

Abstract
Muscular dystrophy arises from ongoing muscle degeneration and insufficient regeneration. This imbalance leads to loss of muscle with replacement by scar or fibrosis resulting in muscle weakness and, eventually, loss of muscle function. Human muscular dystrophy is characterized by a wide range of disease severity, even when the same genetic mutation is present. This variability implies that other factors, both genetic and environmental, modify the disease outcome. There has been an ongoing effort to define the genetic and molecular bases that influence muscular dystrophy onset and progression. Modifier genes for muscle disease have been identified through candidate gene approaches as well as genomewide surveys. Multiple lines of experimental evidence have now converged on the TGFβ pathway as a modifier for muscular dystrophy. TGFβ signaling is upregulated in dystrophic muscle as a result of a destabilized plasma membrane and/or altered extracellular matrix. Given the important biological role of the TGFβ pathway, and its role beyond muscle homeostasis, we review modifier genes that alter the TGFβ pathway and approaches to modulate TGFβ activity to ameliorate muscle disease.
Keywords: muscular dystrophy, genetic modifier, extracellular matrix, TGFβ
Muscular dystrophy (MD) is genetically diverse and arises from mutations in many different single genes leading to progressive loss of muscle mass, weakness, and eventually loss of muscle function. Like most Mendelian disorders, the primary genetic defect has the most significant effect in determining the age of onset of muscle degeneration and symptoms, the muscle groups most targeted, and, most importantly, the pace at which degeneration and loss of function occur. Some aspects of MD differ with the specific primary gene mutation, but recently there has been increasing focus on understanding the genetic and molecular basis of disease variability observed with the same identical genetic mutation. Evidence for genetic modifiers of muscular dystrophy derives from both human and animal models, and this review will focus on data from the clinical arena as well as from models of MD with focus on Duchenne and Becker MD (DMD and BMD) and the mechanisms by which the TGFβ pathway has emerged as critical pathway that determines outcome in MD. The identification of genetic modifier pathways informs not only the prognosis of individual patients, but also uncovers approaches that can be used to treat disorders for which there is presently no cure.
Skeletal muscle fibers are single elongated, multinucleate cells that are arranged in parallel. Within a given muscle, myofibers tend to have a uniform fiber diameter that appears polygonal in cross section (Fig. 1). Skeletal muscle fibers are organized into fascicles, which are separated by epimysial connective tissue. Myofiber nuclei are generally peripherally located, below the sarcolemmal membrane. Skeletal muscle is dynamic and capable of regeneration after injury. In contrast to normal muscle, regenerating muscle is characterized by internally positioned nuclei and muscle fibers of varying diameter. Regeneration is central to repairing damage and restoring muscle function. However, regeneration has limits and may not be sufficient to restore fully the normal muscle architecture. In dystrophic muscle, regeneration is often outpaced by degeneration. Reflecting this, dystrophic muscle appears disorganized; in cross section, skeletal muscle fiber diameter includes both small and large fibers, and the myofiber nuclei are misplaced and centrally located within a myofiber. Disruption of the dystrophin complex by dystrophin or sarcoglycan gene mutations produces a leaky sarcolemma with markedly increased intracellular calcium. In advanced DMD muscle, fibroblasts and adipocytes replace the myofibers and the inflammatory response is exacerbated leading to fibrosis, muscle weakness, and muscle wasting (Fig. 1). The inflammatory response is seen in young DMD muscle and therefore is an early feature of disease [1]. The mdx mouse model of DMD similarly has a chronic and persistent inflammatory response [2]. In addition to cytokines, there is a marked upregulation of mRNA of macrophage products responsible for extracellular matrix synthesis and turnover in the mdx limb [2]. The TGFβ pathway was seen in these gene expression profiles as a major proinflammatory and profibrotic cytokine important for regulating DMD.
Fig. 1. Fibrosis, fatty and inflammatory infiltrate replace myofibers in advanced dystrophic muscle.
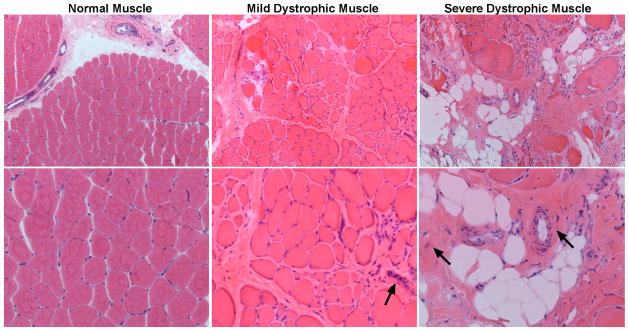
H&E staining of human skeletal muscle reveals increased fatty and inflammatory infiltrate and replacement of skeletal muscle fibers by fibrotic tissue (arrows) as Duchenne muscular dystrophy (DMD) pathogenesis progresses from a mild to an advanced state.
Modifiers in multiple forms of human MD
Dystrophin gene (DMD) mutations underlie Duchenne MD, and the degree to which dystrophin protein remains expressed accounts for the milder allelic Becker MD [3][4]. Even in the presence of premature stop codons, the primary mutation can be bypassed in some muscle fibers through a process known as exon skipping resulting in a milder phenotype [5]. However, exon skipping and the production of revertant fibers does not entirely explain disease severity, because some patients with no detectable dystrophin production may express a milder phenotype than what is predicted from their DMD mutation [6]. Many dystrophin mutations predictably produce no dystrophin, but for at least 16% of DMD mutations it is possible that small amounts of dystrophin protein can be produced [3]. The wide range of DMD mutations associated with muscular dystrophy has made it challenging to detect genetic modifiers since it is easier to identify modifiers with a more homogeneous primary genetic defect.
Limb-girdle muscular dystrophy (LGMD) type 2C is caused by mutations in the dystrophin associated protein γ-sarcoglycan encoded by the SGCG gene. Although there are a number of different SGCG mutations responsible for LGMD 2C, there are at least two more frequent SGCG mutations documented in many LGMD 2C patients. The first mutation, Δ521-T, disrupts the reading frame of SGCG resulting in the absence of protein production; this mutation has been documented in many LGMD 2C patients [7–9]. A second SGCG mutation is a G to A point mutation in codon 283 that substitutes a conserved cysteine with a tyrosine in the EGF-like motif found at the extreme carboxy-terminus in the extracellular domain of γ-sarcoglycan [10]. With these single mutations, individuals with the same identical mutation vary considerably in their age of onset of muscle weakness, age of ambulatory loss, the degree to which there is associated cardiomyopathy, and rate at which they lose respiratory muscle function [8, 9].
Other forms of muscular dystrophy are also associated with a range of phenotypic expression that cannot be explained by the primary mutation. For example, LGMD 2B and Miyoshi myopathy both arise from mutations in dysferlin, a membrane-associated protein implicated in muscle membrane repair [11, 12]. LGMD 2B has a variable age of onset but often results in significant loss of muscle mass and reduced function, including loss of ambulation. In contrast, Miyoshi myopathy causes muscle wasting only of the gastrocnemius muscle in the calf and typically does not cause loss of ambulation. The same identical dysferlin mutation has been associated with either LGMD 2B or Miyoshi myopathy [13, 14]. Similarly, LMNA mutations in the gene encoding lamin A/C, protein of the nuclear membrane produce a range of muscle dysfunction and associated cardiomyopathy as well as electrical conduction system defects affecting the heart [15]. Through a genomewide analysis of a large French family with a single LMNA mutation in 19 affected individuals, a locus on chromosome 2 was mapped that acted as a modifier [16]. Within this chromosome 2 locus is the DES gene encoding the intermediate filament protein desmin. DES mutations also cause cardiomyopathy and muscular dystrophy [17]. Cumulatively, these human genetic observations emphasize the high degree of variability in muscle disease severity seen in patients carrying an identical gene mutation and suggest the presence of modifier genes underscoring the disease outcome.
Experimental models for MD also support the presence of modifiers
Both naturally occurring mutations and engineered mutations in MD animal models have been used to demonstrate that genetic backgrounds alter the pace and tempo of disease. There is evidence for genetic modifiers of MD in multiple species including in dogs, mice, fish and flies. For example, Golden Retriever muscular dystrophy dogs (GRMD) show a great clinical variability in the severity of disease. Affected dogs have a frameshift point mutation in a splice site in intron 6 of the dystrophin gene, which results in complete absence of the dystrophin protein [18]. Despite carrying the same primary DMD mutation, dystrophic dogs show a wide range of clinical manifestations including dogs that are severely affected and die perinatally, as well as those that are less severely affected and live longer [18]. Furthermore, GRMD dogs in a colony established from a single female carrier show a varying degree of exercise capability including significant variation in mean step length, maximum jumping height and the time required to change position [18].
Mouse models of MD have been helpful in identifying genetic modifiers of disease. It has been shown that the most commonly used model of DMD, the mdx mouse exhibits an enhanced phenotype when bred into the DBA/2J background [19]. The hindlimb muscles of DBA/2-mdx showed reduced muscle weight, fewer myofibers, and elevated fat and fibrosis compared to C57Bl/10-mdx mice [19], indicating that the DBA/2J genome contributed to the severity of disease compared to the mild phenotype of the mdx mice alone. However, mice are not the only dystrophic model that has been useful for identifying strain-induced differences in phenotype. Genetic manipulations in Drosophila melanogaster have also helped identify modifier genes that interact with the dystrophin glycoprotein complex (DGC) and alter the fly wing phenotype [20]. A genetic screen was performed on three different mutant fly lines inspecting phenotypic differences in the fly wing cross-vein phenotype, a visible phenotype easy to identify. Through this genetic screen, 37 genes were identified as modifiers of wing vein phenotype [20]. The 37 modifier genes clustered into six functional groups including genes important for muscle development, neuronal/cell migration, motor function, cytoskeletal components and as well as genes involved in the TGFβ and EGFR signaling pathway [20]. These findings provide a broader understanding of the DGC and interacting proteins.
Zebrafish express many of the same DGC proteins as humans, and the MD phenotype in zebrafish has been used for genetic interaction studies and for chemical screening [21]. The sapje fish mutant harbors a dystrophin gene mutation in a splice site at the end of exon 63 [21]. This mutation results in the destabilization of the sarcoglycan complex, a common molecular finding observed in DMD patients and results in disrupted muscle birefringence, a pattern that is highly visible and useful for chemical and genetic screens.
Mapping genetic modifiers using a genomewide approach
Because the evidence from humans strongly supported the presence of genetic for LGMD 2C, we took advantage of a mouse model for this disorder, the Sgcg mouse [22]. The Sgcg mouse model was generated by deleting exon 2 of Sgcg, which encodes the cytoplasmic and transmembrane domains resulting in the absence of any detectable γ-sarcoglycan protein. This Sgcg mouse recapitulates the phenotype seen in LGMD 2C patients with progressive muscle degeneration and a shortened lifespan. Like the mdx mouse, Sgcg mice are less severely impaired than their human equivalents, since mice remain ambulatory unlike humans with these forms of MD. The Sgcg null mutation was bred through ten generations into four different genetic backgrounds where it was shown that three of these backgrounds, 129/SVEMS+/J JAX, C57BL/6J JAX, and CD41 VAF+(CD1) conferred a milder phenotype and one background, the DBA2J JAX strain, enhanced the phenotype of MD [23]. To determine MD severity, two different pathogenic traits were measured. The amount of Evans blue dye uptake by dystrophic muscle was determined as a measure of membrane leakiness, and fibrosis was quantified by the assessing the amount of hydroxyproline present in the muscle.
Using an unbiased, genomewide analysis of SNPs that differed between the mild and severe mouse strains carrying the Sgcg mutation, the latent TGFβ binding protein 4 (Ltbp4) was identified as a modifier of muscle disease [24]. Importantly, Ltbp4 genotype was linked to both traits, muscle membrane leakiness and fibrosis, two features that were previously not thought to be directly related. The idea that the same genetic polymorphism regulates both muscle membrane stability and the amount of fibrosis in muscle indicates that these two features are more intimately related than previously appreciated. The specific mutation responsible for modifying MD is an insertion/deletion polymorphism that affects a domain that alters TGFβ release and therefore TGFβ activity. These results were replicated by conducting a genomewide analysis in a larger, independent cohort of mice [25]. This study confirmed that the region on chromosome 7 containing the Ltbp4 gene correlated with enhanced disease phenotype in all the limb-based skeletal muscles studied. Additional genomic intervals were identified that influenced the severity of disease as it affects heart and trunk muscles [25]. These data point to different genetic bases for disease progression in limb-based skeletal muscle versus the trunk based muscles.
Most recently, it was shown that LTBP4 polymorphisms translate to human dystrophinopathy. The human LTBP4 gene has two major haplotypes that differ by four amino acids, and these residues are hypothesized to modify TGFβ affinity [4]. The human and mouse LTBP4 gene polymorphisms are different but elicit the same net effect of regulating TGFβ activity. Those polymorphisms that increase TGFβ signaling are associated with greater disease intensity, while those that reduce TGFβ signaling ameliorate disease. The murine Ltbp4 polymorphisms fall in the proline rich “hinge” region of LTBP4, while the human LTBP4 SNPs are distributed at several points along the LTBP4 protein but not in the hinge region (Fig 2). In humans, LTBP4 SNPs have also been linked to Chronic Obstructive Pulmonary Disease (COPD) [26], colorectal cancer [27, 28] and abdominal aortic aneurysm [27]. The association of LTBP4 SNPs with multiple human diseases reflects the tissue distribution of LTBP4 protein expression. This constellation of LTBP4 target tissues, muscle, lung, and colon, is also reflected in mice engineered with a hypomorphic allele of Ltbp4 (Ltbp4S−/ −) [29]. The Ltbp4S−/− mouse model was generated through a gene trap within the 5′ end of Ltbp4 that markedly reduced LTBP4 protein expression [29]. These mice develop cardiomyopathy, pulmonary dysfunction and fibrosis and colon cancer, and these traits reflect the high level LTBP4 protein expression in heart, lung and colon [30].
Fig. 2. LTBP4 isoforms.

LTBP4 differs at the 5′ end. Shown are common LTBP4 proteins produced from alternative promoters. The proline rich “hinge” region is shown in red. Arrows indicate common single nucleotide polymorphisms that modify disease outcome in patients with COPD and DMD.
LTBPs and Fibrillin Superfamily of Proteins are Essential ECM proteins
LTBPs belong to the fibrillin superfamily of proteins. There are three fibrillins (fibrillin 1, 2, and 3) and four LTBPs (LTBP1 through 4) in this superfamily. Features common to all family members include a large number of epidermal growth factor (EGF) repeats, many of which contain calcium-binding sequences [31] (Fig. 3). A distinguishing feature between the LTBPs and the fibrillins is the ability of LTBPs, specifically LTBP1, 3, and 4, to bind directly TGFβ. Fibrillins do not bind TGFβ directly and instead bind LTBPs, and in doing so play a critical role in the signaling capacity of the extracellular matrix (ECM). Like LTBPs, the fibrillins are also secreted to the extracellular matrix and contribute to the assembly and integrity of the ECM [31, 32]. Ltbp4S−/ − mice also contribute to ECM formation because elastin and its associated proteins cannot be integrated into the microfibril fibers [31]. These findings further confirm the intimate association between LTBPs, fibrillins, and the integrity of the ECM.
Fig. 3. Fibrillin superfamily of proteins.
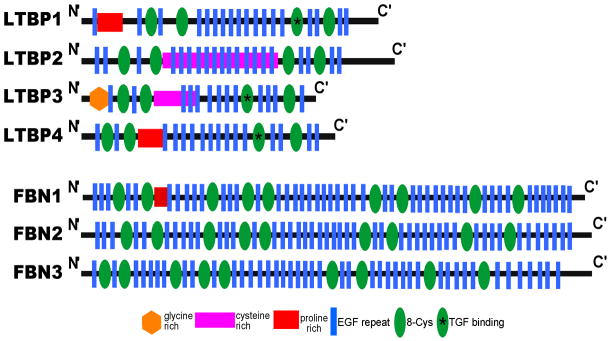
LTBPs 1 through 4, and fibrillins 1 through 3 together form the Fibrillin superfamily of proteins. LTBPs and the fibrillins share a similar protein structure and play an integral role in the stability of the extracellular matrix.
Autosomal dominant FBN1 mutations cause Marfan syndrome, an inherited connective tissue disorder [33]. Marfan syndrome is characterized by cardiovascular, musculoskeletal, and ocular defects that associate with aberrant TGFβ activity [34]. Fbn1 mutant mice with a Marfan associated “knock-in” substitution (Fbn1 C1039G/+) have elevated TGFβ signaling and elevated pSmad2/3 activity in the lung, skeletal muscle, and heart [34]. The angiotensin II receptor antagonist losartan was shown to reduce TGFβ activity and protect against aortic root aneurysm progression, aortic wall thickening and mitral valve degeneration in Fbn1C1039G/+ mice [35]. Furthermore, losartan administration protects Marfan patients against aortic root dilation [36], and TGFβ receptor antagonist and losartan administration both restore muscle architecture in Fbn1C1039G/+ mice [37].
LTBPs show tissue-enriched expression patterns [38]. LTBP1 is predominantly expressed in the heart, placenta, lung, spleen, kidney and stomach [38, 39]. LTBP2 is expressed in the lung, skeletal muscle, placenta and liver [38]. LTBP3 and LTBP4 are both expressed in the heart, skeletal muscle, small intestine and ovaries [30, 38, 40]. The expression pattern among the LTBPs suggests distinct functional roles in different tissues, and expression overlap in some tissues may provide redundancy to ensure biological function. Exogenously added LTBP1 was shown to stimulate aortic smooth muscle cell migration and thickening of arteries in diabetic rats [41] and elevated LTBP1 mRNA and protein levels have been found upregulated in atherosclerotic plaques [42]. In humans, LTBP2 mutations segregate with primary congenital glaucoma [43, 44] and have been shown to disrupt the extracellular matrix [45]. Interestingly Ltbp2 null mice die very early in development consistent with a role in embryo implantation [46]. Ltbp3 null mice have reduced alveolar formation and reduced TGFβ signaling [47], and Ltbp3 function is required for proliferation and osteogenic differentiation of human stem cells [48].
In humans, rare loss of function recessive alleles of LTBP4 result in a syndrome of impaired pulmonary function, gastrointestinal, musculoskeletal and dermal development [49]. These features are reminiscent of the features described in the Ltbp4S−/− [29]. These findings support that the developmental requirements for LTBP4 function likely relate to its importance for the activity of multiple TGFβ family members. In particular, bone morphogenetic protein 4 is increased in the absence of LTBP4, consistent with a role of LTBP4 in regulating not only TGFβ activity, but that of other TGFβ superfamily members as well [50]. The findings from loss of function allele are in contrast to the common LTBP4 polymorphisms that associate with expansion of abdominal aortic aneurysm and impaired exercise tolerance in patients with COPD [26, 27]. We found that the two common LTBP4 alleles associated with differential TGFβ signaling [4]. Given the chronic injury and inflammation that underlie DMD [2], it is fair to view DMD as a “hyper-TGFβ” state. Reduced TGFβ signaling in both humans and mouse LTBP4 modifiers is associated with decreased pathogenesis, especially fibrosis [4, 24]. The observation that a variant in Ltbp4 segregates with worse membrane damage and fibrosis in mice with LGMD [24] is consistent with this model since reduction in TGFβ was associated with the improved phenotype. Similarly, a specific allele of human LTBP4 was associated with prolonged ambulation in DMD patients [4]. These data are highly consistent that TGFβ levels are elevated in DMD and that partial reduction of this hyper-TGFβ signaling improves outcome by stabilizing the plasma membrane of muscle and reducing fibrosis (Fig. 4).
Fig. 4. TGFβsecretion and bioavailability are tightly regulated.

Secretion and activity of TGFβ are tightly regulated. In a basal state, TGFβ is bound to the LTBPs and kept inactive in the extracellular matrix. Upon muscle injury TGFβ is activated and elicits downstream SMAD signaling, to repair the injury and restore muscle function. Dystrophic muscle is characterized by elevated TGFβ activity, which exacerbates the inflammatory response and aggravates the fibrotic response.
Association of LTBPs with TGFβ
LTBPs bind to and mediate the secretion of inactive TGFβ into the ECM. TGFβ is synthesized as a latent dimerized complex unable to engage its membrane bound TGFβ receptor. The amino-terminal pro domain, known as the TGFβ latency associated protein (LAP), binds to the TGFβ dimer via non-covalent interactions and keeps this small latent TGFβ complex (SLC) inactive. The SLC is covalently linked to LTBPs, forming the large latent TGFβ complex (LLC) [51]. The association of the LAP-TGFβ with an LTBP is essential for the secretion and activation of TGFβ. LTBPs mediate not only the secretion of TGFβ into the ECM, but assure their recruitment to the ECM microfibrils. The LLC associates with the extracellular matrix fibers and keeps TGFβ inactive until its activity is needed [31, 51]. Furthermore, the association of LLC with the ECM may provide cells with a readily available TGFβ pool that is positioned to respond to injury [38]. LTBPs are synthesized in molecular excess of TGFβ, further suggesting that most secreted cellular TGFβ occurs through the LLC [52–55].
LTBPs associate with the ECM via an amino-terminal ECM-binding domain. The ECM binding domain contains transglutaminase substrate motifs, and transglutaminase is required for the covalent association of LTBPs with the ECM [56]. The amino-termini of human LTBP1 and 4 are alternatively spliced, conferring differential affinity for the ECM [30, 57]. There are two amino-terminal isoforms for LTBP1 and LTBP2 specifically [39, 57, 58] and for LTBP1 the two isoforms have been shown to utilize different promoters [57, 59]. In the case of LTBP4, there are four LTBP4 isoforms that differ at the amino-terminus (Fig. 3). The carboxy-terminus of the LTBPs has also been implicated in ECM binding, but it is less well understood [60].
The LTBPs have a highly repetitive structure, composed primarily of two distinct cysteine-rich motifs. The first motif is characterized by six cysteines, which is similar to the EGF-like domains, and the second consists of eight intramolecularly-bound cysteines, commonly referred to as the 8-cys or TB-domain for TGFβ-binding domains [31]. Each LTBP contains four TGFβ binding domains, separated by multiple EGF-like repeats. The first TB domain is commonly referred to as the “hybrid domain”, because it shares similarities with both, the TB domain and the EGF-like repeats. The second and third TB domains are separated by the proline-rich hinge region and multiple EGF-like repeats. The proline-rich hinge region is divergent among the four LTBPs [61]. LTBPs are cleaved at the “hinge” region by serine proteases in order to release TGFβ from the ECM [55, 60, 62, 63].
In order for TGFβ to be activated, cleavage of LTBPs must occur. Direct TGFβ activation has been demonstrated in vitro by proteolysis, enzymatic deglycosylation, and acid treatment [38]. The hinge domains of LTBP1 and 3 are alternatively spliced, further affecting susceptibility to cleavage [42, 64, 65]. A splice variant of LTBP1 lacks 53 amino acids in the hinge region, including a heparin-binding site, shows diminished proteolytic cleavage [64]. The third TB domain directly binds to the LAP domain of TGFβ via disulfide bonds, and this mechanism is described for TGFβ binding in LTBP1, 3 and 4 [58, 66]. How proteolytic cleavage in one region of LTBP alters TGFβ at a more distal region of the protein is not clear and requires further study.
TGFβ signaling and implications in muscular dystrophy
TGFβ superfamily members transduce their signal from the membrane to the nucleus through distinct combinations of transmembrane type I and type II serine/threonine receptors and their downstream effectors, the Smad proteins [67]. Ligand binding induces the type I and type II receptors to associate, which leads to a unidirectional phosphorylation event in which the type II receptor phosphorylates the type I receptor, thereby activating its kinase domain [68]. Phosphorylated type I TGFβ receptor recruits regulatory Smad proteins (R-Smads) via interactions with Smad anchor for receptor activation (SARA), which results in R-Smad phosphorylation. Phosphorylated R-Smads form a complex with common Smad (Co-Smad), a cytosolic protein containing a nuclear localization signal, and migrate to the nucleus to initiate gene transcription [68]. TGFβ also activates other signaling cascades, including Erk, JNK, TGFβ-activated kinase 1 (TAK1), c-Abl and MAPK pathways. TGFβ family members are multifunctional polypeptide growth factors involved in the regulation of many important biological processes such as growth, differentiation, immune response, secretion and maintenance of the extracellular matrix components and these effects are paramount during injury response and especially during fibrosis [69, 70]. TGFβ is rapidly induced upon cutaneous injury and is consistently present in wound fluid throughout the repair process [71]. TGFβ release attracts neutrophils, macrophages, and fibroblasts, which in turn releases more TGFβ. Expression of TGFβ and TGFβ receptors are elevated in fibroblasts of human post-burn hypertrophic scars, in keloids that result from an excessive wound healing response, and in keloid-derived fibroblasts [72, 73]. TGFβ induces excess matrix synthesis when injected subcutaneously in mice [74]. Moreover, wound treatment with TGFβ promotes wound closure and scarring in vivo [74]. Incisional wounds, if treated with anti-TGFβ antibodies or antisense oligonucleotides, suppress ECM synthesis and scarring [75, 76]. Consistent with these observations, TGFβ activity has been shown to aggravate muscle disease states.
In the muscular dystrophies, enhanced TGFβ has been described in human DMD [77, 78]. Increased TGFβ protein and mRNA is associated with increased canonical (SMAD) and noncanonical (non-SMAD) signaling in both human muscle and mouse models [78]. Increased TGFβ signaling is best described in DMD, but has also been described in other forms of muscular dystrophy [79]. How TGFβ signaling mediates adverse consequences on muscle pathology has not been shown. It is known that JNK and ERK pathways participate in muscular dystrophy [80, 81]. But the relationship to this signaling as a downstream consequence of TGFβ activation has not been determined. Systemic administration of neutralizing TGFβ antibody or the angiotensin II type 1 receptor blocker losartan was found to normalize muscle architecture, repair, and function in the mdx model suggesting a direct role of excessive TGFβ signaling in muscle disease [37]. Inhibiting TGFβ activity using the same neutralizing TGFβ antibody 1D11, losartan, or a combination of both therapies improved respiratory function in mdx mice [82]. TGFβ inhibition resulted in improved functional respiratory parameters including normalized Penh values, increased peak respiratory flow, and decreased inspiration time and breathing frequency. In addition, administration of both, 1D11 and losartan, improved grip strength. 1D11 treatment proved effective at improving grip strength as early as 2 months of age, compared to losartan that proved effective at 9 months of age [82]. Serum creatine kinase levels and hydroxyproline levels significantly decreased following 1D11 treatment, and diaphragm muscle fiber density increased, suggesting improved muscle function [82].
Genetic manipulation of the periostin gene, Postn, also ameliorates the MD phenotype and restores muscle function in mice lacking the δ-sarcoglycan gene (Sgcd−/−) [83]. Periostin is upregulated by TGFβ normally expressed in low amounts in adult tissue, however its expression is significantly increased in disease and during fibrogenesis [84]. Circulating levels of periostin are elevated in (Sgcd−/−) mice and immunohistochemical analysis reveals accumulation of periostin in the ECM [83]. Mice lacking both δ-sarcoglycan and the periostin gene (Sgcd−/−)Postn−/−) show improved histopathology across multiple muscle groups, with no significant change in central nucleation, suggesting that loss of periostin does not interfere with myofiber regeneration [83]. In addition, loss of periostin results in reduced serum creatine kinase levels in Sgcd−/− Postn−/− mice compared to Sgcd−/− mice, and improved exercise performance [83]. Ablation of TGFβ signaling using TGFβ blocking monoclonal antibody worsened muscle function in Sgcd−/− Postn−/− mice compared to mice receiving vehicle treatment, pointing out to the conundrum associated with TGFβ activity observed in MD models. TGFβ is tightly regulated and depending on the amount of active TGFβ that is biologically active, both beneficial and adverse effects have been reported.
Osteopontin is an extracellular matrix protein that regulates TGFβ. The deletion of the SPP gene encoding osteopontin has little phenotype in muscle. When crossed into the mdx mouse model of DMD, there was a marked reduction in fibrosis and improvement in muscle strength [85]. In addition, deletion of the SPP gene reduced neutrophils but not macrophage invasion into dystrophic muscle and reduced TGFβ mRNA levels [85]. These data identify SPP as a regulator of inflammatory response, a contributing factor to promoting disease progression in dystrophic muscle. A polymorphism within the SPP promoter was associated with prolonged ambulation in a cohort of DMD patients [86]. The “g” allele of rs28357094 altered gene promoter function and associated with reduced osteopontin mRNA in HeLa cells, and paradoxically with reduced levels of CD4 and CD68-positive cells in DMD [87]. TGFβ has been shown to activate the promoter of SPP1 gene [88] and a polymorphism in the TGFBR2 promoter correlated with osteopontin mRNA levels, further confirming interplay between osteopontin and TGFβ [87].
Genetic manipulation of TGFβ and SMAD signaling helps restore normal heart and muscle function in γ/δ-sarcoglycan null flies (Sgcd[840]) [89]. Partial reduction of the SMAD signaling using haploinsufficient alleles that reduced SMAD activity in the Sgcd[840] flies improved negative geotaxis, the ability of the flies to walk upwards against gravity, an ability that is lost in Sgcd mutant flies as well as those lacking dystrophin [89–91]. Furthermore, optical coherence tomography showed that reducing TGFβ and SMAD signaling in the Sgcd[840] flies, restored heart function to wildtype levels. Interestingly, genetic manipulation of various downstream targets of the TGFβ signaling revealed that TGFβ signaling involving the BMP has a direct role in improved skeletal muscle function but not heart tube function [89]. These findings further emphasize the intricate nature of TGFβ signaling and the crosstalk between various signaling mechanisms in MD.
Future therapeutic directions
Human patients and animal models with MD confirm the integral role of TGFβ and SMAD signaling in the progression and severity of muscle disease. Mechanisms to reduce this signaling have focused on pharmacological approaches through angiotensin receptor blockade. Interestingly, in the Marfan model, it has been suggested that the noncanonical TGFβ signaling may be most beneficial [92, 93]. These data are in contrast to what has been shown using the Drosophila model of muscular dystrophy where canonical TGFβ signaling was shown to be important for the progression of heart and muscle disease [89]. Whether these differences reflect the underlying differences in the invertebrate system or the differences between vascular tissues and striated muscle is yet unclear. Further studies are needed to address whether reduction in TGFβ signaling is required, in order to more fully dissect which intracellular pathways are most beneficial for treating MD.
Acknowledgments
Support: NIH NS071848, HL61322, AR052646, NS072027, and the Doris Duke Charitable Foundation
Abbreviations
- MD
Muscular dystrophy
- DGC
Dystrophin Glycoprotein Complex
- DMD
Duchenne muscular dystrophy
- ECM
extracellular matrix
- EGF
epidermal growth factor
- GRMD
Golden Retriever Muscular Dystrophy
- LLC
large latent complex
- LAP
latency associated peptide
- LGMD
Limb girdle muscular dystrophy
- SLC
small latent complex
- TGFβ
transforming growth factor beta
References
- 1.Chen YW, Nagaraju K, Bakay M, McIntyre O, Rawat R, Shi R, Hoffman EP. Early onset of inflammation and later involvement of TGFbeta in Duchenne muscular dystrophy. Neurology. 2005;65:826–34. doi: 10.1212/01.wnl.0000173836.09176.c4. [DOI] [PubMed] [Google Scholar]
- 2.Porter JD, Khanna S, Kaminski HJ, Rao JS, Merriam AP, Richmonds CR, Leahy P, Li J, Guo W, Andrade FH. A chronic inflammatory response dominates the skeletal muscle molecular signature in dystrophin-deficient mdx mice. Hum Mol Genet. 2002;11:263–72. doi: 10.1093/hmg/11.3.263. [DOI] [PubMed] [Google Scholar]
- 3.Flanigan KM, Dunn DM, von Niederhausern A, Soltanzadeh P, Gappmaier E, Howard MT, Sampson JB, Mendell JR, Wall C, King WM, Pestronk A, Florence JM, Connolly AM, Mathews KD, Stephan CM, Laubenthal KS, Wong BL, Morehart PJ, Meyer A, Finkel RS, Bonnemann CG, Medne L, Day JW, Dalton JC, Margolis MK, Hinton VJ, Weiss RB. Mutational spectrum of DMD mutations in dystrophinopathy patients: application of modern diagnostic techniques to a large cohort. Hum Mutat. 2009;30:1657–66. doi: 10.1002/humu.21114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Flanigan KM, Ceco E, Lamar K, Kaminoh Y, Dunn DM, Mendell JR, King WM, Pestronk A, Florence JM, Mathews KD, Finkel RS, Swoboda KJ, Gappmaier E, Howard MT, Day JW, McDonald C, McNally EM, Weiss RB. LTBP4 genotype predicts age of ambulatory loss in Duchenne Muscular Dystrophy. Ann of Neurolol. 2013 doi: 10.1002/ana.23819. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ginjaar IB, Kneppers AL, vd Meulen JD, Anderson LV, Bremmer-Bout M, van Deutekom JC, Weegenaar J, den Dunnen JT, Bakker E. Dystrophin nonsense mutation induces different levels of exon 29 skipping and leads to variable phenotypes within one BMD family. Eur J Hum Genet. 2000;8:793–6. doi: 10.1038/sj.ejhg.5200535. [DOI] [PubMed] [Google Scholar]
- 6.Hattori N, Kaido M, Nishigaki T, Inui K, Fujimura H, Nishimura T, Naka T, Hazama T. Undetectable dystrophin can still result in a relatively benign phenotype of dystrophinopathy. Neuromuscul Disord. 1999;9:220–6. doi: 10.1016/s0960-8966(99)00005-x. [DOI] [PubMed] [Google Scholar]
- 7.Noguchi S, McNally EM, Ben Othmane K, Hagiwara Y, Mizuno Y, Yoshida M, Yamamoto H, Bonnemann CG, Gussoni E, Denton PH, Kyriakides T, Middleton L, Hentati F, Ben Hamida M, Nonaka I, Vance JM, Kunkel LM, Ozawa E. Mutations in the dystrophin-associated protein gamma-sarcoglycan in chromosome 13 muscular dystrophy. Science. 1995;270:819–22. doi: 10.1126/science.270.5237.819. [DOI] [PubMed] [Google Scholar]
- 8.McNally EM, Passos-Bueno MR, Bonnemann CG, Vainzof M, de Sa Moreira E, Lidov HG, Othmane KB, Denton PH, Vance JM, Zatz M, Kunkel LM. Mild and severe muscular dystrophy caused by a single gamma-sarcoglycan mutation. Am J Hum Genet. 1996;59:1040–7. [PMC free article] [PubMed] [Google Scholar]
- 9.Kefi M, Amouri R, Driss A, Ben Hamida C, Ben Hamida M, Kunkel LM, Hentati F. Phenotype and sarcoglycan expression in Tunisian LGMD 2C patients sharing the same del521-T mutation. Neuromuscul Disord. 2003;13:779–87. doi: 10.1016/s0960-8966(03)00136-6. [DOI] [PubMed] [Google Scholar]
- 10.Piccolo F, Jeanpierre M, Leturcq F, Dode C, Azibi K, Toutain A, Merlini L, Jarre L, Navarro C, Krishnamoorthy R, Tome FM, Urtizberea JA, Beckmann JS, Campbell KP, Kaplan JC. A founder mutation in the gamma-sarcoglycan gene of gypsies possibly predating their migration out of India. Hum Mol Genet. 1996;5:2019–22. doi: 10.1093/hmg/5.12.2019. [DOI] [PubMed] [Google Scholar]
- 11.Glover L, Brown RH., Jr Dysferlin in membrane trafficking and patch repair. Traffic. 2007;8:785–94. doi: 10.1111/j.1600-0854.2007.00573.x. [DOI] [PubMed] [Google Scholar]
- 12.Mastaglia FL, Lamont PJ, Laing NG. Distal myopathies. Curr Opin Neurol. 2005;18:504–10. doi: 10.1097/01.wco.0000175936.23945.b6. [DOI] [PubMed] [Google Scholar]
- 13.Weiler T, Bashir R, Anderson LV, Davison K, Moss JA, Britton S, Nylen E, Keers S, Vafiadaki E, Greenberg CR, Bushby CR, Wrogemann K. Identical mutation in patients with limb girdle muscular dystrophy type 2B or Miyoshi myopathy suggests a role for modifier gene(s) Hum Mol Genet. 1999;8:871–7. doi: 10.1093/hmg/8.5.871. [DOI] [PubMed] [Google Scholar]
- 14.Illarioshkin SN, Ivanova-Smolenskaya IA, Greenberg CR, Nylen E, Sukhorukov VS, Poleshchuk VV, Markova ED, Wrogemann K. Identical dysferlin mutation in limb-girdle muscular dystrophy type 2B and distal myopathy. Neurology. 2000;55:1931–3. doi: 10.1212/wnl.55.12.1931. [DOI] [PubMed] [Google Scholar]
- 15.Bonne G, Mercuri E, Muchir A, Urtizberea A, Becane HM, Recan D, Merlini L, Wehnert M, Boor R, Reuner U, Vorgerd M, Wicklein EM, Eymard B, Duboc D, Penisson-Besnier I, Cuisset JM, Ferrer X, Desguerre I, Lacombe D, Bushby K, Pollitt C, Toniolo D, Fardeau M, Schwartz K, Muntoni F. Clinical and molecular genetic spectrum of autosomal dominant Emery-Dreifuss muscular dystrophy due to mutations of the lamin A/C gene. Ann Neurol. 2000;48:170–80. [PubMed] [Google Scholar]
- 16.Granger B, Gueneau L, Drouin-Garraud V, Pedergnana V, Gagnon F, Ben Yaou R, Tezenas du Montcel S, Bonne G. Modifier locus of the skeletal muscle involvement in Emery-Dreifuss muscular dystrophy. Hum Genet. 2011;129:149–59. doi: 10.1007/s00439-010-0909-1. [DOI] [PubMed] [Google Scholar]
- 17.Goldfarb LG, Park KY, Cervenakova L, Gorokhova S, Lee HS, Vasconcelos O, Nagle JW, Semino-Mora C, Sivakumar K, Dalakas MC. Missense mutations in desmin associated with familial cardiac and skeletal myopathy. Nat Genet. 1998;19:402–3. doi: 10.1038/1300. [DOI] [PubMed] [Google Scholar]
- 18.Zucconi E, Valadares MC, Vieira NM, Bueno CR, Jr, Secco M, Jazedje T, da Silva HC, Vainzof M, Zatz M. Ringo: discordance between the molecular and clinical manifestation in a golden retriever muscular dystrophy dog. Neuromuscul Disord. 2010;20:64–70. doi: 10.1016/j.nmd.2009.10.011. [DOI] [PubMed] [Google Scholar]
- 19.Fukada S, Morikawa D, Yamamoto Y, Yoshida T, Sumie N, Yamaguchi M, Ito T, Miyagoe-Suzuki Y, Takeda S, Tsujikawa K, Yamamoto H. Genetic background affects properties of satellite cells and mdx phenotypes. Am J Pathol. 2010;176:2414–24. doi: 10.2353/ajpath.2010.090887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kucherenko MM, Pantoja M, Yatsenko AS, Shcherbata HR, Fischer KA, Maksymiv DV, Chernyk YI, Ruohola-Baker H. Genetic modifier screens reveal new components that interact with the Drosophila dystroglycan-dystrophin complex. PLoS One. 2008;3:e2418. doi: 10.1371/journal.pone.0002418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guyon JR, Steffen LS, Howell MH, Pusack TJ, Lawrence C, Kunkel LM. Modeling human muscle disease in zebrafish. Biochimica et biophysica acta. 2007;1772:205–15. doi: 10.1016/j.bbadis.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 22.Hack AA, Ly CT, Jiang F, Clendenin CJ, Sigrist KS, Wollmann RL, McNally EM. Gamma-sarcoglycan deficiency leads to muscle membrane defects and apoptosis independent of dystrophin. J Cell Biol. 1998;142:1279–87. doi: 10.1083/jcb.142.5.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heydemann A, Huber JM, Demonbreun A, Hadhazy M, McNally EM. Genetic background influences muscular dystrophy. Neuromuscul Disord. 2005;15:601–9. doi: 10.1016/j.nmd.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 24.Heydemann A, Ceco E, Lim JE, Hadhazy M, Ryder P, Moran JL, Beier DR, Palmer AA, McNally EM. Latent TGF-beta-binding protein 4 modifies muscular dystrophy in mice. J Clin Invest. 2009;119:3703–12. doi: 10.1172/JCI39845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Swaggart KA, Heydemann A, Palmer AA, McNally EM. Distinct genetic regions modify specific muscle groups in muscular dystrophy. Physiol Genomics. 2011;43:24–31. doi: 10.1152/physiolgenomics.00172.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hersh CP, Demeo DL, Lazarus R, Celedon JC, Raby BA, Reilly JJ, Silverman EK. Genetic determinants of functional impairment in chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2006;3:476. doi: 10.1513/pats.200603-039MS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thompson AR, Cooper JA, Jones GT, Drenos F, van Bockxmeer FM, Biros E, Walker PJ, van Rij AM, Golledge J, Norman PE, Hafez H, Humphries SE. Assessment of the association between genetic polymorphisms in transforming growth factor beta, and its binding protein (LTBP), and the presence, and expansion, of Abdominal Aortic Aneurysm. Atherosclerosis. 2010;209:367–73. doi: 10.1016/j.atherosclerosis.2009.09.073. [DOI] [PubMed] [Google Scholar]
- 28.Forsti A, Li X, Wagner K, Tavelin B, Enquist K, Palmqvist R, Altieri A, Hallmans G, Hemminki K, Lenner P. Polymorphisms in the transforming growth factor beta 1 pathway in relation to colorectal cancer progression. Genes Chromosomes Cancer. 2010;49:270–81. doi: 10.1002/gcc.20738. [DOI] [PubMed] [Google Scholar]
- 29.Sterner-Kock A, Thorey IS, Koli K, Wempe F, Otte J, Bangsow T, Kuhlmeier K, Kirchner T, Jin S, Keski-Oja J, von Melchner H. Disruption of the gene encoding the latent transforming growth factor-beta binding protein 4 (LTBP-4) causes abnormal lung development, cardiomyopathy, and colorectal cancer. Genes Dev. 2002;16:2264–73. doi: 10.1101/gad.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Giltay R, Kostka G, Timpl R. Sequence and expression of a novel member (LTBP-4) of the family of latent transforming growth factor-beta binding proteins. FEBS Lett. 1997;411:164–8. doi: 10.1016/s0014-5793(97)00685-6. [DOI] [PubMed] [Google Scholar]
- 31.Todorovic V, Rifkin DB. LTBPs, more than just an escort service. J Cell Biochem. 2012;113:410–8. doi: 10.1002/jcb.23385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dallas SL, Chen Q, Sivakumar P. Dynamics of assembly and reorganization of extracellular matrix proteins. Curr Top Dev Biol. 2006;75:1–24. doi: 10.1016/S0070-2153(06)75001-3. [DOI] [PubMed] [Google Scholar]
- 33.Dietz HC, Cutting GR, Pyeritz RE, Maslen CL, Sakai LY, Corson GM, Puffenberger EG, Hamosh A, Nanthakumar EJ, Curristin SM, et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature. 1991;352:337–9. doi: 10.1038/352337a0. [DOI] [PubMed] [Google Scholar]
- 34.Doyle JJ, Gerber EE, Dietz HC. Matrix-dependent perturbation of TGFbeta signaling and disease. FEBS Lett. 2012;586:2003–15. doi: 10.1016/j.febslet.2012.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Habashi JP, Judge DP, Holm TM, Cohn RD, Loeys BL, Cooper TK, Myers L, Klein EC, Liu G, Calvi C, Podowski M, Neptune ER, Halushka MK, Bedja D, Gabrielson K, Rifkin DB, Carta L, Ramirez F, Huso DL, Dietz HC. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006;312:117–21. doi: 10.1126/science.1124287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brooke BS, Habashi JP, Judge DP, Patel N, Loeys B, Dietz HC., 3rd Angiotensin II blockade and aortic-root dilation in Marfan’s syndrome. N Engl J Med. 2008;358:2787–95. doi: 10.1056/NEJMoa0706585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cohn RD, van Erp C, Habashi JP, Soleimani AA, Klein EC, Lisi MT, Gamradt M, ap Rhys CM, Holm TM, Loeys BL, Ramirez F, Judge DP, Ward CW, Dietz HC. Angiotensin II type 1 receptor blockade attenuates TGF-beta-induced failure of muscle regeneration in multiple myopathic states. Nat Med. 2007;13:204–10. doi: 10.1038/nm1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koli K, Saharinen J, Hyytiainen M, Penttinen C, Keski-Oja J. Latency, activation, and binding proteins of TGF-beta. Microsc Res Tech. 2001;52:354–62. doi: 10.1002/1097-0029(20010215)52:4<354::AID-JEMT1020>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 39.Moren A, Olofsson A, Stenman G, Sahlin P, Kanzaki T, Claesson-Welsh L, ten Dijke P, Miyazono K, Heldin CH. Identification and characterization of LTBP-2, a novel latent transforming growth factor-beta-binding protein. J Biol Chem. 1994;269:32469–78. [PubMed] [Google Scholar]
- 40.Saharinen J, Taipale J, Monni O, Keski-Oja J. Identification and characterization of a new latent transforming growth factor-beta-binding protein, LTBP-4. J Biol Chem. 1998;273:18459–69. doi: 10.1074/jbc.273.29.18459. [DOI] [PubMed] [Google Scholar]
- 41.Kanzaki T, Otabe M. Latent transforming growth factor-beta binding protein-1, a component of latent transforming growth factor-beta complex, accelerates the migration of aortic smooth muscle cells in diabetic rats through integrin-beta3. Diabetes. 2003;52:824–8. doi: 10.2337/diabetes.52.3.824. [DOI] [PubMed] [Google Scholar]
- 42.Oklu R, Hesketh R, Wicky S, Metcalfe JC. Localization of latent transforming growth factor-beta binding protein-1 in human coronary atherosclerotic plaques. Circ J. 2011;75:196–200. doi: 10.1253/circj.cj-10-0334. [DOI] [PubMed] [Google Scholar]
- 43.Sharafieh R, Child AH, Khaw PT, Fleck B, Sarfarazi M. LTBP2 gene analysis in the GLC3C-linked family and 94 CYP1B1-negative cases with primary congenital glaucoma. Ophthalmic Genet. 2012 doi: 10.3109/13816810.2012.716486. [DOI] [PubMed] [Google Scholar]
- 44.Abu-Amero KK, Osman EA, Mousa A, Wheeler J, Whigham B, Allingham RR, Hauser MA, Al-Obeidan SA. Screening of CYP1B1 and LTBP2 genes in Saudi families with primary congenital glaucoma: genotype-phenotype correlation. Mol Vis. 2011;17:2911–9. [PMC free article] [PubMed] [Google Scholar]
- 45.Haji-Seyed-Javadi R, Jelodari-Mamaghani S, Paylakhi SH, Yazdani S, Nilforushan N, Fan JB, Klotzle B, Mahmoudi MJ, Ebrahimian MJ, Chelich N, Taghiabadi E, Kamyab K, Boileau C, Paisan-Ruiz C, Ronaghi M, Elahi E. LTBP2 mutations cause Weill-Marchesani and Weill-Marchesani-like syndrome and affect disruptions in the extracellular matrix. Hum Mutat. 2012;33:1182–7. doi: 10.1002/humu.22105. [DOI] [PubMed] [Google Scholar]
- 46.Shipley JM, Mecham RP, Maus E, Bonadio J, Rosenbloom J, McCarthy RT, Baumann ML, Frankfater C, Segade F, Shapiro SD. Developmental expression of latent transforming growth factor beta binding protein 2 and its requirement early in mouse development. Mol Cell Biol. 2000;20:4879–87. doi: 10.1128/mcb.20.13.4879-4887.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Colarossi C, Chen Y, Obata H, Jurukovski V, Fontana L, Dabovic B, Rifkin DB. Lung alveolar septation defects in Ltbp-3-null mice. Am J Pathol. 2005;167:419–28. doi: 10.1016/S0002-9440(10)62986-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Koli K, Ryynanen MJ, Keski-Oja J. Latent TGF-beta binding proteins (LTBPs)-1 and -3 coordinate proliferation and osteogenic differentiation of human mesenchymal stem cells. Bone. 2008;43:679–88. doi: 10.1016/j.bone.2008.06.016. [DOI] [PubMed] [Google Scholar]
- 49.Urban Z, Hucthagowder V, Schurmann N, Todorovic V, Zilberberg L, Choi J, Sens C, Brown CW, Clark RD, Holland KE, Marble M, Sakai LY, Dabovic B, Rifkin DB, Davis EC. Mutations in LTBP4 cause a syndrome of impaired pulmonary, gastrointestinal, genitourinary, musculoskeletal, and dermal development. Am J Hum Genet. 2009;85:593–605. doi: 10.1016/j.ajhg.2009.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Koli K, Wempe F, Sterner-Kock A, Kantola A, Komor M, Hofmann WK, von Melchner H, Keski-Oja J. Disruption of LTBP-4 function reduces TGF-beta activation and enhances BMP-4 signaling in the lung. J Cell Biol. 2004;167:123–33. doi: 10.1083/jcb.200403067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sinha S, Nevett C, Shuttleworth CA, Kielty CM. Cellular and extracellular biology of the latent transforming growth factor-beta binding proteins. Matrix Biol. 1998;17:529–45. doi: 10.1016/s0945-053x(98)90106-8. [DOI] [PubMed] [Google Scholar]
- 52.Miyazono K, Thyberg J, Heldin CH. Retention of the transforming growth factor-beta 1 precursor in the Golgi complex in a latent endoglycosidase H-sensitive form. J Biol Chem. 1992;267:5668–75. [PubMed] [Google Scholar]
- 53.Miyazono K, Olofsson A, Colosetti P, Heldin CH. A role of the latent TGF-beta 1-binding protein in the assembly and secretion of TGF-beta 1. EMBO J. 1991;10:1091–101. doi: 10.1002/j.1460-2075.1991.tb08049.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Taipale J, Koli K, Keski-Oja J. Release of transforming growth factor-beta 1 from the pericellular matrix of cultured fibroblasts and fibrosarcoma cells by plasmin and thrombin. J Biol Chem. 1992;267:25378–84. [PubMed] [Google Scholar]
- 55.Taipale J, Lohi J, Saarinen J, Kovanen PT, Keski-Oja J. Human mast cell chymase and leukocyte elastase release latent transforming growth factor-beta 1 from the extracellular matrix of cultured human epithelial and endothelial cells. J Biol Chem. 1995;270:4689–96. doi: 10.1074/jbc.270.9.4689. [DOI] [PubMed] [Google Scholar]
- 56.Munger JS, Harpel JG, Gleizes PE, Mazzieri R, Nunes I, Rifkin DB. Latent transforming growth factor-beta: structural features and mechanisms of activation. Kidney Int. 1997;51:1376–82. doi: 10.1038/ki.1997.188. [DOI] [PubMed] [Google Scholar]
- 57.Olofsson A, Ichijo H, Moren A, ten Dijke P, Miyazono K, Heldin CH. Efficient association of an amino-terminally extended form of human latent transforming growth factor-beta binding protein with the extracellular matrix. J Biol Chem. 1995;270:31294–7. doi: 10.1074/jbc.270.52.31294. [DOI] [PubMed] [Google Scholar]
- 58.Saharinen J, Taipale J, Keski-Oja J. Association of the small latent transforming growth factor-beta with an eight cysteine repeat of its binding protein LTBP-1. EMBO J. 1996;15:245–53. [PMC free article] [PubMed] [Google Scholar]
- 59.Koski C, Saharinen J, Keski-Oja J. Independent promoters regulate the expression of two amino terminally distinct forms of latent transforming growth factor-beta binding protein-1 (LTBP-1) in a cell type-specific manner. J Biol Chem. 1999;274:32619–30. doi: 10.1074/jbc.274.46.32619. [DOI] [PubMed] [Google Scholar]
- 60.Unsold C, Hyytiainen M, Bruckner-Tuderman L, Keski-Oja J. Latent TGF-beta binding protein LTBP-1 contains three potential extracellular matrix interacting domains. J Cell Sci. 2001;114:187–197. doi: 10.1242/jcs.114.1.187. [DOI] [PubMed] [Google Scholar]
- 61.Annes JP, Munger JS, Rifkin DB. Making sense of latent TGFbeta activation. J Cell Sci. 2003;116:217–24. doi: 10.1242/jcs.00229. [DOI] [PubMed] [Google Scholar]
- 62.Ge G, Greenspan DS. BMP1 controls TGFbeta1 activation via cleavage of latent TGFbeta-binding protein. J Cell Biol. 2006;175:111–20. doi: 10.1083/jcb.200606058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dallas SL, Rosser JL, Mundy GR, Bonewald LF. Proteolysis of latent transforming growth factor-beta (TGF-beta )-binding protein-1 by osteoclasts. A cellular mechanism for release of TGF-beta from bone matrix. J Biol Chem. 2002;277:21352–60. doi: 10.1074/jbc.M111663200. [DOI] [PubMed] [Google Scholar]
- 64.Michel K, Roth S, Trautwein C, Gong W, Flemming P, Gressner AM. Analysis of the expression pattern of the latent transforming growth factor beta binding protein isoforms in normal and diseased human liver reveals a new splice variant missing the proteinase-sensitive hinge region. Hepatology. 1998;27:1592–9. doi: 10.1002/hep.510270619. [DOI] [PubMed] [Google Scholar]
- 65.Yin W, Smiley E, Bonadio J. Alternative splicing of LTBP-3. Biochem Biophys Res Commun. 1998;245:454–8. doi: 10.1006/bbrc.1998.8456. [DOI] [PubMed] [Google Scholar]
- 66.Saharinen J, Keski-Oja J. Specific sequence motif of 8-Cys repeats of TGF-beta binding proteins, LTBPs, creates a hydrophobic interaction surface for binding of small latent TGF-beta. Mol Biol Cell. 2000;11:2691–704. doi: 10.1091/mbc.11.8.2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bujak M, Frangogiannis NG. The role of TGF-beta signaling in myocardial infarction and cardiac remodeling. Cardiovasc Res. 2007;74:184–95. doi: 10.1016/j.cardiores.2006.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Leask A, Abraham DJ. TGF-beta signaling and the fibrotic response. FASEB J. 2004;18:816–27. doi: 10.1096/fj.03-1273rev. [DOI] [PubMed] [Google Scholar]
- 69.Piek E, Heldin CH, Ten Dijke P. Specificity, diversity, and regulation in TGF-beta superfamily signaling. FASEB J. 1999;13:2105–24. [PubMed] [Google Scholar]
- 70.Taipale J, Saharinen J, Keski-Oja J. Extracellular matrix-associated transforming growth factor-beta: role in cancer cell growth and invasion. Adv Cancer Res. 1998;75:87–134. doi: 10.1016/s0065-230x(08)60740-x. [DOI] [PubMed] [Google Scholar]
- 71.Kane CJ, Hebda PA, Mansbridge JN, Hanawalt PC. Direct evidence for spatial and temporal regulation of transforming growth factor beta 1 expression during cutaneous wound healing. J Cell Physiol. 1991;148:157–73. doi: 10.1002/jcp.1041480119. [DOI] [PubMed] [Google Scholar]
- 72.Chin GS, Liu W, Peled Z, Lee TY, Steinbrech DS, Hsu M, Longaker MT. Differential expression of transforming growth factor-beta receptors I and II and activation of Smad 3 in keloid fibroblasts. Plast Reconstr Surg. 2001;108:423–9. doi: 10.1097/00006534-200108000-00022. [DOI] [PubMed] [Google Scholar]
- 73.Schmid P, Itin P, Cherry G, Bi C, Cox DA. Enhanced expression of transforming growth factor-beta type I and type II receptors in wound granulation tissue and hypertrophic scar. Am J Pathol. 1998;152:485–93. [PMC free article] [PubMed] [Google Scholar]
- 74.Lin RY, Sullivan KM, Argenta PA, Meuli M, Lorenz HP, Adzick NS. Exogenous transforming growth factor-beta amplifies its own expression and induces scar formation in a model of human fetal skin repair. Ann Surg. 1995;222:146–54. doi: 10.1097/00000658-199508000-00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cordeiro MF. Transforming growth factor-beta function blocking already effective as therapeutic strategy. Circulation. 2003;107:E37–7. doi: 10.1161/01.cir.0000053951.16591.20. author reply E37–7. [DOI] [PubMed] [Google Scholar]
- 76.Shah M, Foreman DM, Ferguson MW. Neutralisation of TGF-beta 1 and TGF-beta 2 or exogenous addition of TGF-beta 3 to cutaneous rat wounds reduces scarring. J Cell Sci. 1995;108(Pt 3):985–1002. doi: 10.1242/jcs.108.3.985. [DOI] [PubMed] [Google Scholar]
- 77.Yamazaki M, Minota S, Sakurai H, Miyazono K, Yamada A, Kanazawa I, Kawai M. Expression of transforming growth factor-beta 1 and its relation to endomysial fibrosis in progressive muscular dystrophy. Am J Pathol. 1994;144:221–6. [PMC free article] [PubMed] [Google Scholar]
- 78.Bernasconi P, Torchiana E, Confalonieri P, Brugnoni R, Barresi R, Mora M, Cornelio F, Morandi L, Mantegazza R. Expression of transforming growth factor-beta 1 in dystrophic patient muscles correlates with fibrosis. Pathogenetic role of a fibrogenic cytokine. J Clin Invest. 1995;96:1137–44. doi: 10.1172/JCI118101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Burks TN, Cohn RD. Role of TGF-beta signaling in inherited and acquired myopathies. Skelet Muscle. 2011;1:19. doi: 10.1186/2044-5040-1-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Barton ER. Impact of sarcoglycan complex on mechanical signal transduction in murine skeletal muscle. Am J Physiol Cell Physiol. 2006;290:C411–9. doi: 10.1152/ajpcell.00192.2005. [DOI] [PubMed] [Google Scholar]
- 81.Kolodziejczyk SM, Walsh GS, Balazsi K, Seale P, Sandoz J, Hierlihy AM, Rudnicki MA, Chamberlain JS, Miller FD, Megeney LA. Activation of JNK1 contributes to dystrophic muscle pathogenesis. Curr Biol. 2001;11:1278–82. doi: 10.1016/s0960-9822(01)00397-9. [DOI] [PubMed] [Google Scholar]
- 82.Nelson CA, Hunter RB, Quigley LA, Girgenrath S, Weber WD, McCullough JA, Dinardo CJ, Keefe KA, Ceci L, Clayton NP, McVie-Wylie A, Cheng SH, Leonard JP, Wentworth BM. Inhibiting TGF-beta activity improves respiratory function in mdx mice. Am J Pathol. 2011;178:2611–21. doi: 10.1016/j.ajpath.2011.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lorts A, Schwanekamp JA, Baudino TA, McNally EM, Molkentin JD. Deletion of periostin reduces muscular dystrophy and fibrosis in mice by modulating the transforming growth factor-beta pathway. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:10978–83. doi: 10.1073/pnas.1204708109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Oka T, Xu J, Kaiser RA, Melendez J, Hambleton M, Sargent MA, Lorts A, Brunskill EW, Dorn GW, 2nd , Conway SJ, Aronow BJ, Robbins J, Molkentin JD. Genetic manipulation of periostin expression reveals a role in cardiac hypertrophy and ventricular remodeling. Circ Res. 2007;101:313–21. doi: 10.1161/CIRCRESAHA.107.149047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Vetrone SA, Montecino-Rodriguez E, Kudryashova E, Kramerova I, Hoffman EP, Liu SD, Miceli MC, Spencer MJ. Osteopontin promotes fibrosis in dystrophic mouse muscle by modulating immune cell subsets and intramuscular TGF-beta. J Clin Invest. 2009;119:1583–94. doi: 10.1172/JCI37662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pegoraro E, Hoffman EP, Piva L, Gavassini BF, Cagnin S, Ermani M, Bello L, Soraru G, Pacchioni B, Bonifati MD, Lanfranchi G, Angelini C, Kesari A, Lee I, Gordish-Dressman H, Devaney JM, McDonald CM. SPP1 genotype is a determinant of disease severity in Duchenne muscular dystrophy. Neurology. 2011;76:219–26. doi: 10.1212/WNL.0b013e318207afeb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Piva L, Gavassini BF, Bello L, Fanin M, Soraru G, Barp A, Ermani M, Angelini C, Hoffman EP, Pegoraro E. TGFBR2 but not SPP1 genotype modulates osteopontin expression in Duchenne muscular dystrophy muscle. J Pathol. 2012;228:251–9. doi: 10.1002/path.4026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hullinger TG, Pan Q, Viswanathan HL, Somerman MJ. TGFbeta and BMP-2 activation of the OPN promoter: roles of smad- and hox-binding elements. Exp Cell Res. 2001;262:69–74. doi: 10.1006/excr.2000.5074. [DOI] [PubMed] [Google Scholar]
- 89.Goldstein JA, Kelly SM, LoPresti PP, Heydemann A, Earley JU, Ferguson EL, Wolf MJ, McNally EM. SMAD signaling drives heart and muscle dysfunction in a Drosophila model of muscular dystrophy. Hum Mol Genet. 2011;20:894–904. doi: 10.1093/hmg/ddq528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Allikian MJ, Bhabha G, Dospoy P, Heydemann A, Ryder P, Earley JU, Wolf MJ, Rockman HA, McNally EM. Reduced life span with heart and muscle dysfunction in Drosophila sarcoglycan mutants. Hum Mol Genet. 2007;16:2933–43. doi: 10.1093/hmg/ddm254. [DOI] [PubMed] [Google Scholar]
- 91.Shcherbata HR, Yatsenko AS, Patterson L, Sood VD, Nudel U, Yaffe D, Baker D, Ruohola-Baker H. Dissecting muscle and neuronal disorders in a Drosophila model of muscular dystrophy. EMBO J. 2007;26:481–93. doi: 10.1038/sj.emboj.7601503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Holm TM, Habashi JP, Doyle JJ, Bedja D, Chen Y, van Erp C, Lindsay ME, Kim D, Schoenhoff F, Cohn RD, Loeys BL, Thomas CJ, Patnaik S, Marugan JJ, Judge DP, Dietz HC. Noncanonical TGFbeta signaling contributes to aortic aneurysm progression in Marfan syndrome mice. Science. 2011;332:358–61. doi: 10.1126/science.1192149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Habashi JP, Doyle JJ, Holm TM, Aziz H, Schoenhoff F, Bedja D, Chen Y, Modiri AN, Judge DP, Dietz HC. Angiotensin II type 2 receptor signaling attenuates aortic aneurysm in mice through ERK antagonism. Science. 2011;332:361–5. doi: 10.1126/science.1192152. [DOI] [PMC free article] [PubMed] [Google Scholar]