

Abstract
Recently we have shown that, as a versatile ionization technique, desorption electrospray ionization (DESI) can serve as a useful interface to combine electrochemistry (EC) with mass spectrometry (MS). In this study, the EC/DESI-MS method has been further applied to investigate some aqueous phase redox reactions of biological significance, including the reduction of peptide disulfide bonds and nitroaromatics as well as the oxidation of phenothiazines. It was found that knotted/enclosed disulfide bonds in the peptides apamin and endothelin could be electrochemically cleaved. Subsequent tandem MS analysis of the resulting reduced peptide ions using collision-induced dissociation (CID) and electron-capture dissociation (ECD) gave rise to extensive fragment ions, providing a fast protocol for sequencing peptides with complicated disulfide bond linkages. Flunitrazepam and clonazepam, a class of nitroaromatic drugs, are known to undergo reduction into amines which was proposed to involve nitroso and N-hydroxyl intermediates. Now in this study, these corresponding intermediate ions were successfully intercepted and their structures were confirmed by CID. This provides mass spectrometric evidence for the mechanism of the nitro to amine conversion process during nitroreduction, an important redox reaction involved in carcinogenesis. In addition, the well-known oxidation reaction of chlorpromazine was also examined. The putative transient one-electron transfer product, the chlorpromazine radical cation (m/z 318), was captured by MS, for the first time, and its structure was also verified by CID. In addition to these observations, some features of the DESI-interfaced electrochemical mass spectrometry were discussed, such as simple instrumentation and the lack of background signal. These results further demonstrate the feasibility of EC/DESI-MS for the study of the biology-relevant redox chemistry and would find applications in proteomics and drug development research.
Keywords: Mass spectrometry, Electrochemistry, Desorption electrospray ionization, Disulfide bond reduction, Nitroreduction mechanism, Chlorpromazine radical cation
Introduction
Electrochemical mass spectrometry refers to the online combined electrochemistry (EC) with mass spectrometry (MS) [1]. This technique is powerful in the identification of the electrochemical reaction products or intermediates, leading to extensive applications in bioanalysis and mechanistic studies of redox reactions. The advantage of adopting MS as an EC detector stems from the fact that MS is sensitive and can provide molecular weight information and that tandem MS can be also used for structural analysis based on ion dissociation. Following the seminal work of the first EC/MS coupling carried out by Bruckenstein [2], differential electrochemical mass spectrometry was developed for the online MS detection of gaseous and volatile electrochemical products in real time [3]. Later, the combination of EC with MS was further realized using such ionization methods as thermospray [4], fast atom bombardment [5], and electrospray ionization (ESI) [6–8]. The strength of EC/ESI-MS coupling is that ESI is advantageous in ionizing nonvolatile, polar, thermally labile compounds, and large biological molecules. This method has been used for mimicking biologically relevant electrochemical reactions [9], for the coupling with chromatographic separation [10, 11], for online chemical tagging [12], and for oxidative cleavage of peptides/proteins [9, 13]. Recently, ESI-MS has been combined with scanning electrochemical microscopy to provide not only the mass and structure information of the redox products, but also their spatial distribution near the electrode [14]. The analytical aspects of EC/ESI-MS have been recently reviewed [1, 15, 16]. In general, oxidation reactions were commonly investigated and little attention was given to reduction reactions [17].
Ambient mass spectrometry is a recent advance in the field of MS, which has been introduced to provide direct ionization of analytes with little or no sample preparation. Desorption electrospray ionization (DESI) [18] developed by Cooks and co-workers and direct analysis in real time [19] developed by Cody and co-workers are the first two of this new family of technologies. It has been shown that DESI can be applied to the fast analysis of a variety of different analytes including pharmaceuticals, explosives, drugs of abuse, and tissue imaging [20, 21]. In DESI experiments, ionization of samples occurs via the interactions with charged microdroplets generated in a pneumatically assisted electrospray of an appropriate solvent. DESI has been typically used for solid sample analysis. Recently, DESI has been extended to directly ionize liquid samples in our and other laboratories [22–32]. Owing to its direct sampling capability, several interesting applications of liquid sample DESI have been found, including the coupling of MS with chromatography [27], microfluidics [24], and microextraction [28] as well as probing protein conformation in solution [23]. It was also shown that liquid sample DESI has capabilities for direct ionization of large noncovalent protein complexes and proteins (e.g., 150 kDa IgG antibodies) [29]. It also allows for the development of sub-millisecond time resolved mass spectrometry for the study of fast reaction kinetics [30]. In particular, liquid sample DESI is useful in coupling MS with electrochemical cells [22, 27, 31, 32].
By using the direct sampling DESI as the interface, our EC/DESI-MS method has several unique and valuable features. First, contrary to the previously reported studies for the combination of EC with ESI-MS where the EC system usually needed to be either floated or decoupled from the ESI source, there is no conflict between the small potential applied to the electrochemical cell and the high voltage used for spray ionization in the EC/DESI-MS as the two potentials are physically separated (refer to one EC/DESI-MS apparatus shown in Fig. S1, Electronic Supplementary Material). This leads to simple instrumentation and also the freedom to use either positive or negative ionization modes while performing either reduction or oxidation reactions in the EC cells. Second, as DESI can ionize aqueous samples without addition of organic additives, there is more freedom to employ traditional aqueous solvent compared with EC/ESI-MS. Third, DESI appears to have high salt tolerance. It was shown that, when 10 mM NaCl was used as electrolyte, good MS signal of the analyte could be still detected [31]. Fourth, in some cases like dopamine oxidation [31], there is no background signal from oxidation of the analyte caused by DESI ionization. This feature is beneficial for the detection of reaction intermediates or products from the electrochemical event in the cell with no interference from the MS ionization step [31].
Employing such an EC/DESI-MS coupling device, we investigated several electrochemical reactions involving biological molecules in aqueous solution, including dopamine and thiol oxidation, peptide and protein disulfide bond reduction, and online derivatization of peptides and proteins [22, 27, 31, 32]. In this study, we further implemented our online EC/DESI-MS method for the investigation of several biologically relevant reduction and oxidation reactions, including elucidating redox mechanisms and exploring their analytical applications. Specifically, we aimed to examine the reduction of complicated disulfide linkages in peptides (e.g., two knotted/enclosed disulfide bonds in a peptide), the reduction of aromatic drugs and the oxidation of phenothiazine compounds such as chlorpromazine. The emphasis in this study was given to reduction reactions, for the aforementioned reason that reduction was rarely studied in previous electrochemical mass spectrometric investigations.
Experimental
Chemicals
Apamin (MW, 2,027.3 Da), endothelin (MW, 2,491.9 Da), flunitrazepam, clonazepam, chlorpromazine hydrochloride, and formic acid were purchased from Sigma-Aldrich (St. Louis, MO). Acetic acid and HPLC-grade methanol were obtained from Fisher Scientific (Fair Lawn, NJ) and GFS Chemicals (Columbus, OH), respectively. The deionized water used for sample preparation was obtained using a Nanopure Diamond Barnstead purification system (Barnstead International, Dubuque, IA).
Online EC/DESI-MS apparatus
A home-built apparatus (shown in Fig. S1, Electronic Supplementary Material) for coupling a thin-layer electrochemical flow cell with a Thermo Finnigan LCQ DECA ion trap mass spectrometer (San Jose, CA) by liquid sample DESI was used and described previously in detail [31, 32]. The thin-layer electrochemical flow cell used was either μ-PrepCell or ReactorCell (ANTEC BV, Zoeterwoude, the Netherlands). The μ-PrepCell equipped with a magic diamond (boron-doped diamond) working electrode (30×12 mm2, ANTEC BV, the Netherlands) was employed for the peptide reduction experiments. For electroreduction of disulfide bonds, mercury–gold amalgam electrode was used in our previous study [31, 32]. Now the magic diamond electrode was used to replace the amalgam in this study, for avoiding using toxic mercury electrode. The ReactorCell employed a glassy carbon working electrode (i.d., 8 mm, ANTEC BV, the Netherlands) and a Pt working electrode (i.d., 8 mm, ANTEC BV, the Netherlands) for nitroreduction and for chlorpromazine oxidation, respectively. The selection of glassy carbon and platinum working electrodes in this study followed the literature work [33, 34]. These electrodes were not pre-treated and they were used as received from commercial sources. A ROXY potentiostat (ANTEC BV, the Netherlands) was used to apply potentials to the electrochemical cell for triggering redox conversion of the analytes that flowed through the cell. Sample solutions were prepared in methanol/water containing formic acid as electrolyte and degassed by argon for 20 min to remove dissolved oxygen prior to the injection to the cell by syringe pump for electrolysis. The flow rate of sample solutions passing through the electrochemical cell for electrolysis was 4~8 μL/min unless specified otherwise. The product flowed out of the cell via a short piece of fused silica connection capillary (i.d., 0.1 mm, length 4.0 cm) and underwent interactions with the charged microdroplets from DESI solvent spray for ionization. The DESI spray probe was aimed at the mass spectrometer’s inlet orifice and kept 3~4 cm away from the mass spectrometer inlet (Fig. S1, Electronic Supplementary Material). The capillary outlet was placed about 1 mm downstream from the DESI spray probe tip and kept in line with the sprayer tip and the mass spectrometer’s inlet. The spray solvent for DESI was methanol/water (1:1 by volume) containing 1% acetic acid and injected at a rate of 5 μL/min with a high voltage of 5 kV applied to the sprayed solvent. The nebulizing gas (N2) pressure for DESI spray probe was 160 psi.
Electron-capture dissociation (ECD) spectra were collected using a Bruker 12 Tesla SolariX Fourier-Transfer Ion Cyclotron mass spectrometer (FT-ICR-MS, Bruker Daltonics, Bremen, Germany). In the experiment, the cathode filament of the FT-ICR instrument was conditioned at 1.6 A. To record the spectra, the ECD lens was set to 10 V, and a pulse length of 100 ms and ECD bias of 1.2 V were employed. Each acquired spectrum was the average of 100~120 broadband 1 M time-domain transient.
Results and discussion
Reduction of peptides containing two disulfide bonds
Redox-active disulfide bonds are one of the most common protein post-translational modifications (PTMs) and provide reversible covalent cross-linkages in native proteins for maintaining the three-dimensional structures of proteins and their biological activities [35]. Such linkages play a critical role in determining the activity of enzymes and are also a key structural feature of biologically active peptide hormones. The correct connection configuration of disulfide bonds is critical for the development of functional protein pharmaceuticals [36]. The presence of the disulfide linkages increases the complexity for the protein structure elucidation by MS. The cleavage of disulfide bonds is often essential for the protein/peptide analysis as dissociation of a reduced protein/peptide ion can give rise to more structurally informative fragment ions than that of the intact counterpart [35]. The traditional approach for breaking disulfide involves using an excess amount of reagents like dithiothreitol (DTT) or tris (2-carboxyethyl)phosphine (TCEP). However, the reduction usually takes one half to several hours and the removal of the excess amount of reductant is time-consuming and troublesome. An alternative way for reducing disulfide bonds without involving chemical reductants is electrolytic reduction. Previous electrochemical investigations showed that such a reduction followed by electrolytic oxidation of the resulting thiols back to disulfides in an electrochemical flow cell can be coupled with either HPLC or electrophoresis separation, which are useful for the simultaneous detection of thiol- and disulfide-containing peptides in mixtures [37].
Our recent study showed that the peptides containing disulfide bonds could be quickly identified from enzymatic digestion mixtures using this EC/DESI-MS method, simply based on the abrupt decrease in their relative ion abundances after electrolysis [32]. Peptide mass mapping and tandem MS analysis of the ions of the resulting free peptides can potentially establish the disulfide linkage pattern and sequence the precursor peptides. Alternatively, peptides in an enzymatic digest can be first separated by liquid chromatography and then undergo electrochemical reduction followed by online MS detection. Using the latter approach, the reduced products can be explicitly correlated to their precursor peptides and further tandem MS analysis of the resulting products provides the precursor peptide structure [27]. Although some interesting results were obtained, peptides with complicated disulfide linkage patterns (e.g., multiple knotted or enclosed disulfides in a peptide) were not well explored previously, which were examined in this work. In the present study, the peptides apamin and endothelin were chosen for investigation using the EC/DESI-MS method.
Reduction of apamin
Apamin (MW, 2,027.3 Da) has 18 amino acid residues in a single peptide chain and carries two knotted disulfide bonds located at Cys1–Cys11 and Cys3–Cys15 (see its sequence in the inset of Fig. 1a). The peptide C terminus was modified with an amide group to replace the carboxylic acid group. Figure 1a shows the DESI-MS spectrum acquired when a solution of 20 μM apamin in methanol/water (1:1 by volume) containing 0.5% formic acid flowed through the thin-layer electrochemical cell with no potential applied. Peaks corresponding to the quardruply ([apamin+4H]4+), triply ([apamin+3H]3+), and doubly ([apamin+2H]2+) charged peptide ions were detected at m/z 507.8, 676.7 and 1014.4, respectively. When a negative potential of −1.5 V was applied to the working electrode to trigger reduction, these peaks shifted in mass to m/z of 508.9, 678.1, and 1,016.0, respectively (Fig. 1b). The calculated mass difference of the peptide before and after the reduction is approximately 4 Da, indicating the two disulfide bonds of apamin were fully reduced. In this experiment, the complete reduction of the peptide in the flow cell is probably ascribed to the large surface area (30×12 mm2) and good electrochemical performance of the magic diamond working electrode.
Fig. 1.

DESI-MS spectra acquired when a solution of 20 μM apamin in methanol/water (1:1 by volume) containing 0.5% formic acid flowed through the thin-layer electrochemical cell with an applied potential of (a) 0.0 V and (b) −1.5 V; CID MS/MS spectra of (c) [apamin+4H]4+ (m/z 507.9) before reduction and (d) [reduced apamine+4H]4+ (m/z 508.8) after reduction; ECD MS/MS spectra of (e) [apamin+4H]4+ (m/z 507.8) before reduction and (f) [reduced apamine+4H]4+ (m/z 508.5) after reduction
The quardruply charged peptide ion [apamin+4H]4+ was chosen for tandem MS analysis. When the cell was off, the fragmentation of [apamin+4H]4+ (m/z 507.9) was very limited and only b173+ ion was observed (Fig. 1c). There are some other peaks present in the MS/MS spectrum that could not be assigned and might be from sample impurity. After reduction, in the CID spectrum of [reduced apamin+4H]4+ (m/z 508.8; Fig. 1d), many more fragment ions were observed and identified, including b2, b3, b4, b6, b7, b163+, b173+, y92+, y102+, y112+, y113+, y122+, y132+, y133+, y142+, y143+, y153+, y162+, y163+, and y164+ (these fragment ions are also summarized in Table 1). The formation of these fragment ions further confirms the complete reduction of the two disulfide bonds during electrolysis because CID is typically not able to cleave the disulfide bond in the positive ion mode. In other words, these fragments ions result from the cleavage of the backbone enclosed by the two disulfide bonds otherwise would be missing in the MS/MS spectrum.
Table 1.
Summary of the fragmentation of apamin and endothelin ions before and after reduction
| Peptide ions | Fragment ions observed
|
||
|---|---|---|---|
| Before electrolysis | After electrolysis | ||
| +4 apamin ion | CID | b173+ | b2, b3, b4, b6, b7, b163+, b173+, y92+, y102+, y112+, y113+, y122+, y132+, y133+, y142+, y143+, y153+, y162+, y163+, y164+ |
| ECD | c152+, c162+, c172+ | c3, c4, c6, c7, c8, c9, c10, c11, c12, c13, c132+, c14, c142+, c152+, c162+, c172+ | |
| +4 endothelin ion | CID | b172+, b182+, b192+, b202+ | b5, b8, b12, b13, b14, b15, b16, b17, b172+, b182+, b192+, b202+, y4, y5, y6, y7, y8, y9, y10, y11, y12, y13, y15 |
With both intact and reduced peptide ions at hand, one could also adopt a different tandem MS approach, for instance, ECD [38]. ECD is a powerful dissociation technique which often gives rise to extensive backbone cleavages for multiply charged protein or peptide ions. In ECD experiments, low-energy electrons are reacted with peptide cations in the ICR cell located in the center of the magnetic field of a FT-ICR-MS [38]. In this study, ECD was also employed to dissociate the intact and reduced +4 apamin ions. As shown in Fig. 1e, ECD of the intact apamin ion [apamin+4H]4+ (m/z 507.8) also produces limited number of fragment ions c152+, c162+, and c172+, in addition to the charge reduced product +3 apamin ion. Although ECD is able to cleave both disulfide bonds and protein backbone bonds [39], the dissociation efficiency of the backbone in this case is low. It is likely that the presence of the knotted disulfide bonds preferentially capture the electrons and thereby prevents the effective dissociation of the peptide bonds [40]. In contrast, after reduction, ECD of [reduced apamin+4H]4+ (m/z 508.5) gives rise to extensive backbone cleavage, yielding a nearly complete set of c ions such as c3, c4, c6, c7, c8, c9, c10, c11, c12, c13, c132+, c14, c142+, c152+, c162+, and c172+ (see Fig. 1f and also Table 1). The absence of c5 is due to the presence of the proline in the 6th position of the peptide sequence and the absence of c1 and c2 is probably due to their m/z that are out of the recorded range in the ICR instrument. With the combined CID and ECD data of [reduced apamin+4H]4+ obtained, the full sequence of the peptide except for the first two residues in the N terminus can be determined, suggesting the utility of the EC/DESI-MS method in conjunction with tandem MS analysis in peptide sequencing. This result demonstrates that EC/DESI-MS is applicable for the analysis of peptides with knotted disulfide bonds. Compared with the chemical reduction method using DTT, it is much faster and more convenient with no need to get rid of excess reductant, which might be of high value in proteomics applications. Note that a small negative potential was used for EC reduction and a high positive potential was used for ionizing the products by MS. The EC/DESI-MS allows for such a combination as the two potentials are physically separated, as mentioned before.
Reduction of endothelin
Endothelin (MW: 2491.9 Da) is another single chain peptide carrying two disulfide bonds Cys1–Cys15 and Cys3–Cys11, in which the latter is enclosed by the former disulfide bond (see the peptide sequence in the inset of Fig. 2a). Figure 2a shows the DESI-MS spectrum acquired when a solution of 20 μM endothelin in methanol/water (1:1 by volume) containing 0.5% formic acid flowed through the cell with no potential applied. Peaks corresponding to the doubly ([endothelin+2H]2+) and triply ([endothelin +3H]3+) charged peptide were detected at m/z 1,246.6 and 831.6, respectively. When a negative potential of −1.5 V was applied to the working electrode, a positive mass shift occurred and the doubly and triply reduced peptide ions appeared at m/z 1,248.9 and 833.2, respectively (Fig. 2b). The mass shift of approximately 4 Da again indicates that both disulfide bonds of the peptide were reduced. The reduction yield was also high in this case. Figure 2c displays the CID spectrum of [endothelin +2H]2+ (m/z 1246.6) in which only few fragment ions b172+, b182+, b192+, and b202+ appeared. The cleavage of the region enclosed by the two disulfide bonds was missing. After reduction, CID of [reduced endothelin +2H]2+ (m/z 1248.9) evidently provides more fragment ions b5, b8, b12, b13, b14, b15, b16, b17, b172+, b182+, b192+, b202+, y4, y5, y6, y7, y8, y9, y10, y11, y12, y13, and y15 (Fig. 2d and Table 1). Again, in this case, more sequence information was obtained using the online EC/DESI-MS method.
Fig. 2.

DESI-MS spectra acquired when a solution of 20 μM endothelin in methanol/water (1:1 by volume) containing 0.5% formic acid flowed through the thin-layer electrochemical cell with an applied potential of (a) 0.0 V and (b) −1.5 V; CID MS/MS spectra of (c) [endothelin +2H]2+ (m/z 1,246.6) and (d) [reduced endothelin +2H]2+ (m/z 1,248.9)
Reduction of nitroaromatics
Nitroreduction is an important step in N-directed metabolic activation and is responsible for covalent binding of nitroarenes to macromolecules, leading to carcinogenesis [41]. It is believed that the nitro group can either be partially reduced to form a nitroso or an N-hydroxyl intermediate or be completely reduced to an amine [42]. The amine can be reoxidized to form N-hydroxyl products and be further oxidized in the erythrocyte to form a nitroso compound. Electrochemical reduction of the nitro group was extensively studied previously and the similar mechanism was proposed [33, 43]. However, no attempt has been made yet to detect the reduction intermediates/products by electrochemical mass spectrometry.
Flunitrazepam (refer to its structure in Fig. 3a) is a potent sedative and powerful hypnotic used in the treatment of insomnia around the world. About ten times more potent than diazepam, this drug reduces anxiety, inhibition, and muscular control and can also cause anterograde amnesia and loss of consciousness [44]. Figure 3a shows the DESI-MS spectrum acquired when a solution of 20 μM flunitrazepam in methanol/water (1:1 by volume) containing 0.5% formic acid and 20 mM NH4OAc flowed through the thin-layer electrochemical cell with no potential applied. The dominant protonated flunitrazepam at m/z 314 was observed and the peak at m/z 310 was from background contribution (Fig. 3a). CID of m/z 314 yields the fragment ions m/z 268 and 240 by consecutive losses of CO and NO2, consistent with the flunitrazepam ion structure (Fig. S2a, Electronic Supplementary Material). Direct loss of CO due to ring contraction was also noted to form fragment ion of m/z 286. When a negative potential of −1.5 V was applied to the glassy carbon working electrode, the protonated ion of m/z 314 decreased and some new ions of m/z 284, 298, and 300 appeared (Fig. 3b). The formation of these ions can be rationalized using the electroreduction mechanism proposed in Scheme 1. First, the nitro group of flunitrazepam 1a undergoes a two e− reduction process with loss of one molecule of water to form a nitroso intermediate 2a. The newly produced ion of m/z 298 in Fig. 3b corresponds to the protonated nitroso intermediate. This can be confirmed from the characteristic loss of NO from this ion upon CID (Fig. S2c, Electronic Supplementary Material). MS3 also shows the further loss of CH2=NH from the resulting fragment ion of m/z 268 into m/z 239 (Fig. S2d, Electronic Supplementary Material). 2a further undergoes two e− reduction to yield the N-hydroxyl intermediate 3a, as evidenced by the appearance of the corresponding protonated ion of m/z 300 in Fig. 3b. The m/z 300 dissociates by losses of OH. and further NH2. radicals (Fig. S2e and f, Electronic Supplementary Material), which might be due to the weak bond of N–O in the hydroxylamine group of 3a. Finally, further reduction followed by another water loss from 3a to yield the amine product 4a (Scheme 1) and the corresponding ion of 4a is shown at m/z 284 (Fig. 3b). Fragmentation of m/z 284 displays characteristic fragmentation behaviors such as loss of HF, CO, CH2=NH, and PhNH2, confirming its structure (Fig. S2b, Electronic Supplementary Material). These results clearly show that all the nitroreduction intermediates and the final amine product can be intercepted in this EC/DESI-MS experiment, providing mass spectrometric evidence to support the widely accepted nitroreduction mechanism.
Fig. 3.

DESI-MS spectra acquired when a solution of 20 μM flunitrazepam in methanol/water (1:1 by volume) containing 0.5% formic acid and 20 mM NH4OAc flowed through the thin-layer electrochemical cell with an applied potential of (a) 0.0 V and (b) −1.5 V; DESI-MS spectra acquired when a solution of 20 μM clonazepam in methanol/water (1:1 by volume) containing 0.5% formic acid and 20 mM NH4OAc flowed through the thin-layer electrochemical cell with an applied potential of (c) 0.0 V and (d) −1.5 V
Scheme 1.

Proposed mechanism for the electroreduction of flunitrazepam
Clonazepam has a similar structure to flunitrazepam and the only structural difference is that F is replaced by Cl and there is no CH3 substituent group in clonazepam. In order to testify the electrolytic reduction mechanism proposed above for the reduction of nitro compounds, clonazepam was also studied using the online EC-DESI/MS method. The DESI-MS spectra of clonazepam before and after the potential are shown in Fig. 3c, d, respectively. Very analogous reaction outcome to that of the flunitrazepam reduction was observed in the case of clonazepam. Before the cell was turned on, there was only the protonated clonazepam ion (m/z 316) observed in Fig. 3c in addition to the background peak at m/z 310. When −1.5 V potential was applied for reduction, the nitroso intermediate 2b, N-hydroxyl intermediate 3b and amine final reduction product 4b (Scheme 2) from the electrolytic reduction were all observed and the corresponding ions appeared at m/z 300, 302, and 286, respectively (Fig. 3d). The assignment of these newly generated ions was confirmed by the acquired CID spectra (see the spectra and discussion in Fig. S3, Electronic Supplementary Material). This result further confirms the nitroaromatic compound reduction process involving nitroso, N-hydroxyl, and amine products.
Scheme 2.

Proposed mechanism for the electroreduction of clonazepam
Oxidation of chlorpromazine
As one of the most important compounds in the phenothiazine class of drugs, chlorpromazine has been widely used in the treatment of schizophrenia. It was proposed [33, 45, 46] that chlorpromazine can easily undergo oxidation in acid medium, in which a one e− oxidation process leads to the formation of a radical cation intermediate. It is known that, while the radical cation of phenothiazine is comparably stable, the radical cations of the substituted phenothiazines are believed to be unstable and could further react with solvent to give rise to sulfoxide [47]. Previously, Van Berkel et al. reported the observation of radical cations of analytes including phenothiazine generated from the electrospray needle [48]. Karst et al. [49] used LC/EC/MS to study phenothiazine and its derivatives, in which some radical cations and other oxidation products were detected by MS and the oxidation pathways were identified. Online EC/ESI-MS [50] was also used to investigate the electrochemical oxidation of zotepine and chlorpromazine. However, no detection of chlorpromazine radical cation by MS was reported, which might be due to the instability and short lifetime of the intermediate. In this experiment, the online EC/DESI-MS apparatus was utilized to investigate the electrochemical oxidation of chlorpromazine in conjunction with tandem MS analysis.
When 20 μM chlorpromazine in methanol/water (1:1 by volume) containing 0.5% formic acid was allowed to flow through the thin-layer electrochemical cell without any potential applied, the acquired DESI-MS spectrum (Fig. 4a) shows only the protonated chlorpromazine at m/z 319 and its chlorine isotope peak at m/z 321. There was no background oxidation signal of the chlorpromazine radical cation caused by DESI ionization. This is advantageous for using MS to probe electrochemical reactions as the background signal is unwanted. Recently, there has been account reporting the unexpected oxidation of analytes observed during ionization by traditional DESI when solid samples were ionized [51]. In our experiment of liquid sample DESI, the oxidation from ionization due to the high voltage is inhibited probably due to the presence of solvents in the sample, which may not favor electron transfer between the analyte molecule and the impacting charged droplets. Another reason for absence of the background signal in the DESI experiment is that, unlike traditional ESI experiments, the analyte sample was not flowed through the DESI sprayer where a high voltage was applied. This observation is in agreement with our previous EC/DESI-MS study of dopamine [31].
Fig. 4.

DESI-MS spectra acquired when a solution of 20 μM chlorpromazine in methanol/water (1:1 by volume) containing 0.5% formic acid flowed through the thin-layer electrochemical cell with an applied potential of (a) 0.0 V and (b) +1.5 V; (c) CID MS/MS spectrum of the chlorpromazine radical cation (m/z 318)
When the electrochemical cell was turned on with an applied potential of +1.5 V, the chlorpromazine radical cation of m/z 318 arose (Fig. 4b). CID dissociation of m/z 318 (Fig. 4c) produces mainly the fragment ions of m/z 283, 273, and 233 by losses of Cl, NH(CH3)2, and CH2=CHCH2N (CH3)2, respectively, confirming its structure. To the best of our knowledge, this result represents the detection of the transient chlorpromazine radical cation by MS for the first time. The success in the detection of m/z 318 is presumably ascribed to the online experiment of EC/DESI-MS in which the response time is short (in seconds depending on the flow rate of the sample infused into the electrochemical cell). Indeed, when we tried an offline experiment using ESI to ionize the electrochemically oxidized chlorpromazine, it failed to detect this radical cation. We further investigated the influence of the sample flow rate on the detection of m/z 318 in this EC/DESI-MS experiment. When the flow rate was lowered from 5 to 2 or 4 μl/min, no radical cation was observed (Fig. S4 shows the acquired DESI-MS spectra at different sample flow rates, Electronic Supplementary Material). As the delay time of EC/DESI-MS (i.e., the time interval between the onset of the electrochemical oxidation and the detection of the product ions by MS) for 5 μl/min flow rate was approximately 0.25 min, the lifetime of the chlorpromazine radical cation is estimated to be around 15 seconds, under the experimental conditions that was used in our experiment. This estimated life time of the chlorpromazine radical cation is in line with the previous report [46]. In Fig. 4b, another major oxidation product corresponding to the protonated chlorpromazine sulfoxide at m/z 335 was observed, probably due to the subsequent chemical reaction between the chlorpromazine radical cation and water (a typical EC mechanism, see the mechanism proposed in Scheme 3). In addition, another peak at m/z 349 was seen, corresponding to a methoxylated chlorpromazine ion. It is likely the result of the reaction between chlorpromazine radical cation and methanol. When the solvent was changed to ethanol, the corresponding peak was observed at m/z 363. The high reactivity of the chlorpromazine radical cation explains its short lifetime. These results clearly show that the electrochemically oxidized chlorpromazine, the radical cation, along with other oxidation products can be clearly studied using this EC/DESI-MS approach.
Scheme 3.
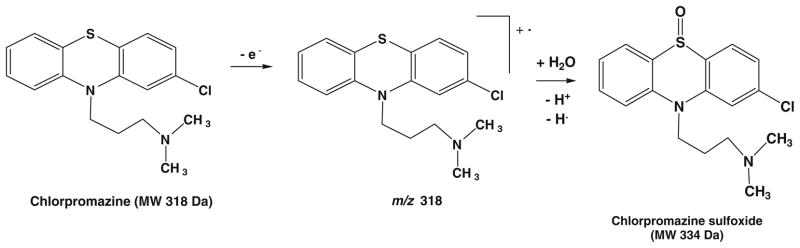
Proposed mechanism for the electro-oxidation of chlorpromazine involving the formation of chlorpromazine radical cation which subsequently reacts with water to produce chlorpromazine sulfoxide
Conclusions
In conclusion, biologically important redox reactions, including the reduction of peptide disulfide bonds and nitroaromatics and the oxidation of chlorpromazine, were examined using electrochemical mass spectrometry interfaced by DESI. Several interesting findings in this study have been uncovered. In the stark contrast to intact peptide ions of apamin and endothelin which display few backbone dissociation cleavages upon CID and ECD, fragmentation was greatly enhanced following the reduction, which is useful for the peptide sequencing. This experiment is fast and does not involve the use of any chemical reductants. In addition to disulfide reductive cleavage, nitroreduction was also tested as exemplified by the reduction of nitroaromatic, flunitrazepam, and clonazepam. Remarkably, all nitroso intermediates, N-hydroxyl intermediates and amine final products from the electroreduction were successfully detected, proving a solid mass spectrometric evidence for the nitroreduction mechanism. Likewise, the putative transient one-electron transfer product, the chlorpromazine radical cation (m/z 318), was captured by MS, for the first time. These results suggest that electrochemical mass spectrometry, including the one interfaced by DESI as demonstrated in this study, opens a door to use MS to probe transient species involved in electrochemical redox process. It is expected that the EC/DESI-MS method would find important applications in proteomics and drug development research. As the intensity of ion signal in EC/DESI-MS experiments can be affected by positions of the DESI sprayer and the sample transfer capillary outlet, future optimization of the apparatus would include the integration of positioning elements such as x, y, z-stages into the DESI ion source to precisely control those positions for improved performance.
Supplementary Material
Acknowledgments
The support of this work by NSF (CHE-0911160) and ASMS Research Award was gratefully acknowledged. We are thankful to Prof. Michael L. Gross for the access to the NIH/NCRR Mass Spectrometry Resources at Washington University in St. Louis (Grant number 2P41RR000954) and High-End Instrument Program of the NCRR (Grant No. 1S10 025101). In particular, we thank ANTEC BV Company and Mr. Martin Eysberg very much for their generous support in this electrochemical mass spectrometry study.
Footnotes
Published in the special paper collection on Electrochemistry–Mass Spectrometry with guest editors Uwe Karst and Martin Vogel.
Electronic supplementary material The online version of this article (doi:10.1007/s00216-011-5679-7) contains supplementary material, which is available to authorized users.
Contributor Information
Mei Lu, Center for Intelligent Chemical Instrumentation, Department of Chemistry and Biochemistry, Ohio University, Athens, OH 45701, USA.
Chloe Wolff, Center for Intelligent Chemical Instrumentation, Department of Chemistry and Biochemistry, Ohio University, Athens, OH 45701, USA.
Weidong Cui, Department of Chemistry, Washington University, St. Louis, MO 63130, USA.
Hao Chen, Email: chenh2@ohio.edu, Center for Intelligent Chemical Instrumentation, Department of Chemistry and Biochemistry, Ohio University, Athens, OH 45701, USA.
References
- 1.Gun J, Bharathi S, Gutkin V, Rizkov D, Voloshenko A, Shelkov R, Sladkevich S, Kyi N, Rona M, Wolanov Y, Rizkov D, Koch M, Mizrahi S, Pridkhochenko PV, Modestov A, Lev O. Highlights in coupled electrochemical flow cell-mass spectrometry, EC/MS. Israel J Chem. 2010;50:360–373. [Google Scholar]
- 2.Bruckenstein S, Gadde RR. Use of a porous electrode for in situ mass spectrometric determination of volatile electrode reaction products. J Am Chem Soc. 1971;93:793–794. [Google Scholar]
- 3.Wolter O, Heitbaum J. Differential electrochemical mass-spectroscopy (Dems)—a new method for the study of electrode processes. Ber Bunsen-Ges Phys Chem. 1984;88:2–6. [Google Scholar]
- 4.Hambitzer G, Heitbaum J. Electrochemical thermospray mass spectrometry. Anal Chem. 1986;58:1067–1070. doi: 10.1021/ac970753c. [DOI] [PubMed] [Google Scholar]
- 5.Bartmess JE, Phillips LR. Electrochemically assisted fast atom bombardment mass spectrometry. Anal Chem. 1987;59:2012–2014. doi: 10.1002/bms.1200181007. [DOI] [PubMed] [Google Scholar]
- 6.Zhou F, Berkel GJV. Electrochemistry combined online with electrospray mass spectrometry. Anal Chem. 1995;67:3643–3649. [Google Scholar]
- 7.Bond AM, Colton R, D’Agostino A, Downard AJ, Traeger JC. A role for electrospray mass spectrometry in electrochemical studies. Anal Chem. 1995;67:1691–1695. [Google Scholar]
- 8.Xu X, Lu W, Cole RB. On-line probe for fast electrochemistry/electrospray mass spectrometry. Investigation of polycyclic aromatic hydrocarbons. Anal Chem. 1996;68:4244–4253. doi: 10.1021/ac960362i. [DOI] [PubMed] [Google Scholar]
- 9.Permentier HP, Jurva U, Barroso B, Bruins AP. Electrochemical oxidation and cleavage of peptides analyzed with on-line mass spectrometric detection. Rapid Commun Mass Spectrom. 2003;17:1585–1592. doi: 10.1002/rcm.1090. [DOI] [PubMed] [Google Scholar]
- 10.Deng H, Van Berkel GJ, Takano H, Gazda D, Porter MD. Electrochemically modulated liquid chromatography coupled on-line with electrospray mass spectrometry. Anal Chem. 2000;72:2641–2647. doi: 10.1021/ac991461+. [DOI] [PubMed] [Google Scholar]
- 11.Lohmann W, Baumann A, Karst U. Electrochemistry and LC-MS for metabolite generation and identification: tools, technologies, and trends. LC GC North America. 2010;28:470–476. [Google Scholar]
- 12.Roussel C, Dayon L, Lion N, Rohner TC, Josserand J, Rossier JS, Jensen H, Girault HH. Generation of mass tags by the inherent electrochemistry of electrospray for protein mass spectrometry. J Am Soc Mass Spectrom. 2004;15:1767–1779. doi: 10.1016/j.jasms.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 13.Permentier HP, Bruins AP. Electrochemical oxidation and cleavage of proteins with on-line mass spectrometric detection: development of an instrumental alternative to enzymatic protein digestion. J Am Soc Mass Spectrom. 2004;15:1707–1716. doi: 10.1016/j.jasms.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 14.Modestov AD, Srebnik S, Lev O, Gun J. Scanning capillary microscopy/mass spectrometry for mapping spatial electrochemical activity of electrodes. Anal Chem. 2001;73:4229–4240. doi: 10.1021/ac001369+. [DOI] [PubMed] [Google Scholar]
- 15.Karst U. Electrochemistry/mass spectrometry (EC/MS)—a new tool to study drug metabolism and reaction mechanisms. Angew Chem Intl Ed. 2004;43:2476–2478. doi: 10.1002/anie.200301763. [DOI] [PubMed] [Google Scholar]
- 16.Permentier HP, Bruins AP, Bischoff R. Electrochemistry-mass spectrometry in drug metabolism and protein research. Mini-Rev Med Chem. 2008;8:46–56. doi: 10.2174/138955708783331586. [DOI] [PubMed] [Google Scholar]
- 17.Johnson KA, Shira BA, Anderson JL, Amster IJ. Chemical and on-line electrochemical reduction of metalloproteins with high-resolution electrospray ionization mass spectrometry detection. Anal Chem. 2001;73:803–808. doi: 10.1021/ac001004p. [DOI] [PubMed] [Google Scholar]
- 18.Takats ZW, Wiseman JM, Gologan B, Cooks RG. Mass spectrometry sampling under ambient conditions with desorption electrospray ionization. Science. 2004;306:471–473. doi: 10.1126/science.1104404. [DOI] [PubMed] [Google Scholar]
- 19.Cody RB, Laramee JA, Durst HD. Versatile new ion source for the analysis of materials in open air under ambient conditions. Anal Chem. 2005;77:2297–2302. doi: 10.1021/ac050162j. [DOI] [PubMed] [Google Scholar]
- 20.Wiseman JM, Puolitaival SM, Takats Z, Cooks RG, Caprioli RM. Mass spectrometric profiling of intact biological tissue by using desorption electrospray ionization. Angew Chem Int Ed. 2005;44:7094–7097. doi: 10.1002/anie.200502362. [DOI] [PubMed] [Google Scholar]
- 21.Denes J, Katona M, Hosszu A, Czuczy N, Takats Z. Analysis of biological fluids by direct combination of solid phase extraction and desorption electrospray ionization mass spectrometry. Anal Chem. 2009;81:1669–1675. doi: 10.1021/ac8024812. [DOI] [PubMed] [Google Scholar]
- 22.Miao Z, Chen H. Direct analysis of liquid samples by desorption electrospray ionization-mass spectrometry (DESI-MS) J Am Soc Mass Spectrom. 2009;20:10–19. doi: 10.1016/j.jasms.2008.09.023. [DOI] [PubMed] [Google Scholar]
- 23.Miao Z, Wu S, Chen H. Probing protein conformations in solution via direct sampling by desorption electrospray ionization mass spectrometry. J Am Soc Mass Spectrom. 2010;21:1730–1736. doi: 10.1016/j.jasms.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ma X, Zhao M, Lin Z, Zhang S, Yang C, Zhang X. Versatile platform employing desorption electrospray ionization mass spectrometry for high-throughput analysis. Anal Chem. 2008;80:6131–6136. doi: 10.1021/ac800803x. [DOI] [PubMed] [Google Scholar]
- 25.Chipuk JE, Brodbelt JS. Transmission mode desorption electrospray ionization. J Am Soc Mass Spectrom. 2008;19:1612–1620. doi: 10.1016/j.jasms.2008.07.002. [DOI] [PubMed] [Google Scholar]
- 26.Zhang Y, Chen H. Detection of saccharides by reactive desorption electrospray ionization (DESI) using modified phenyl-boronic acids. Int J Mass Spectrom. 2010;289:98–107. [Google Scholar]
- 27.Zhang Y, Yuan Z, Dewald HD, Chen H. Coupling of liquid chromatography with mass spectrometry by desorption electrospray ionization (DESI) Chem Commun. 2011;47:4171–4173. doi: 10.1039/c0cc05736c. [DOI] [PubMed] [Google Scholar]
- 28.Sun X, Miao Z, Yuan Z, Harrington PB, Colla J, Chen H. Coupling of single-droplet microextraction with desorption electrospray ionization mass spectrometry. Int J Mass Spectrom. 2010;301:102–110. [Google Scholar]
- 29.Ferguson CN, Benchaar SA, Miao ZX, Loo JA, Chen H. Direct ionization of large proteins and protein complexes by desorption electrospray ionization-mass spectrometry. Anal Chem. 2011;83:6468–6473. doi: 10.1021/ac201390w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miao Z, Chen H, Liu P, Liu Y. Development of submilli-second time-resolved mass spectrometry using desorption electrospray ionization. Anal Chem. 2011;83:3994–3997. doi: 10.1021/ac200842e. [DOI] [PubMed] [Google Scholar]
- 31.Li J, Dewald HD, Chen H. Online coupling of electrochemical reactions with liquid sample desorption electrospray ionization-mass spectrometry. Anal Chem. 2009;81:9716–9722. doi: 10.1021/ac901975j. [DOI] [PubMed] [Google Scholar]
- 32.Zhang Y, Dewald HD, Chen H. On-line mass spectrometric analysis of proteins/peptides following electrolytic reduction of disulfide bonds. J Proteome Res. 2011;10:1293–1304. doi: 10.1021/pr101053q. [DOI] [PubMed] [Google Scholar]
- 33.Morales A, Richter P, Toral MI. Voltammetric behavior of nitrofurazone, furazolidone and other nitro-derivatives of biological importance. Analyst. 1987;112:965–970. doi: 10.1039/an9871200965. [DOI] [PubMed] [Google Scholar]
- 34.Merkle FH, Discher CA. Electrochemical oxidation of chlorpromazine hydrochloride. J Pharmaceut Sci. 1964;53:620–623. doi: 10.1002/jps.2600530610. [DOI] [PubMed] [Google Scholar]
- 35.Bilusich D, Bowie JH. Feagmentations of (M-H)-anions of underivatised peptides. Part 2: characteristic cleavages of ser and Cys and of disulfides and other post-translational modifications, together with unusual internal processes. Mass Spectrom Rev. 2009;28:20–34. doi: 10.1002/mas.20206. [DOI] [PubMed] [Google Scholar]
- 36.Zhang M, Kaltashov IA. Mapping of protein disulfide bonds using negative ion fragmentation with a broadband precursor selection. Anal Chem. 2006;78:4820–4829. doi: 10.1021/ac060132w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhong M, Lunte SM. Tubular-wire dual electrode for detection of thiols and disulfides by capillary electrophesis/electrochemistry. Anal Chem. 1999;71:251–255. doi: 10.1021/ac9806198. [DOI] [PubMed] [Google Scholar]
- 38.Zubarev RA, Kelleher NL, McLafferty FW. Electron capture dissociation of multiply charged protein cations. A nonergodic process. J Am Chem Soc. 1998;120:3265. doi: 10.1021/ac990811p. [DOI] [PubMed] [Google Scholar]
- 39.Zubarev RA, Kruger NA, Fridriksson EK, Lewis MA, Horn DM, Carpenter BK, McLafferty FW. Electron capture dissociation of gaseous multiply-charged proteins is favored at disulfide bonds and other sites of high hydrogen atom affinity. J Am Chem Soc. 1999;121:2857–2862. [Google Scholar]
- 40.Chen J, Shiyanov P, Zhang L, Schlager JJ, Green-Church KB. Top-down characterization of a native highly intralinked protein: concurrent cleavages of disulfide and protein backbone bonds. Anal Chem. 2010;82:6079–6089. doi: 10.1021/ac1006766. [DOI] [PubMed] [Google Scholar]
- 41.Malaveille C, Croisy A, Brun G, Bartsch H. Hydroxylation and nitroreduction are required to activate dimethylnitramine into alkylating and mutagenic agents. Carcinogenesis. 1983;4:1477–1481. doi: 10.1093/carcin/4.11.1477. [DOI] [PubMed] [Google Scholar]
- 42.Suzuki J, Meguro SI, Morita HS, Suzuki S. Comparison of in vivo binding of aromatic nitro compounds to rat hemoglobin. Biochem Pharmacol. 1989;38:3511–3519. doi: 10.1016/0006-2952(89)90122-6. [DOI] [PubMed] [Google Scholar]
- 43.Honeychurch KC, Hart JP. Determination of flunitrazepam and nitrazepam in beverage samples by liquid chromatography with dual electrode detection using a carbon fibre veil electrode. J Solid State Electrochem. 2008;12:1317–1324. [Google Scholar]
- 44.Gahlinger P. Club drugs: MDMA, gamma-hydroxybutyrate (GHB), rohypnol, and ketamine. Am Fam Physician. 2004;69:2619–2627. [PubMed] [Google Scholar]
- 45.Cheng HY, Sackett PH, McCreery RL. Kinetics of chlorpromazine cation radical decomposition in aqueous buffers. J Am Chem Soc. 1978;100:962–967. [Google Scholar]
- 46.Madej E, Wardman P. Pulse radiolysis and cyclic voltammetry studies of redox properties of phenothiazine radicals. Radiation Phys Chem. 2006;75:990–1000. [Google Scholar]
- 47.Pankratov AN, Uchaeva IM, Stepanov AN. Chemical and electrochemical oxidation of phenothiazine. Can J Chem-Rev Can Chim. 1993;71:674–677. [Google Scholar]
- 48.Vanberkel GJ, McLuckey SA, Glish GL. Electrochemical origin of radical cations observed in electrospray ionization mas sspectra. Anal Chem. 1992;64:1586–1593. [Google Scholar]
- 49.Hayen H, Karst U. Analysis of phenothiazine and its derivatives using LC/electrochemistry/MS and LC/electrochemistry/fluorescence. Anal Chem. 2003;75:4833–4840. doi: 10.1021/ac0346050. [DOI] [PubMed] [Google Scholar]
- 50.Nozaki K, Kitagawa H, Kimura S, Kagayama A, Arakawa R. Investigation of the electrochemical oxidation products of zotepine and their fragmentation using on-line electrochemistry/electrospray ionization mass spectrometry. J Mass Spectrom. 2006;41:606–612. doi: 10.1002/jms.1017. [DOI] [PubMed] [Google Scholar]
- 51.Benassi M, Wu C, Nefliu M, Ifa D, Volny M, Cooks RG. Redox transformations in desorption electrospray ionization. Int J Mass Spectrom. 2009;280:235–240. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.