

Abstract
The pancreatic acinar cell is the main parenchymal cell of the exocrine pancreas and plays a primary role in the secretion of pancreatic enzymes into the pancreatic duct. It is also the site for the initiation of pancreatitis. Here we describe how acinar cells are isolated from whole pancreas tissue and intracellular calcium signals are measured. In addition, we describe the techniques of transfecting these cells with adenoviral constructs, and subsequently measuring the leakage of lactate dehydrogenase, a marker of cell injury, during conditions that induce acinar cell injury in vitro. These techniques provide a powerful tool to characterize acinar cell physiology and pathology.
Keywords: Cancer Biology, Issue 77, Cellular Biology, Molecular Biology, Medicine, Biochemistry, Biomedical Engineering, Acinar Cells, Pancreatitis, Transfection, Microscopy, Confocal, Calcium Signaling, Pancreatic Acinar Cells, Pancreatitis, Calcium Signaling, Cytotoxicity, LDH Leakage, cell injury, imaging
Introduction
Dynamic changes in cytosolic calcium are necessary for both physiological and pathological acinar cell events. These divergent effects of calcium are thought to result from distinct spatial and temporal patterns of calcium signaling 1. For example, enzyme and fluid secretion from the acinar cell are linked to calcium spikes from a restricted region of the apical pole where secretion takes place 2. In contrast, a global calcium wave followed by intense non-oscillatory calcium signals is associated with early pathological events which lead to acute pancreatitis 3,4. These include intra-acinar protease activation, reduced enzyme secretion, and acinar cell injury. Our lab uses isolated pancreatic acini to study these early pathological events which lead to disease both in vivo and in vitro 5-7. The methods detailed here describe isolation of primary acinar cells for the purpose of measuring cytosolic calcium levels and cell injury. A method for adenoviral infection of these cells is also provided.
Protocol
1. Preparing Pancreatic Acinar Cells for Calcium Imaging
Prepare HEPES incubation buffer containing 20 mM HEPES, 95 mM NaCl, 4.7 mM KCl, 0.6 mM MgCl2, 1.3 mM CaCl2, 10 mM glucose, 2 mM glutamine, and 1 × minimum Eagle's medium non-essential amino acids. Adjust the final solution to pH 7.4 with NaOH.
Prepare a BSA incubation buffer by adding BSA (1% w/v final) to 25 ml of the HEPES incubation buffer (described above).
Prepare a collagenase digestion buffer by adding 1.1 mg/ml (200 Units/ml) type-4 collagenase and 1 mg/ml soybean trypsin inhibitor to 6 ml of BSA incubation buffer (described above).
Acid-wash 22x22 mm coverslips by immersing in two parts HNO3 and one part HCl for 2 hr. Then decant using DI water and store in 70% ethanol.
Euthanize one mouse by CO2 asphyxiation. Orient the animal in the supine position, and prepare the abdominal surface by cleaning with 70% ethanol. Perform a laparotomy to expose the abdominal cavity. Dissect out the pancreas and immediately rinse in a small weigh boat containing 6 ml of collagenase digestion buffer.
Dissect away any large pieces of fat or visible blood vessels, then remove 5 ml of collagenase digestion buffer and add it back to the 15 ml conical tube.
Using fine dissection scissors mince the pancreas in the small weighing boat containing 1 ml of collagenase digestion buffer. Mince until the resulting solution appears evenly dispersed.
Transfer the minced product to a 125 ml Erlenmeyer plastic flask, and add the remaining 5 ml of collagenase digestion buffer to the container. Make sure the tissue is fully submerged in buffer.
Place the flask in a 37 °C water bath and shake at 90 rpm for 30 min. During this short period of downtime, prepare calcium activating agonists (e.g. caerulein, carbachol) and rinse the 22x22 mm acid-washed coverslips with DI water. Dry, and then place on top of a flat surface lined by laboratory film.
After the 30 min digest is complete, transfer the suspension back to the 15 ml conical tube and allow the cells to settle. Carefully pipette out the collagenase digestion buffer and replace with 6 ml of BSA incubation buffer. Vigorously shake the tube by hand for 10 sec in order to disperse the cells into smaller clusters. Immediately upon shaking, remove any large, floating debris from the unsettled media.
Allow the remaining cells to settle, and exchange the media with fresh BSA incubation buffer.
Repeat step 1.11 twice, until the suspension is comprised of only small clusters which are only finely visible to the naked eye. The cells should resemble those depicted in Figure 1A (top row).
Remove the BSA incubation buffer and replace with HEPES incubation buffer.
Prepare a 750 μM, 100X stock of Fluo-4AM in 10% DMSO.
Load the cells in the 15 ml conical tube containing the cell suspension and BSA-free HEPES incubation buffer. To do this, add an appropriate volume (1:100 dilution) of the stock solution to the cell suspension. Then plate 500 μl of this suspension onto each 22x22 mm coverslip.
Keep coverslips in the dark at room temperature. Take the first Ca2+ measurements 30 min after loading. The dye is automatically removed from the medium upon starting the perfusion, which should occur 30 min after dye loading.
2. Measuring Calcium in Pancreatic Acinar Cells
Rinse each perfusion set-up with DI water and fill with an appropriate amount of buffer or agonist. Prime each syringe with buffer or agonist to insure proper flow. Clamp syringes to a ring stand approximately 1-2 feet above the microscope stage.
Use fine tweezers to carefully grasp from its edge one coverslip containing cell suspension. Tilt the coverslip 45°, and allow the excess buffer to run off. Place the coverslip on top of the rubber gasket with the cells on top of the coverslip, and assemble the chamber (Figures 2A and 2B).
Secure the perfusion chamber to the stage and insert the tubing which contains buffer into the first inlet of the chamber (Figure 2C). Turn the syringe containing buffer on and allow the buffer to perfuse over the entire surface of the coverslip. Once the buffer has reached the opposite end of the chamber, insert a vacuum line into the vacuum inlet. Insert any other tubes (containing agonist, inhibitors, etc.) into their appropriate position along the chamber.
Visualize the cells using either a 200X or 400X, 1.4 numerical aperture objective and an argon laser to excite the Fluo-4AM dye at a wavelength of 488 nm (Figure 1A, bottom). The power settings of the laser should be adjusted such that the photomultiplier tube (PMT) is not saturated by the emitted light. In addition, the voltage across the PMT needs to be optimized for maximum light detection.
Long-pass emission signals of > 515 nm are collected at frame speeds of 2-5 sec/frame for visualizing slow oscillatory patterns and 0.2-0.3 sec/frame for visualizing faster, global calcium waves (Figures 3 and 4).
After imaging is complete, close the syringes and remove all tubing. Disassemble the chamber and repeat steps 2.2-2.4.
Collect data as numerical values of fluorescence intensity over time and transfer to Image J (software package provided as freeware by the National Institutes of Health and can be found at http://rsb.info.nih.gov/ij/).
View real-time cellular images and identify regions of interest (ROIs; i.e. apical, basolateral, nuclear, etc.)
Acquire fluorescence intensities for these ROIs and represent as fluorescence intensity/baseline fluorescence intensity (i.e. F/F0).
Generate tracings by plotting F/F0 vs. time. In addition to tracings, representative images are commonly provided and displayed using pseudocolor.
3. Preparation of Pancreatic Acinar Cells for Cell Injury Assays
Warm DMEM F-12 media (without phenol red) to 37 °C.
Add BSA (0.1% w/v final), and HCl (50 μM final) to the DMEM F-12 media.
Aliquot 50 ml of DMEM into a conical tube.
Prepare a collagenase buffer by adding 0.5 mg/ml (12 U/ml) of collagenase type IV to 10 ml of supplemented DMEM F-12 media.
Euthanize one mouse by CO2 asphyxiation. Orient the animal in the supine position, and prepare the abdominal surface by cleaning with 70% ethanol. Perform a laparotomy to expose the abdominal cavity. Dissect out the pancreas and immediately rinse in a small weighing boat containing 6 ml of collagenase buffer.
Mince the tissue using fine dissection scissors for 3-5 min or until the resulting solution appears evenly dispersed.
Transfer the minced tissue to a 125 ml Erlenmeyer flask with 5 ml of additional collagenase buffer, then place into a 37 °C water bath with shaking at 90 rpm for 5 min.
Remove from the shaker, allow cells to briefly settle, and exchange with 5 ml of fresh collagenase buffer. Then incubate for an additional 35 min in a 37 °C water bath with shaking at 90 rpm.
Vigorously resuspend the digest using a transfer pipette until the suspension appears homogeneous with no obvious cell clumps. Then gently pipette the cell suspension through a pre-wet nylon mesh into a conical flask.
Vigorously pipette an additional 3 ml of fresh DMEM F-12 media (with collagenase) through the mesh, in order to force through any remaining cells.
Add another 6-10 ml of DMEM F-12 media and allow the cells to settle for 2-3 min. Repeat this step twice, and remove the supernatant after the final wash.
Add fresh DMEM F-12 media, resuspend the cells, and plate 500 μl of cell suspension into each well of a 48-well tissue culture plate. The cells should resemble those depicted in Figure 1B.
Allow the plate to sit in a 37 °C water bath at 90 rpm for 5 min before adding agonists.
Stimulate the cells with varying agonists for 2-4 hr.
Tilt the plate at an angle to allow the cells to settle and carefully pipette out an aliquot of media (usually 100 μl) and flash freeze in liquid nitrogen.
Flash freeze the remaining cell suspension. Flash freezing is not essential for LDH measurements. As an alternative samples can be kept on ice if they will be assayed the same day or stored in -20 °C for long term storage.
4. Measuring Lactate Dehydrogenase Leakage
Measure lactate dehydrogenase (LDH) leakage primarily using the reagents and instructions supplied in the Promega CytoTox 96 Non-Radioactive Cytotoxicity Assay kit (see Table of specific reagents and materials).
Reconstitute the LDH substrate (that is provided in the kit) with 12 ml of assay buffer (also provided).
Thaw frozen cell suspension and media samples in a water bath at room temperature.
Plate 50 μl of each media sample in a 96-well plate.
To the cell suspension, add the necessary volume of 10X lysis buffer so that the final concentration is 1X. Vortex briefly and incubate at 37 °C for 60 min.
Centrifuge lysed samples at 250 x g for 4 min to pellet cell debris.
For cell lysates, plate 50 μl of a 1:10 dilution into each well of a 96 well plate.
Add 50 μl of reconstituted substrate to each well, incubate the plate in the dark at room temperature for 30 min.
Stop the reaction by adding 50 μl of the stop solution (provided in the kit) to each well.
Measure absorbance at 490 nm.
Use the following formulae to calculate %LDH leakage. The optical densities below are corrected by subtracting out a blank OD reading from a well containing only PBS, substrate, and stop solution.
LDH in media = (Media OD492) x (media dilution factor) x (vol. of media)*Since it is usually unnecessary to dilute media samples, the dilution factor will most often equal1.
Total LDH = ((Lysate OD490) x (lysate dilution factor) x (vol. in cell suspension))+ ((Media OD490) x (dilution factor) x (vol. of media aliquoted for sampling))
![]()
5. Infecting Pancreatic Acinar Cells with Adenovirus
Prepare pancreatic acinar cells as described in section 3.
Following step 3.12, add 107 infectious units (IFUs) of adenovirus to the cell suspension and incubate for 30 min at 37 °C.
Evenly plate 2 ml of cell suspension in a 6-well plate.
For most expression constructs, cells should be adequately infected within 18 hr; and for some luciferase constructs, just within 6 hr.
Representative Results
An example of acinar cell calcium measurements in response to physiologic stimuli is provided in Figure 3. Acinar cells were loaded with the calcium dye Fluo-4 and perfused with the acetylcholine analogue carbachol (CCh; 1 μM)) 8. Cells responded in the form of a calcium wave which initiates in the apical region and propagates to the basolateral region 3,9. Representative tracings shown in Figure 3B demonstrate the typical peak-plateau pattern commonly observed with 1 μM carbachol. Conversely, using sub-maximal doses of the cholecystokinin analogue caerulein (10 pM) yields oscillatory calcium responses (Figure 4) 10.
Leakage of LDH from acinar cells in response to pancreatitis-inducing agonists is shown in Figure 5. Here we demonstrate concentration-dependent increases in LDH leakage in the presence of caerulein, carbachol, or the bile acid taurolithocholic acid-3 sulfate (TLCS). The concentrations necessary to induce maximal leakage of LDH are consistent with those required to induce intra-acinar protease activation and pathologic calcium signaling, suggesting these events may lead to injury.
Acinar cells were infected with a luciferase reporter adenovirus, that is driven by the promoter for a downstream Cn effector, nuclear factor of activated T cells (NFAT; Figure 6A). We provide representative data validating our infection method, demonstrating concentration- and time-dependent increases in NFAT-luciferase activity in the presence of TLCS (Figures 6B and 6C).
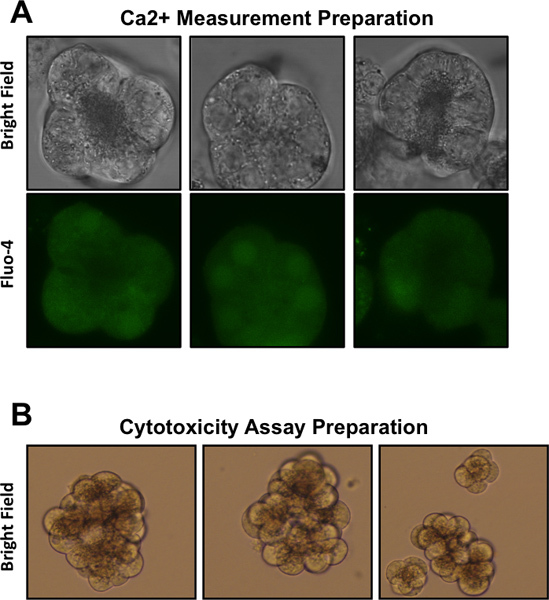 Figure 1. Representative images of pancreatic acinar cells following various preparations. (A) Acinar cells prepared for Ca2+ measurements, as visualized at 630X magnification. Top row depicts bright field microscopy, bottom row depicts acinar cells loaded with the Ca2+ dye Fluo-4 and visualized using fluorescence microscopy. (B) Acinar cells prepared for cytotoxicity assays, as visualized at 400X magnification.
Figure 1. Representative images of pancreatic acinar cells following various preparations. (A) Acinar cells prepared for Ca2+ measurements, as visualized at 630X magnification. Top row depicts bright field microscopy, bottom row depicts acinar cells loaded with the Ca2+ dye Fluo-4 and visualized using fluorescence microscopy. (B) Acinar cells prepared for cytotoxicity assays, as visualized at 400X magnification.
 Figure 2. The perifusion chamber.(A) The chamber consists of three layers: a metal base; a rubber gasket; and a plastic cover. (B) The rubber gasket is placed on top of the metal base and a 22x22 mm coverslip containing cells is placed upwards on top of the rubber gasket. The plastic cover is screwed to the metal base and an 18x18 mm coverslip is placed on top. (C) The chamber is then secured to the microscope stage. Tubing, containing buffer or agonist, is fed into inlets located on the chamber's edge (arrow). Separate tubing leads to a vacuum (arrow head).
Figure 2. The perifusion chamber.(A) The chamber consists of three layers: a metal base; a rubber gasket; and a plastic cover. (B) The rubber gasket is placed on top of the metal base and a 22x22 mm coverslip containing cells is placed upwards on top of the rubber gasket. The plastic cover is screwed to the metal base and an 18x18 mm coverslip is placed on top. (C) The chamber is then secured to the microscope stage. Tubing, containing buffer or agonist, is fed into inlets located on the chamber's edge (arrow). Separate tubing leads to a vacuum (arrow head).
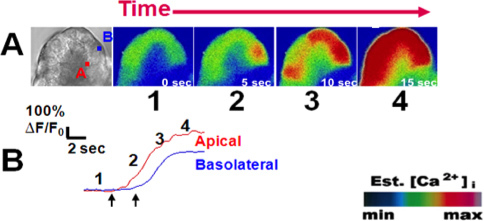 Figure 3. A typical peak-plateau calcium signal upon stimulation with carbachol (1 μM). (A) From left to right; Bright field view of an acinus labeled at the (A)pical and (B)asolateral regions of interest from an acinar cell. Cells were loaded with the calcium indicator Fluo-4 (5 μM). Upon stimulation with physiologic carbachol (1 μM; Ach analogue), subsequent images show the initiation of the calcium signal in the apical region followed by propagation to the basal region. (B) Each paneled image (1-4), corresponds to a frame along a representative tracing of change in fluorescence over time for each region of interest. Images are represented in pseudocolor with a color scale (bottom right). Left and right arrows show time of first calcium rise in the apical and basal regions, respectively. This figure was originally published in the Journal of Biological Chemistry. (Orabi A.I., Shah A.U., Muili K., Luo Y., Mahmood S.M., Ahmad A., Reed A., Husain S.Z. Ethanol enhances carbachol-induced protease activation and accelerates calcium waves in isolated rat pancreatic acini. The Journal of Biological Chemistry, 286, 14090-14097 (2011).
Figure 3. A typical peak-plateau calcium signal upon stimulation with carbachol (1 μM). (A) From left to right; Bright field view of an acinus labeled at the (A)pical and (B)asolateral regions of interest from an acinar cell. Cells were loaded with the calcium indicator Fluo-4 (5 μM). Upon stimulation with physiologic carbachol (1 μM; Ach analogue), subsequent images show the initiation of the calcium signal in the apical region followed by propagation to the basal region. (B) Each paneled image (1-4), corresponds to a frame along a representative tracing of change in fluorescence over time for each region of interest. Images are represented in pseudocolor with a color scale (bottom right). Left and right arrows show time of first calcium rise in the apical and basal regions, respectively. This figure was originally published in the Journal of Biological Chemistry. (Orabi A.I., Shah A.U., Muili K., Luo Y., Mahmood S.M., Ahmad A., Reed A., Husain S.Z. Ethanol enhances carbachol-induced protease activation and accelerates calcium waves in isolated rat pancreatic acini. The Journal of Biological Chemistry, 286, 14090-14097 (2011).
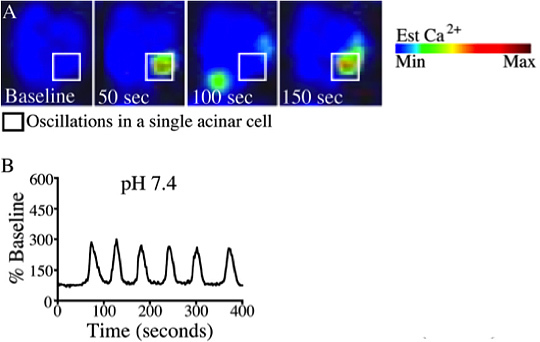 Figure 4. A typical calcium oscillation upon stimulation caerulein (10 pM). (A) Changes in whole cell cytosolic calcium were measured once per second by time-lapse confocal microscopy using the calcium dye Fluo-4/ AM. Images are represented in pseudocolor with a color scale (top right). (B) Representative plot of fluorescence over time were recorded from a single cell treated with caerulein (10 pM) at pH 7.4. This figure was originally published in the Journal of Biological Chemistry. (Reed A.M., Husain S.Z., Thrower E., Alexandre M., Shah A., Gorelick F.S., Nathanson M.H. Low Extracellular pH Induces Damage in the Pancreatic Acinar Cell by Enhancing Calcium Signaling. The Journal of Biological Chemistry, 286, 1919-1926 (2011).
Figure 4. A typical calcium oscillation upon stimulation caerulein (10 pM). (A) Changes in whole cell cytosolic calcium were measured once per second by time-lapse confocal microscopy using the calcium dye Fluo-4/ AM. Images are represented in pseudocolor with a color scale (top right). (B) Representative plot of fluorescence over time were recorded from a single cell treated with caerulein (10 pM) at pH 7.4. This figure was originally published in the Journal of Biological Chemistry. (Reed A.M., Husain S.Z., Thrower E., Alexandre M., Shah A., Gorelick F.S., Nathanson M.H. Low Extracellular pH Induces Damage in the Pancreatic Acinar Cell by Enhancing Calcium Signaling. The Journal of Biological Chemistry, 286, 1919-1926 (2011).
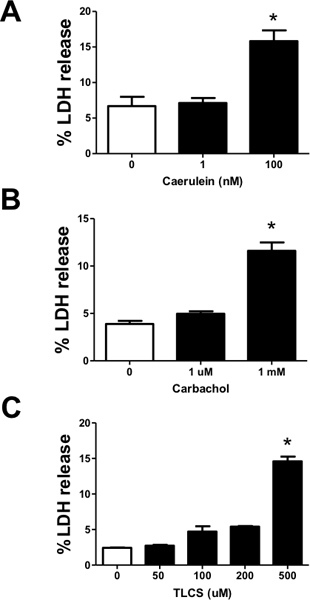 Figure 5. Measuring cell injury from acinar cells treated with various pancreatitis-inducing agonists. In isolated acinar cells, lactate dehydrogenase (LDH) leakage was used as a biochemical indicator of injury. Isolated acinar cells were treated with varying concentrations of (A) caerulein (1-100 nM) (B) carbachol (1 μM -1 mM) and (C) TLCS (50-500 μM), and %LDH leakage was measured after 2 hr. (n=3). #, *, P<0.05 compared to the control and TLCS alone, respectively.
Figure 5. Measuring cell injury from acinar cells treated with various pancreatitis-inducing agonists. In isolated acinar cells, lactate dehydrogenase (LDH) leakage was used as a biochemical indicator of injury. Isolated acinar cells were treated with varying concentrations of (A) caerulein (1-100 nM) (B) carbachol (1 μM -1 mM) and (C) TLCS (50-500 μM), and %LDH leakage was measured after 2 hr. (n=3). #, *, P<0.05 compared to the control and TLCS alone, respectively.
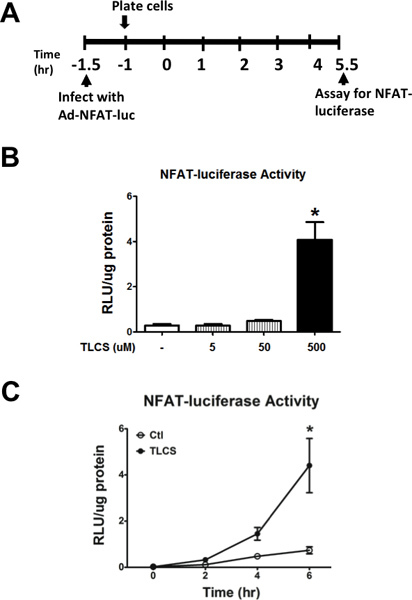 Figure 6. Measuring luciferase activity in pancreatic acinar cells infected with adeno-NFAT-luciferase. (A) Schema for the infection of primary mouse pancreatic acinar cells with adenoviral NFAT-luciferase. (B) Administration of TLCS (5-500 μM) induced NFAT-luciferase activity. (C) Time course demonstrating accumulation of luminescence with TLCS (500 μM) given over a 6 hr period. (n=3). *, #, P<0.05, relative to the control or TLCS alone, respectively.
Figure 6. Measuring luciferase activity in pancreatic acinar cells infected with adeno-NFAT-luciferase. (A) Schema for the infection of primary mouse pancreatic acinar cells with adenoviral NFAT-luciferase. (B) Administration of TLCS (5-500 μM) induced NFAT-luciferase activity. (C) Time course demonstrating accumulation of luminescence with TLCS (500 μM) given over a 6 hr period. (n=3). *, #, P<0.05, relative to the control or TLCS alone, respectively.
Discussion
The cell isolation method and subsequent assays depicted here represent powerful tools with which to study the physiological and pathophysiological features of the exocrine pancreas. The method for isolating dispersed pancreatic acinar cells was first described by Amsterdam and Jamieson in 1972 11. The methods presented here have been adapted from more recent isolation methods described by Van Acker and colleagues 12. Although these techniques are highly reproducible and easily learned, there are several critical features to each method which must be carried out precisely. Understanding these technical details will allow for easier trouble-shooting and improved application.
In preparing the cells for calcium measurements, we've found that the pancreas from younger rodents yields more consistent results. The acini are more evenly distributed, possibly because they contain less fibro-fatty tissue. For Ca2+ measurements, we prepare smaller acini for optimal single cell imaging. This requires a higher collagenase concentration (1.1 mg/ml or 200 U/ml) than that used for LDH measurements. For LDH measurements, we prepare larger acini because this method causes less injury at baseline. The preparation, therefore, receives a lower collagenase concentration (0.5 mg/ml or 91 U/ml).
For Ca2+ measurements, it's critical that cells be plated on acid-washed coverslips. The purpose of the acid-wash is to provide a clean surface as well as electrostatic interactions that will allow maximum adherence of acini. Cell Tak (BD Bioscience) can be used in lieu of or in addition to this method. In addition, it's critical that the Ca2+ dye is loaded in a BSA-free incubation buffer in order (1) to prevent the dye from binding to BSA and (2) to allow the acini to maximally adhere to the acid-washed coverslips.
The perfusion chamber described was custom designed. Commercial chambers and flow systems, however, are available through Warner Instruments. In addition to the perfusion, setting up a properly regulated vacuum is a critical factor in preventing acinar cells from being suctioned off the coverslip.
When measuring calcium signals from pancreatic acinar cells, we describe the use of a confocal microscope. Confocal imaging uses a pinhole in front of a photomultiplier tube in order to eliminate light from above or below the focal plane. In contrast, wide-field imaging detects light throughout the excitation path of the specimen, which allows out of focus light (i.e. light above and below the desired plane of focus) to contaminate the fluorescence image of the specimen. The advantage of confocal microscopy over wide-field is that it allows for thin-section imaging of a sample, as well as 3D reconstruction of optical slices collected along a Z-axis. In addition, confocal microscopy provides high-speed collection systems which can capture fast changes in calcium fluorescence using a spinning disk, line scanner, sweptfield scanner, or even narrowed regions from a point scanner.
Spatial and temporal changes in calcium signaling patterns such as waves and oscillations, respectively, can be reliably measured using the F/F0 calculation, but this is not a true indicator of [Ca2+]. Quantitative measurements of [Ca2+] can be calculated using single wavelength excitation and emission calcium dyes such as Fluo-4, although errors can occur in these measurements due to photobleaching and dye loss 13,14. [Ca2+] can be measured more accurately by using ratiometric dyes, such as Fura2 or Indo115, but this is impractical on most confocal microscopes because of the requirement of a UV laser for excitation. On the other hand, concomitant use of Fluo-3 or Fluo-4 and Fura-Red can allow ratio imaging using the 488 nm excitation line that is present on most confocal microscopes 16.
LDH leakage is a sensitive tool for measuring acinar cell injury 7,17. Another complementary method is propidium iodide uptake 18. Adenovirus infections achieve high infection rates in cultured acini 19. The concentration of virus may need to be titrated. In addition, it is helpful to provide comparable control conditions in which another irrelevant adenoviral construct is also infected. Most of our infection studies last for under 12 hr. However, for longer periods, antibiotics can be added to the DMEM-F12 media at the outset.
Disclosures
No conflict of interest declared.
Acknowledgments
This work was supported by a National Institutes of Health Grant DK083327, and DK093491 (to S.Z.H.).
References
- Toescu EC, Lawrie AM, Petersen OH, Gallacher DV. Spatial and temporal distribution of agonist-evoked cytoplasmic Ca2+ signals in exocrine acinar cells analysed by digital image microscopy. Embo J. 1992;11:1623–1629. doi: 10.1002/j.1460-2075.1992.tb05208.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Miyashita Y, Kasai H. Micromolar and submicromolar Ca2+ spikes regulating distinct cellular functions in pancreatic acinar cells. Embo J. 1997;16:242–251. doi: 10.1093/emboj/16.2.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husain SZ, et al. The ryanodine receptor mediates early zymogen activation in pancreatitis. Proc. Natl. Acad. Sci. U.S.A. 2005;102:14386–14391. doi: 10.1073/pnas.0503215102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raraty M, et al. Calcium-dependent enzyme activation and vacuole formation in the apical granular region of pancreatic acinar cells. Proc. Natl. Acad. Sci. U.S.A. 2000;97:13126–13131. doi: 10.1073/pnas.97.24.13126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orabi AI, et al. Dantrolene mitigates caerulein-induced pancreatitis in vivo in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2009;299:G196–G204. doi: 10.1152/ajpgi.00498.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah AU, et al. Protease Activation during in vivo Pancreatitis is Dependent upon Calcineurin Activation. Am. J. Physiol. Gastrointest. Liver Physiol. 2009. [DOI] [PMC free article] [PubMed]
- Muili KA, et al. Pharmacologic and genetic inhibition of calcineurin protects against carbachol-induced pathologic zymogen activation and acinar cell injury. Am. J. Physiol. Gastrointest. Liver Physiol. 2012. [DOI] [PMC free article] [PubMed]
- Orabi AI, et al. Ethanol enhances carbachol-induced protease activation and accelerates Ca2+ waves in isolated rat pancreatic acini. J. Biol. Chem. 2011;286:14090–14097. doi: 10.1074/jbc.M110.196832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathanson MH, Padfield PJ, O'Sullivan AJ, Burgstahler AD, Jamieson JD. Mechanism of Ca2+ wave propagation in pancreatic acinar cells. J. Biol. Chem. 1992;267:18118–18121. [PubMed] [Google Scholar]
- Reed AM, et al. Low extracellular pH induces damage in the pancreatic acinar cell by enhancing calcium signaling. J. Biol. Chem. 2011;286:1919–1926. doi: 10.1074/jbc.M110.158329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amsterdam A, Jamieson JD. Structural and functional characterization of isolated pancreatic exocrine cells. Proc. Natl. Acad. Sci. U.S.A. 1972;69:3028–3032. doi: 10.1073/pnas.69.10.3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Acker GJ, et al. Tumor progression locus-2 is a critical regulator of pancreatic and lung inflammation during acute pancreatitis. J. Biol. Chem. 2007;282:22140–22149. doi: 10.1074/jbc.M702225200. [DOI] [PubMed] [Google Scholar]
- Ito K, Miyashita Y, Kasai H. Kinetic control of multiple forms of Ca(2+) spikes by inositol trisphosphate in pancreatic acinar cells. J. Cell Biol. 1999;146:405–413. doi: 10.1083/jcb.146.2.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leite MF, Burgstahler AD, Nathanson MH. Ca2+ waves require sequential activation of inositol trisphosphate receptors and ryanodine receptors in pancreatic acini. Gastroenterology. 2002;122:415–427. doi: 10.1053/gast.2002.30982. [DOI] [PubMed] [Google Scholar]
- Paredes RM, Etzler JC, Watts LT, Zheng W, Lechleiter JD. Chemical calcium indicators. Methods. 2008;46:143–151. doi: 10.1016/j.ymeth.2008.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schild D, Jung A, Schultens HA. Localization of calcium entry through calcium channels in olfactory receptor neurones using a laser scanning microscope and the calcium indicator dyes Fluo-3 and Fura-Red. Cell Calcium. 1994;15:341–348. doi: 10.1016/0143-4160(94)90009-4. [DOI] [PubMed] [Google Scholar]
- Saluja AK, et al. Secretagogue-induced digestive enzyme activation and cell injury in rat pancreatic acini. Am. J. Physiol. 1999;276:835–842. doi: 10.1152/ajpgi.1999.276.4.G835. [DOI] [PubMed] [Google Scholar]
- Husain SZ, et al. Ryanodine receptors contribute to bile acid-induced pathological calcium signaling and pancreatitis in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2012. [DOI] [PMC free article] [PubMed]
- Gurda GT, Guo L, Lee SH, Molkentin JD, Williams JA. Cholecystokinin activates pancreatic calcineurin-NFAT signaling in vitro and in vivo. Mol. Biol. Cell. 2008;19:198–206. doi: 10.1091/mbc.E07-05-0430. [DOI] [PMC free article] [PubMed] [Google Scholar]