS at 3, 6, 12 and 24 hrs after LPS challenge (B); * No Copper vs. Low Copper; + No Copper vs. High Copper; # Low Copper vs. High Copper indicate p < 0.05. Data are Mean ± S.E. of duplicate cultures, representative of 2 independent experiments.
To assess the effect of copper on nitrite release, a single dose of either 1 or 100 μM Cu(I) was used as a pre-treatment 8 hrs prior to exposure to 1000 ng/mL LPS. Aliquots from the medium were sampled 3, 6, 12 and 24 hrs after LPS challenge. Exposure to either dose of Cu(I) 8 hrs before LPS stimulation had no effect on nitrite release 3 hrs after LPS exposure (Figure 1B). However, stimulated nitrite release is reduced by both 1 and 100 μM Cu (I) at 6, 12 and 24 hrs post LPS administration. Indeed, 100 μM Cu(I) almost entirely inhibits the release of nitrite. Reduced nitrite release is not attributable to cell death as FACS analysis of Fitc-conjugated propidium iodide (Invitrogen, Carlsbad, CA) revealed that at 24 hrs after LPS stimulation cells are healthy as are those in the LPS + 100 μM Cu(I) condition ( p = 0.704).
3.2 The effects of Cu(I) on nitrite release in BV2 microglia are specific
To demonstrate that the inhibition of nitrite release is specific to Cu(I) and not observed with other biological metals, the effect of 100 μM Cu(I) on LPS-induced nitrite release to the medium was compared to that of 100 μM Cu(II) (CuCl2), 100 μM zinc (ZnCl2) and 100 μM magnesium (MgCl2). All metals purchased from Sigma-Aldrich (St. Louis, MO) Figure 2 shows that nitrite release is only significantly reduced in LPS-treated cells by the presence of Cu(I).
Figure 2. Copper specifically reduces nitrite in LPS-challenged BV2 microglia.

Nitrite reduction is specific to Cu(I) and it is not observed with the non-redox active metals. BV2 microglia were challenged with 100 μM CuCl [Cu(I)], CuCl2 [Cu(II)], ZnCl2 and MgCl2 for 8 hrs prior to stimulation with 1000 ng/mL LPS. Data are Mean ± S.E. of duplicate cultures, representative of 2 independent experiments. * LPS 1000 vs. LPS 1000 + Cu(I) 100 μM indicates p < 0.05.
3.3 Cu(I)-effect on LPS-mediated nitrite release is not due to an impairment in NF-κB activity
The transcription factor NF-κB is a direct downstream mediator of LPS-induced microglial activation. Therefore, we examined the effect of Cu(I) on LPS-induced NF-κB activation and its downstream targets NOS2 and IL1B. 1000 ng/mL LPS increased NF-κB activity when measured 3 hrs after treatment (p = 0.046). However, pre-treatment with Cu(I) at either 1 or 100 μM dose had no effect on this induction (p = 0.21 and p = 0.37, respectively; Figure 3A). As expected, LPS treatment at either 10 or 1000 ng/mL, produced a significant increase in both NOS2 and IL1B mRNA, when measured 6 hrs after treatment (Figure 3 B & C). While 1 μM Cu(I) pre-treatment had little effect on NOS2 induction in microglia treated with both doses of LPS (p = 0.35 and p = 0.92 for LPS 10 and LPS 1000, respectively), 100 μM Cu(I) dramatically inhibited mRNA expression (p = 0.1 and p = 0.005 for LPS 10 and LPS 1000, respectively); expression of IL1β was also only reduced at the highest dose of Cu(I) and irrespective of LPS dose (p = 0.004 and p = 0.05 for LPS 10 and LPS 1000, respectively).
Figure 3. Inhibition of proinflammatory gene expression of iNOS and IL-1β by High dose of copper is NF-κB independent.

A: Cu(I) does not impair NF-κB activity in LPS-treated BV2 cells. BV2 microglia were challenged with 1 or 100 μM Cu(I) and 8 hrs later stimulated with 0, or 1000ng/mL LPS. NF-κB activity was measured 3 hrs later. The data shown are representative of three independent experiments. B: Three hrs after LPS treatment, total RNA was isolated and subjected to real-time RT/PCR. The data were calculated and normalized by 2-ΔΔCt Method and induction of NOS2 and IL1β mRNA was expressed as ratios over β-Actin. The data shown are representative of three independent experiments. * LPS 10 vs LPS 10 + Cu(I) 100; + LPS 1000 vs LPS 1000 + Cu(I) 100; # LPS 1000 + Cu(I) vs. LPS 1000 + Cu(I) 100 indicate p < 0.05.
3.4 LPS-induced iNOS expression is unaltered by Cu(I) pre-treatment
To determine whether iNOS and Arginase I protein levels were altered by Cu(I) treatment, expression was assessed by western blot. Figure 4B shows that both 10 and 1000 ng/mL LPS resulted in significant induction in iNOS protein expression (p = 0.017 and p = 0.025, respectively). Copper treatment, irrespective of dose, had no effect on the expression of iNOS in both the 10 and the 1000 ng/mL LPS challenge (p = 0.34, Figure 4B). Arginase I expression levels were not affected by either dose of Cu(I), confirming that the reduced nitrite release observed when LPS-stimulated BV2 microglia are exposed to either dose of Cu(I) cannot be attributed to a lack of substrate availability (p = 0.061, Figure 4C). Cu(I) is a redox active metal and it is possible that its effect on nitrite release results not from reduced iNOS function but from an alteration in the redox targets of NO. Therefore we chose to examine the production of intracellular nitrosation as a result of LPS-treatment, using the fluorescent dye DAF2-DA. Figure 4D shows that 6 hrs after LPS-treatment there is a significant increase in nitrosation products within the cell as indicated by an increase in DAF2 fluorescence (p = 0.003). Importantly, this increase is maintained in the presence of Cu(I) pre-treatment (p = 0.0001). These results indicate that iNOS induction not only results in nitrite release to the medium but also increases intracellular nitrosation within BV2 cells. Further, that Cu(I) pre-treatment, while reducing nitrite release, does not reduce intracellular nitrosation; indicating that the redox products of iNOS function may be altered by Cu(I).
Figure 4. Copper does not inhibit expression and activity of iNOS and Arginase I in LPS-treated BV2 microglia.
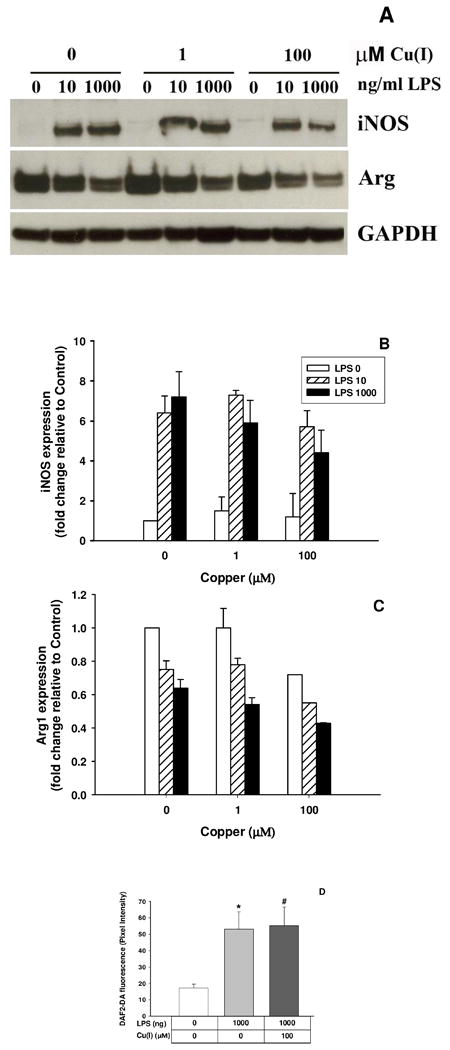
BV2 cells were challenged with 1 or 100 μM Cu(I) and 8 hrs later stimulated with 0, 10 or 1000ng/mL LPS. Cell suspensions were collected 24 hrs after LPS challenge and an equal amount of protein measured for iNOS and Arginase I with GAPDH as internal control by western blot (A). Densitometry analysis of iNOS and Arginase was normalized by GAPDH as total protein loading (B and C). The effects of Cu(I) on intracellular NO content in LPS-treated BV2 microglia are shown in D. Cells were challenged with 100 μM Cu(I) and, 8 hrs later, stimulated with 1000 ng/mL LPS. Six hrs after LPS treatment, the cells were loaded with DAF-2DA for 30 min. * represents LPS significantly different from Control, p=0.003, and # represents Control significantly different from LPS 1000 ng/mL & Cu(I) 100 μM, p=0.0001.
3.5 Cu(I) modulates phenotypic changes in BV2 microglia
In order to determine phenotypic changes in response to Cu(I) in the presence or absence of LPS, BV2 microglia were plated onto glass slides and treated with 1000 ng/mL LPS preceded by 100 μM Cu(I) according to the same conditions as described above. CD11b, Cox2 and NF-κB were selected as markers of classical activation (M1), whereas Dectin 1 and Ym-1 were selected as markers of alternative activation (M2, [19][20]). Antibodies against the above markers were visualized by FITC-conjugation, while cell nuclei were counterstained with DAPI (Figure 5). Under resting conditions (leftmost column, including photomicrographs A, E, I, M and Q), M1 and M2 markers are differentially expressed. While the modest distribution of CD11b immunoreactivity is localized around the nucleus, NF-κB and Cox2 staining is more extensive and less localized; resting BV2 cells also express both M2 markers with Dectin 1 being more localized in the periphery than Ym-1. A dose of 1000ng/mL LPS elicited a pronounced expression of M1 markers as can be seen in photomicrographs of the second column from the left (B, F, J, N and R) the expression of NF-κB is quite pronounced and occurs throughout the cell, including the nucleus as indicated by its turquoise color. A similar, albeit reduced, change is observed for Cox2 staining. In contrast, CD11b is expressed in the cytoplasm and is not stimulated by LPS; Dectin 1 expression is weak and limited to the cytoplasm, whereas Ym-1 expression is pronounced and diffuse. Photomicrographs of the third column from the left (C, G, K, O and S) depict BV2 microglia treated with 100 μM Cu(I). Expression of CD11b is modest, and although the staining of the cytoplasm covers an extensive area, the intensity is moderate and similar to that of control; NF-κB staining is also similar to that of control, while Cox 2 expression remains weak. With respect to M2 markers, Dectin 1 immunoreactivity is intense and primarily localized in the cytoplasm, while Ym-1 staining is weak and diffuse. The pattern of staining in BV2 treated with both LPS and Cu(I), seen in the photomicrographs of the right-most column (D, H, L, P, and T) show that CD11b staining is localized to the cytoplasm and parts of the nucleus, whereas immunostaining for NF-κB and Cox2 is intense and diffuse and includes the nucleus, cytoplasm and processes. M2 markers are differentially expressed, with Dectin 1 staining almost identical to that of control while Ym-1 staining is pronounced and observed both in the nucleus and in the cytoplasm. These expression patterns appear to indicate that while LPS induces a pronounced M1 phenotype, the addition of Cu(I) alters this phenotype such that it appears more M2 like.
Figure 5. Effects of copper on expression of phenotypic markers in LPS-treated BV2 microglia.
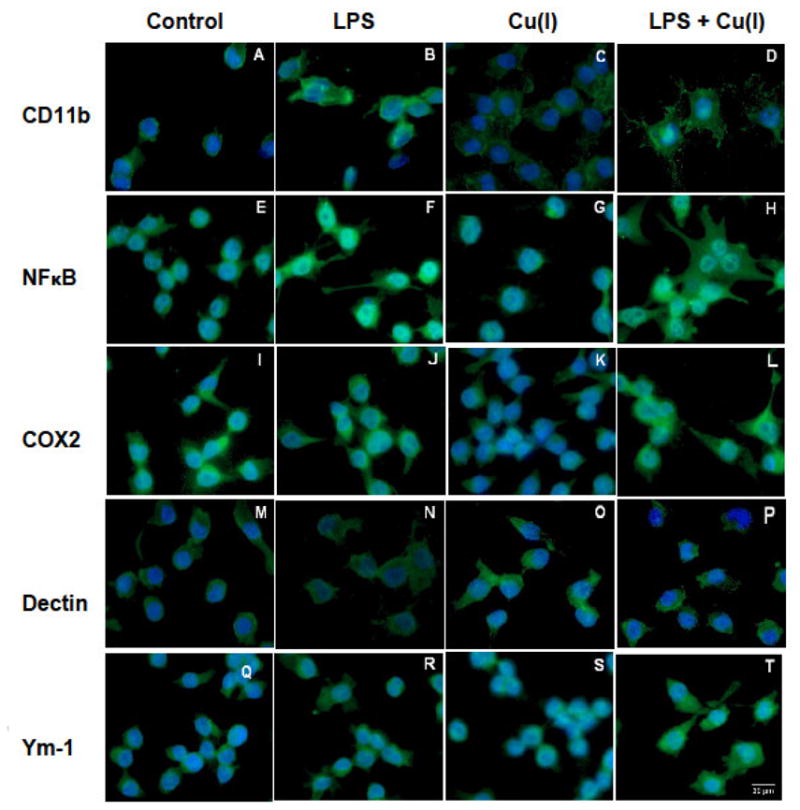
BV2 microglia were cultured on slides and challenged with 100 μM Cu(I) for 8 hrs prior to stimulation with 1000 ng/mL LPS. Twenty-four hrs after LPS stimulation, the cells were fixed and stained with the M1 markers CD11b (photomicrographs A, B, C, D), NF-κB (photomicrographs E, F, G, H) or Cox2 (photomicrographs I, J, K, L); and the M2 markers Dectin 1(M, N, O, P) or YM-1 (photopmicrographs Q, R, S, T) antibodies using FITC-linked secondary antibodies and DAPI. Experimental conditions are noted in the appropriate columns. Fluorescent photomicrographs show morphological changes (magnification for all photomicrographs 400x).
3.6 LPS-treated BV2 microglia express different morphologies in response to Cu(I) and 1400w
The lack of nitrite release upon LPS treatment in the presence of Cu(I) suggests that the change in phenotype observed may result from a lack of NO production. Therefore, we compared the effects of Cu(I) pretreatment to iNOS inhibition using 1400W. Figure 6 shows that CD11b is expressed similarly in the presence of Cu(I) and 1400w (photomicrographs B and C, respectively) indicating that neither condition differentially affects the activation of LPS-stimulated microglia. However, the M1 marker Cox2 is differentially expressed with a diffuse staining observed particularly in the cytosolic aspect of the Cu(I) condition while weak staining is observed in the presence of 1400w (photomicrographs E and F). Expression of Ym-1 reveals that Cu(I) treatment resulted in an enhanced and diffuse staining that comprises both the nuclear and the cytosolic aspect, whereas the 1400w treatment resulted in a staining that was comparable to that of the LPS only condition. These results demonstrate that the alteration in phenotype observed with Cu(I) is not merely a result of reduced NO synthesis.
Figure 6. Effects of iNOS inhibitor, 1400W, on expression of phenotypic markers in LPS-treated BV2 microglia.
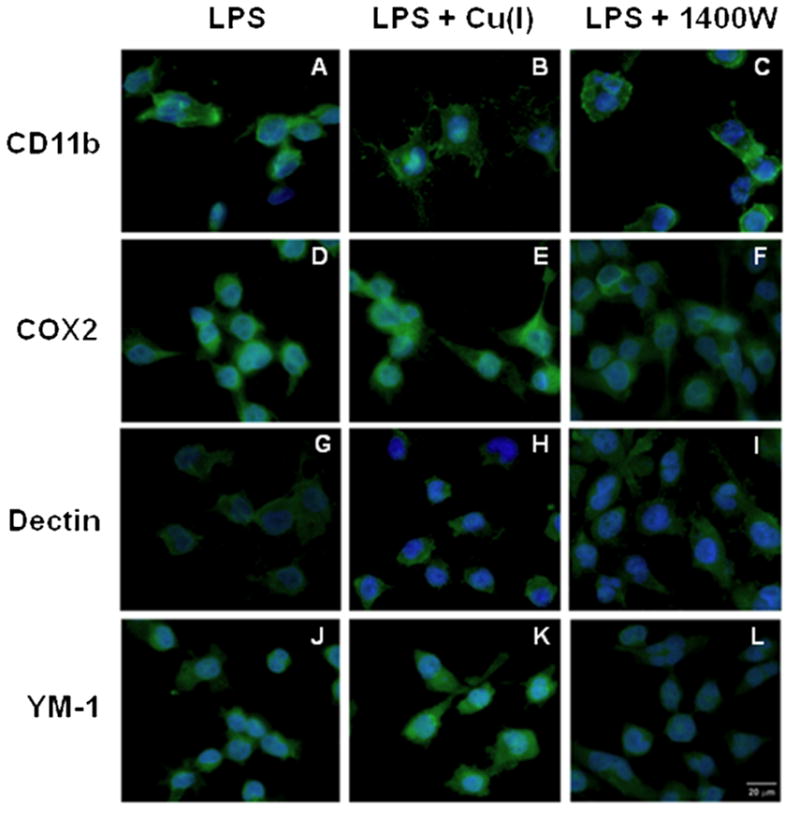
BV2 microglia were cultured on slides and exposed to 100 μM Cu(I) or 20 μM 1400w dihydrochoride followed by 100 ng/mL LPS 8 hrs later. Twenty-four hrs after LPS stimulation, the cells were fixed and stained with CD11b (photomicrographs A, B, C), Cox2 (photomicrographs D, E, F), Dectin 1 (photomicrographs G, H, I), or YM-1 (photomicrographs L, M, N) antibodies using FITC-linked secondary antibodies and DAPI. Experimental conditions are noted in the appropriate columns. Fluorescent photomicrographs show morphological changes (magnification for all micrographs 400x)
4. Discussion
In the present study we show that LPS-dependent changes in the microglial cell BV2 phenotype depend on NO being released from the cell and may be altered by the presence of redox active copper. While Cu(I) alone has no effect on either nitrite release (Figure 1) or iNOS expression in BV2 cells (Figure 3 & 4) it profoundly reduces nitrite release upon LPS-treatment (Figure 1). NF-κB activity, which is a direct downstream mediator of LPS effects, is not altered by Cu(I) treatment (Figure 3). As a result the expression of iNOS, at both the message and protein level, appears to be only marginally affected (Figure 3B and 4B). Furthermore, iNOS appears to be still functional as there is significant intracellular nitrosation in the presence of Cu(I) (Figure 4D). Therefore, it appears that the presence of Cu(I) does not inhibit the ability of BV2 cells to respond to pathogen. Rather, it appears that Cu(I) alters the redox species of NO generated (it is important to note that non-redox active metals do not produce this response, Figure 2). That such an alteration has the potential to change cellular signaling is confirmed by the observation that LPS treatment in the presence of Cu(I) generates a novel BV2 cell phenotype as suggested by the expression of defined markers (Figure 5 & 6). Microglial cells hold multiple functions in the CNS: not only do they constantly survey their local microenvironment, but respond to any disturbance by orchestrating a series of activities to restore homeostasis [7]. Microglia are considered the resident macrophages of the CNS [21], with whom they share many characteristics including modulation of their density, morphology and cell surface receptors. Peripheral macrophages can adopt either an acute inflammatory phenotype, often referred to as ‘classically activated’ or ‘M1’, or an adaptive phenotype that is anti-inflammatory and one that mediates the humoral immune response, often referred to as ‘alternatively activated’ or ‘M2’ [22]. Because of close parallels between macrophages and microglia, this classification and nomenclature has been extended to microglia. The classically activated macrophage is an effector cell in TH1 cellular immune responses [23] that is characterized by the obligatory production of NO [24]. In contrast, the alternatively activated macrophage is involved in immunosuppression and tissue repair, it does not produce NO and expresses higher levels of arginase [25]. An M2 macrophage fails to produce nitrogen radicals [26] and is generally poor at killing intracellular pathogens [27]. NO is an important signaling molecule that controls a wide range of pathways and biological processes. It is produced on demand, has a theoretical diffusion distance of up to 200 μm [28], affects tissue volume [29], and has been proposed to have the ability to mediate multiple post-translational modifications of protein including binding to metal centers, nitrosylation or oxidation of thiols, and nitration of aromatic residues [30]. NO plays a dual role of regulating cellular function and killing pathogens and is a key factor in the response of microglial cells to danger signals, such as toxins and pro-inflammatory cytokines.
Copper levels are elevated in a variety of neurodegenerative diseases where microglia are in a state of constant activation, and Cu(I) is an effective catalyst for the formation and degradation of S-nitrosothiols (SNOs). However, the possibility that Cu(I) may alter the cell signaling function of NO has not been previously considered. In this study we have examined the possibility that Cu(I) by altering NO redox state alters microglial phenotype in response to activation. Nitric Oxide is a potential mediator of the M1 phenotype, and its absence has been suggested to be a factor in the adoption of the M2 phenotype. To this end we have examined the effects of Cu(I) on BV2 polarization towards M1 or M2 phenotype using a battery of previously reported markers. The markers selected for the M1 profile were Cox2, NF-κB and CD11b. Cox 2, which is expressed under physiological conditions [31], is detectible at basal levels in untreated BV2 cell and it increases in response to LPS [32]. Neutralization of Cox 2 in LPS-treated BV2 cells results in the suppression of proinflammatory mediators including IL1β, TNF-α and NOS2 mRNA levels as well as NF-κB protein [33]. NF-κB is a transcription factor that plays an important role in cellular processes, including inflammation [34]. CD11b is expressed in activated microglia when they become motile, and it is traditionally accepted as a reliable marker of microglial activation [35]. Dectin 1 is expressed on the surface of murine primary microglia and is induced by fungal infection; however, unlike for macrophages, β-glucan mediated microglial activation does not result in significant production of cytokines. Dectin-1 also mediates phagocytosis of β-glucan particles and subsequent intracellular production of reactive oxygen species [36]. Ym-1, which is a marker for alternatively activated macrophages [20], is a secretory lectin that may control leukocyte trafficking by competing with leukocytes for binding sites on the local extracellular matrix with the end result of down-regulating inflammation [37]. Ym-1 is produced in microglia under both resting and activated conditions [38].
Our results suggest that when BV2 microglia are exposed to LPS, they acquire an M1 profile, in which Cox2 and NF-κB expression is pronounced and diffuse, and that of CD11b is more localized (Figure 5)., While it is understood that fluorescence images are purely qualitative in nature, these findings are consistent with thoseof NOS2 and IL1B mRNA in which LPS elicited an increase of their expression (Figure 3B and 3C). As expected, the expression of the M2 markers under these conditions is comparable to that of untreated BV2 cells (Figure 5, panels N, M & R, Q). In contrast, Cu(I) appears to have a minimal effect on the expression of M1 and M2 markers. In the presence of Cu(I), LPS-stimulated cells still display increased expression of M1 markers relative to control, but this expression appears reduced compared to LPS alone (Figure 5, panels B, D; F, H; & J, L); worth of note is the robust expression of the M2 marker Ym-1, which is considerably more pronounced than that of either the control or the Cu(I) only condition. Therefore, it appears that exposure to LPS in the presence of Cu(I) leads to the development of a ‘mixed’ phenotype with characteristics reminiscent of both M1 and M2 (Figures 5 & 6). The intracellular signaling mechanisms involved in the development of this ‘mixed’ phenotype are unclear. However, we propose that this alteration in phenotype is dependent upon changes in the redox reactions of NO. Cu(I), potentially through interaction with thiol residues, alters the redox species of NO generated by NOS. This is evidenced by the presence of intracellular nitrosation (Figure 4) in the absence of nitrite release (Figure 1); and that these effects cannot be reproduced by non-redox active metals (Figure 2). Furthermore, the effects of Cu(I) on LPS-induced phenotypic change are clearly different from NOS inhibition, where 1400W appears to reduce activation as a whole rather than change its endpoint.
At present, it is unclear what the functional effect of an altered NO redox state by Cu(I) is in BV2cells. It is also unknown whether this ‘mixed’ morphology and phenotype are transient or long-lived, and, most importantly, what sequelae they could have on brain disease. It is important to keep in mind that BV2 cells derive from an immortalized cell line whose rate of proliferation differs from that of primary microglia, therefore, one should be cautious in generalizing these findings to primary microglia for which phenotypic plasticity might be different. Whether this altered morphology can be extended to primary microglia is being investigated, as is the role of NO in the mechanism that underlies such changes.
In conclusion, microglia are capable of producing and releasing cytokines, chemokines and radicals that can be either beneficial or detrimental to their microenvironment depending on the magnitude and duration of their release. Therefore, understanding the conditions that elicit phenotypic changes may be beneficial both in the development of therapeutic approaches whereby microglial phenotype could be manipulated and in understanding the functional role of microglia.
-
-
Cu(I) has minimal morphologic and phenotypic effects on unstimulated BV2 microglia
-
-
Cu(I) inhibits nitrite release in LPS-stimulated microglia
-
-
Cu(I)-induced NO2 inhibition is not due to altered protein or RNA NOS expression
-
-
Cu(I) affects the morphology of LPS-stimulated BV2 cells
Acknowledgments
This investigation was supported by a grant from the National Institutes of Health 1K99ES018891-01 to ARG.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bessis A, et al. Microglial control of neuronal death and synaptic properties. Glia. 2007;55(3):233–8. doi: 10.1002/glia.20459. [DOI] [PubMed] [Google Scholar]
- 2.Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308(5726):1314–8. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- 3.Hanisch UK. Microglia as a source and target of cytokines. Glia. 2002;40(2):140–55. doi: 10.1002/glia.10161. [DOI] [PubMed] [Google Scholar]
- 4.Banati RB. Visualising microglial activation in vivo. Glia. 2002;40(2):206–17. doi: 10.1002/glia.10144. [DOI] [PubMed] [Google Scholar]
- 5.Davalos D, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8(6):752–8. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- 6.Gehrmann J, Matsumoto Y, Kreutzberg GW. Microglia: intrinsic immuneffector cell of the brain. Brain Res Brain Res Rev. 1995;20(3):269–87. doi: 10.1016/0165-0173(94)00015-h. [DOI] [PubMed] [Google Scholar]
- 7.Ransohoff RM, Perry VH. Microglial physiology: unique stimuli, specialized responses. Annu Rev Immunol. 2009;27:119–45. doi: 10.1146/annurev.immunol.021908.132528. [DOI] [PubMed] [Google Scholar]
- 8.Ullrich O, et al. Regulation of microglial expression of integrins by poly(ADP-ribose) polymerase-1. Nat Cell Biol. 2001;3(12):1035–42. doi: 10.1038/ncb1201-1035. [DOI] [PubMed] [Google Scholar]
- 9.Honda S, et al. Extracellular ATP or ADP induce chemotaxis of cultured microglia through Gi/o-coupled P2Y receptors. J Neurosci. 2001;21(6):1975–82. doi: 10.1523/JNEUROSCI.21-06-01975.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim SU, de Vellis J. Microglia in health and disease. J Neurosci Res. 2005;81(3):302–13. doi: 10.1002/jnr.20562. [DOI] [PubMed] [Google Scholar]
- 11.Frei K, et al. Antigen presentation and tumor cytotoxicity by interferon-gamma-treated microglial cells. Eur J Immunol. 1987;17(9):1271–8. doi: 10.1002/eji.1830170909. [DOI] [PubMed] [Google Scholar]
- 12.Gottfried-Blackmore A, et al. Acute in vivo exposure to interferon-gamma enables resident brain dendritic cells to become effective antigen presenting cells. Proc Natl Acad Sci U S A. 2009;106(49):20918–23. doi: 10.1073/pnas.0911509106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.O'Keefe GM, V, Nguyen T, Benveniste EN. Regulation and function of class II major histocompatibility complex, CD40, and B7 expression in macrophages and microglia: Implications in neurological diseases. J Neurovirol. 2002;8(6):496–512. doi: 10.1080/13550280290100941. [DOI] [PubMed] [Google Scholar]
- 14.Gaetke LM, Chow CK. Copper toxicity, oxidative stress, and antioxidant nutrients. Toxicology. 2003;189(1-2):147–63. doi: 10.1016/s0300-483x(03)00159-8. [DOI] [PubMed] [Google Scholar]
- 15.Stubauer G, Giuffre A, Sarti P. Mechanism of S-nitrosothiol formation and degradation mediated by copper ions. J Biol Chem. 1999;274(40):28128–33. doi: 10.1074/jbc.274.40.28128. [DOI] [PubMed] [Google Scholar]
- 16.Huang X, et al. Redox-active metals, oxidative stress, and Alzheimer's disease pathology. Ann N Y Acad Sci. 2004;1012:153–63. doi: 10.1196/annals.1306.012. [DOI] [PubMed] [Google Scholar]
- 17.Vonk WI, Klomp LW. Role of transition metals in the pathogenesis of amyotrophic lateral sclerosis. Biochem Soc Trans. 2008;36(Pt 6):1322–8. doi: 10.1042/BST0361322. [DOI] [PubMed] [Google Scholar]
- 18.Marshall HE, Merchant K, Stamler JS. Nitrosation and oxidation in the regulation of gene expression. FASEB J. 2000;14(13):1889–1900. doi: 10.1096/fj.00.011rev. [DOI] [PubMed] [Google Scholar]
- 19.Willment JA, et al. Dectin-1 expression and function are enhanced on alternatively activated and GM-CSF-treated macrophages and are negatively regulated by IL-10, dexamethasone, and lipopolysaccharide. J Immunol. 2003;171(9):4569–73. doi: 10.4049/jimmunol.171.9.4569. [DOI] [PubMed] [Google Scholar]
- 20.Raes G, et al. Differential expression of FIZZ1 and Ym1 in alternatively versus classically activated macrophages. J Leukoc Biol. 2002;71(4):597–602. [PubMed] [Google Scholar]
- 21.Streit WJ, Kreutzberg GW. Response of endogenous glial cells to motor neuron degeneration induced by toxic ricin. J Comp Neurol. 1988;268(2):248–63. doi: 10.1002/cne.902680209. [DOI] [PubMed] [Google Scholar]
- 22.Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3(1):23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 23.Mosser DM. The many faces of macrophage activation. J Leukoc Biol. 2003;73(2):209–12. doi: 10.1189/jlb.0602325. [DOI] [PubMed] [Google Scholar]
- 24.MacMicking J, Xie QW, Nathan C. Nitric oxide and macrophage function. Annu Rev Immunol. 1997;15:323–50. doi: 10.1146/annurev.immunol.15.1.323. [DOI] [PubMed] [Google Scholar]
- 25.Goerdt S, Orfanos CE. Other functions, other genes: alternative activation of antigen-presenting cells. Immunity. 1999;10(2):137–42. doi: 10.1016/s1074-7613(00)80014-x. [DOI] [PubMed] [Google Scholar]
- 26.Stein M, et al. Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. J Exp Med. 1992;176(1):287–92. doi: 10.1084/jem.176.1.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Modolell M, et al. Reciprocal regulation of the nitric oxide synthase/arginase balance in mouse bone marrow-derived macrophages by TH1 and TH2 cytokines. Eur J Immunol. 1995;25(4):1101–4. doi: 10.1002/eji.1830250436. [DOI] [PubMed] [Google Scholar]
- 28.Lancaster JR., Jr A tutorial on the diffusibility and reactivity of free nitric oxide. Nitric Oxide. 1997;1(1):18–30. doi: 10.1006/niox.1996.0112. [DOI] [PubMed] [Google Scholar]
- 29.Florenzano F, et al. Do ATP and NO interact in the CNS? Prog Neurobiol. 2008;84(1):40–56. doi: 10.1016/j.pneurobio.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 30.Gow AJ, et al. Biological significance of nitric oxide-mediated protein modifications. Am J Physiol Lung Cell Mol Physiol. 2004;287(2):L262–8. doi: 10.1152/ajplung.00295.2003. [DOI] [PubMed] [Google Scholar]
- 31.Herschman HR. Prostaglandin synthase 2. Biochim Biophys Acta. 1996;1299(1):125–40. doi: 10.1016/0005-2760(95)00194-8. [DOI] [PubMed] [Google Scholar]
- 32.Minghetti L, Levi G. Microglia as effector cells in brain damage and repair: focus on prostanoids and nitric oxide. Prog Neurobiol. 1998;54(1):99–125. doi: 10.1016/s0301-0082(97)00052-x. [DOI] [PubMed] [Google Scholar]
- 33.Li P, et al. Expression of cyclooxygenase-2 and microsomal prostaglandin-E synthase in amoeboid microglial cells in the developing brain and effects of cyclooxygenase-2 neutralization on BV-2 microglial cells. J Neurosci Res. 2010;88(7):1577–94. doi: 10.1002/jnr.22319. [DOI] [PubMed] [Google Scholar]
- 34.Bakkar N, Guttridge DC. NF-kappaB signaling: a tale of two pathways in skeletal myogenesis. Physiol Rev. 90(2):495–511. doi: 10.1152/physrev.00040.2009. [DOI] [PubMed] [Google Scholar]
- 35.Lynch MA. The multifaceted profile of activated microglia. Mol Neurobiol. 2009;40(2):139–56. doi: 10.1007/s12035-009-8077-9. [DOI] [PubMed] [Google Scholar]
- 36.Shah VB, et al. Beta-glucan activates microglia without inducing cytokine production in Dectin-1-dependent manner. J Immunol. 2008;180(5):2777–85. doi: 10.4049/jimmunol.180.5.2777. [DOI] [PubMed] [Google Scholar]
- 37.Chang NC, et al. A macrophage protein, Ym1, transiently expressed during inflammation is a novel mammalian lectin. J Biol Chem. 2001;276(20):17497–506. doi: 10.1074/jbc.M010417200. [DOI] [PubMed] [Google Scholar]
- 38.Ponomarev ED, et al. CNS-derived interleukin-4 is essential for the regulation of autoimmune inflammation and induces a state of alternative activation in microglial cells. J Neurosci. 2007;27(40):10714–21. doi: 10.1523/JNEUROSCI.1922-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]