

Abstract
Background
Oxaliplatin-based chemotherapy has been linked to the development of sinusoidal obstruction syndrome (SOS), which is detrimental to outcome after liver resection for colorectal liver metastases (CLM). The aim of this study was to determine how the expression of genes involved in the transport and metabolism of FOLFOX chemotherapy impacts on tissue injury in a murine model of CLM.
Methods
Experimental CLM was established in C57/B16 mice and treated with FOLFOX chemotherapy. After 3 weeks, the animals were killed and RNA extracted from liver, spleen and tumour tissue. DNA damage was assessed by immunohistochemistry for γH2AX. Gene expression was determined by reverse transcriptase polymerase chain reaction.
Results
FOLFOX treatment was associated with an increase in the number of γH2AX-positive cells in both the spleen (P < 0.01) and tumour tissue (P < 0.01), but not the liver. Tissue resistance to injury following FOLFOX was associated with high expression of the copper transporter ATP7B. Differences in the expression of genes related to 5-fluorouracil metabolism or DNA repair did not correlate with the severity of tissue injury.
Conclusions
High levels of expression of ATP7B are associated with resistance to tissue injury following FOLFOX chemotherapy. Polymorphisms in the ATP7B gene may explain varying susceptibility to SOS among patients following oxaliplatin-based chemotherapy.
Introduction
The use of oxaliplatin-based chemotherapy prior to resection of colorectal liver metastases (CLM) has been associated with the development of sinusoidal obstruction syndrome (SOS) in the non-tumour-bearing liver in up to 45% of patients.1–4 The presence of SOS is a cause for concern in major liver resection and the use of oxaliplatin-based chemotherapy prior to resection of CLM has been associated with a significant increase in perioperative morbidity.1,5 More recently, it has been suggested that the presence of SOS is associated with poorer disease-specific survival and, in particular, an increased risk for intrahepatic recurrence.2 Thus, there is an urgent need to identify those patients with particular risk for developing SOS in order to ensure that their exposure to oxaliplatin-based chemotherapy is minimized.
Over the last few years, several attempts have been made to develop a murine model of oxaliplatin-induced SOS. However, to date, none of these have been successful in inducing histological changes in the liver despite the administration of high doses of chemotherapy for prolonged periods.6,7 One potential explanation for this observation is found in early pharmacological studies conducted in healthy mice, which showed the spleen to be the primary organ to take up oxaliplatin and indicated that reduced concentrations are taken up by the liver.8 Identification of the factors responsible for the relative resistance of the murine liver to oxaliplatin may represent an important step towards establishing why some patients seem particularly vulnerable to the toxic effects of this drug.
Oxaliplatin is a third-generation platinum compound consisting of a platinum atom conjugated to diaminocyclohexane and oxalate groups.9 Work by Katano et al. and Safaei et al. demonstrated, in cancer cell lines, that the cellular influx and efflux kinetics of platinum compounds mirror those of copper.10,11 Cellular uptake of copper is regulated by the plasma membrane copper transporters CTR1 and CTR2. Cell lines which do not express CTR1 demonstrate high levels of resistance to platinum-based chemotherapeutics, which strongly suggests that this transporter plays a critical role in the cellular uptake of these drugs.12,13 In support of this observation, it has been demonstrated in vivo that dorsal root ganglia with high CTR1 expression are particularly vulnerable to oxaliplatin toxicity.14 By contrast, high levels of expression of the copper uptake transporter CTR2 have been shown to be associated with relative resistance to platinum-based chemotherapy both in vitro and in vivo in ovarian cancer.15–17
The cellular export of copper is regulated by the transmembrane ATPases ATP7A and ATP7B. Cancer cell lines which demonstrate resistance to platinum-based chemotherapy commonly express high levels of these transporters, thereby preventing the intracellular accumulation of platinum.10,18,19 The validity of these in vitro observations has been confirmed in vivo: patients with colorectal cancer, in which high levels of ATP7B are expressed, are more likely to experience disease progression during treatment than those with low levels of ATP7B expression (hazard ratio 3.56; P < 0.01).20
Oxaliplatin is rarely administered as monotherapy, but, rather, is used alongside a thymidylate synthase inhibitor such as 5-flourouracil (5-FU) or leucovorin (in FOLFOX) or the oral pro-drug capecitabine (in CAPOX/XELOX).21 Upon cellular entry, 5-FU is converted into its active metabolites by the action of the enzyme thymidine phosphorylase (TP). These metabolites exhibit a cytotoxic effect through the inhibition of the enzyme thymidylate synthase (TS). Many cells also possess the capability to convert 5-FU into biologically inactive metabolites via a pathway that utilizes the enzyme dihydropyrimidine dehydrogenase (DPD).22
The aim of the current study was to determine the differential expression of genes involved in the cellular transport of platinum compounds and tissue injury following exposure to FOLFOX chemotherapy.
Materials and methods
The use of animals in this study was approved by the local research ethics committee and the UK Home Office.
Experimental model of CLM
Following isoflurane anaesthesia, C57/Bl6 mice underwent midline laparotomy. Subcapsular injection of the syngeneic MCA38 colorectal cancer cell line, stably transfected with the pGL4.51 luciferase reporting vector (Promega UK, Southampton, UK), was then performed in the left lobe of the liver (105 cells per animal suspended in 10 μl of Hank's buffered salt solution). The injection track was sealed with manual compression to achieve haemostasis followed by application of butylcyanoacrylate glue (Indermil®; Henkel Ireland Ltd, Dublin, Ireland). The abdomen was closed in two layers with vicryl.
Five days following surgery, the presence of viable tumour was confirmed using the IVIS® Imaging System (Caliper Life Sciences, Hopkinton, MA, USA). At this point, treatment was commenced with intraperitoneal FOLFOX chemotherapy, which consisted of 6 mg/kg oxaliplatin followed 2 h later by 50 mg/kg 5-FU and 90 mg/kg folinic acid. This drug regimen was based on a review of the literature and the present group's own preliminary toxicity studies. Control animals received vehicle (10% DMSO) alone.
Animals were treated weekly for 3 weeks with FOLFOX and killed 24 h after the final dose of chemotherapy. There were five animals in each experimental group.
Histology and immunohistochemistry
Sections of 5 μm were cut from formalin-fixed, paraffin-embedded blocks of liver, spleen and tumour tissue from each animal. One of these sections was stained with haematoxylin and eosin (H&E) in order to facilitate an assessment of drug-induced tissue injury.
To assess chemotherapy-induced DNA damage in each of these tissues, immunohistochemistry was performed for γH2AX. In brief, sections were deparaffinized and peroxidase activity blocked. Antigen retrieval was performed with 1 mμ EDTA pH 8.0, following which endogenous biotin was blocked by use of a commercial kit (Vector Laboratories, Inc., Burlingame, CA, USA) before incubation in 20% swine serum for 30 min. The primary antibody (#9718; Cell Signalling Technology, Inc., Danvers, MA, USA) was added at a concentration of 1 : 100 and sections incubated overnight at 4 °C. Sections were then incubated for 90 min with a biotin-conjugated secondary antibody (Dako UK Ltd, Ely, UK) and for 45 min with streptavidin biotin-peroxidase conjugate (Vector Laboratories, Inc.). 3,3′-diaminobenzidine tetrahydrochloride (DAB; Vector Laboratories, Inc.) was used as the chromogen. The numbers of positive stained cells in 15 separate high-power fields (HPF; magnification ×20) were counted in a blinded manner.
Reverse transcription polymerase chain reaction
RNA was extracted from snap frozen samples of liver, spleen and tumour from each animal using the RNeasy system according to the manufacturer's instructions (Qiagen Ltd, Crawley, UK). RNA was quantified on a spectrophotometer and 1 μg used to generate cDNA using Moloney murine leukaemia virus reverse transcriptase (RT) and a random hexamer primer (Promega UK) to a final total volume of 50 μl.
Reactions consisted of 12.5 μl polymerase chain reaction (PCR) mastermix, 400 nM each of forward and reverse primers and 1 μl of cDNA to a final volume of 25 μl. Amplification was performed using 35 cycles consisting of denaturation at 95 °C, annealing at 55 °C and elongation at 70 °C, each step of which lasted 30 s. This was followed by a final elongation step at 70 °C for 7 min. The PCR product was then run on 1% agarose gel and detected by staining with ethidium bromide. β-Actin served as the internal control. Primer sequences are listed in Table S1 (online).
Statistical analysis
Data are presented as the mean ± standard error. Statistical significance was assessed using Kruskall–Wallis one-way analysis of variance (anova) and the Mann–Whitney U-test as appropriate. A P-value of <0.05 was considered to indicate statistical significance.
Results
FOLFOX-induced tissue injury
Intraperitoneal administration of FOLFOX demonstrated antitumour efficacy in this model, with significant areas of cell death seen on the review of H&E-stained sections (Fig. 1a). In keeping with previous reports, there was no histological evidence of SOS. As the spleen has been reported to be the main organ to take up oxaliplatin-based chemotherapy in pharmacological studies, sections of this organ were also reviewed. These revealed atrophy of the germinal centre of the spleen and relative expansion of the red pulp (Fig. 1b).
Figure 1.
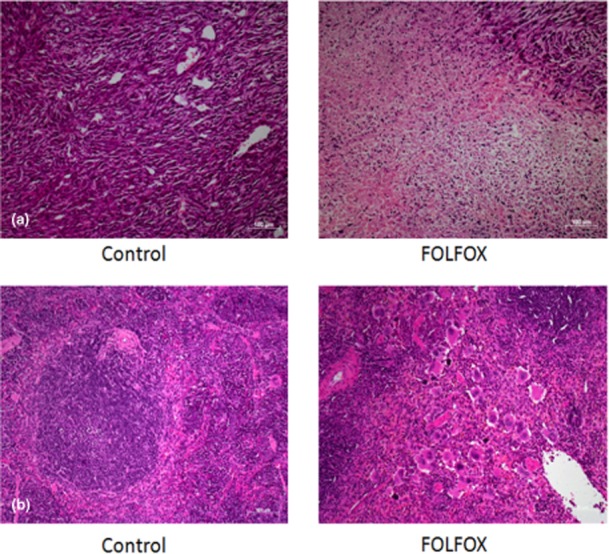
(a) Histopathology demonstrated areas of tumour death in FOLFOX-treated animals compared with control animals, confirming the efficacy of this regimen. (b) Evidence of splenic injury with loss of germinal centres and relative expansion of the red pulp was also apparent. (Haematoxylin and eosin stain; original magnification ×10)
The toxic effects of oxaliplatin are mediated through the binding of platinum to DNA, which results in the formation of DNA double-strand breaks and either activation of the cellular DNA repair mechanisms or, if sufficient damage is present, programmed cell death. In order to determine whether the histological changes observed in the tumour and spleen were a consequence of DNA damage, immunohistochemistry was performed for γH2AX. There was a significant increase in the number of γH2AX-positive cells in both the spleen (38 versus nine cells per HPF; P < 0.01) (Fig. 2a) and tumour tissue (51 versus 38 cells per HPF; P < 0.01) (Fig. 2b) of FOLFOX-treated animals, whereas no such increase was demonstrated in the liver (four versus five cells per HPF) (Fig. 2c). Together, these observations would appear to support the assertion that the murine liver is relatively resistant to FOLFOX-induced injury, whereas the spleen and tumour tissue are more vulnerable.
Figure 2.
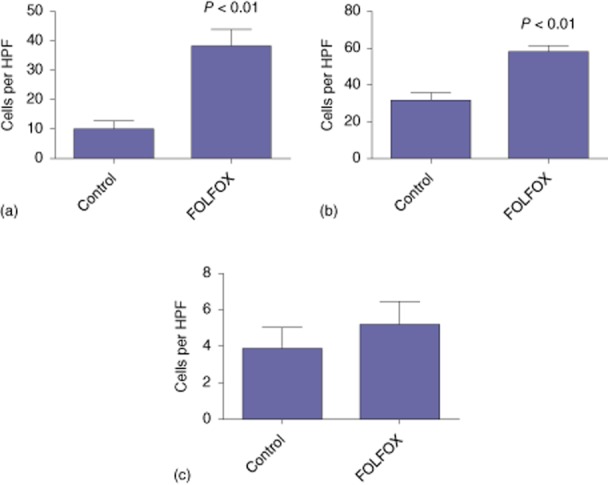
Immunohistochemistry for γH2AX revealed FOLFOX-induced DNA damage in the (a) spleen and (b) tumour tissue. (c) This damage was not seen in the liver. HPF, high-power field
Copper transporter expression
To assess the relationship between the ability of tissues to take up oxaliplatin-based chemotherapy and the development of histological injury, RT-PCR was performed to determine the expression of the copper influx (CTR1 and CTR2) and efflux (ATP7A and ATP7B) transporters. Despite the absence of injury to the liver, mRNA expression of the influx transporters was such as might be expected in an oxaliplatin-sensitive organ (i.e. high expression of CTR1 and relatively low expression of CTR2) (Fig. 3). Overall, the transcript level of these transporters was similar to that seen in oxaliplatin-sensitive tumour tissue. Together, these findings would suggest that the resistance of the murine liver to FOLFOX-induced injury in this model is not a consequence of a deficiency in oxaliplatin uptake.
Figure 3.
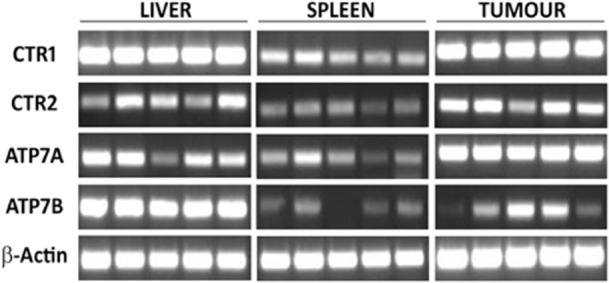
Expression of copper transporter genes in liver, spleen and tumour tissue
The mRNA expression of the efflux transporter ATP7A in the liver was somewhere between those of the tumour and spleen, again suggesting that this is not pivotal in mediating resistance to oxaliplatin-induced injury. By contrast, mRNA expression of ATP7B was markedly higher in the liver than in the other tissues, which suggests that the liver may be able to rapidly export oxaliplatin, thereby providing a mechanism through which intracellular exposure to the drug is limited and thus minimizing toxicity (Fig. 3).
5-FU metabolism
In order to assess the relative sensitivity of the liver to the 5-FU component of the chemotherapy regimen, expression of TS, TP and DPD were compared across all three tissues. The liver was found to express the highest amount of TP transcript, suggesting there should be no deficiency in converting 5-FU into its active metabolites. The expression of DPD was similar in liver and tumour tissue, and the expression of TS was similar across all three tissues (Fig. 4). It therefore seems unlikely that 5-FU resistance prevents the development of histological liver injury in this model.
Figure 4.

Expression of genes related to 5-fluorouracil metabolism in liver, spleen and tumour tissue. TS, thymidylate synthase; TP, thymidine phosphorylase; DPD, dihydropyrimidine dehydrogenase
Nucleotide excision repair pathway
The cellular response to DNA–platinum adducts is to attempt repair by making use of the nucleotide excision repair (NER) pathway in which, following recognition of DNA damage, the DNA helix is unwound to allow excision of the damaged DNA by proteins including ERCC1/2 and XPF. Following excision, the damaged DNA is resynthesized by DNA polymerase and the helix rewinds. Impairment of the NER pathway is associated with increased sensitivity to oxaliplatin-mediated DNA damage.23,24 It has recently been suggested that decreased expression of ERCC2 transcript may be associated with the development of SOS in patients with CLM treated with preoperative chemotherapy.25
In the current model, mRNA expression of the NER components ERCC1/2 and XPF in the liver was generally lower than that seen in tumour tissue, but was broadly similar to that seen in the spleen (Fig. 5). This would seem to suggest that differences in this regard are not responsible for the relative resistance of the murine liver to FOLFOX.
Figure 5.
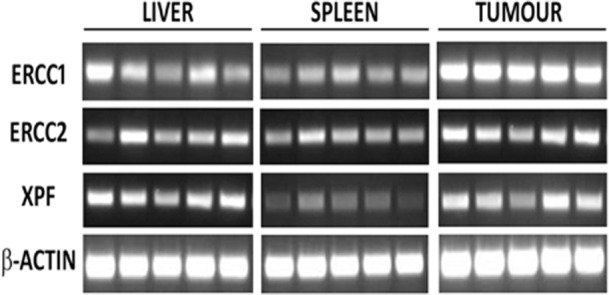
Expression of genes from the nucleotide excision repair pathway
Discussion
This study has confirmed the observation that the murine liver appears to be relatively resistant to the development of injury following exposure to FOLFOX chemotherapy. By contrast, the spleen, along with implanted tumour tissue, appears to be particularly sensitive. By comparing the expression of various genes in these sensitive tissues with that of the liver, this study has been able to demonstrate that this resistance appears to be related to high expression of the copper efflux transporter ATP7B, which enables the rapid clearance of oxaliplatin from the liver. There were no differences in the expression of genes related to 5-FU metabolism or the NER pathway which would otherwise explain this resistance.
The liver plays a central role in copper homeostasis; in particular, its export is tightly regulated by the liver through the actions of ATP7B, which transfers copper to the secretory pathway whereby it is conjugated to caeruloplasmin or, when copper is in excess, is regulated through biliary excretion.26,27 For example, Wilson's disease is characterized by excessive hepatic accumulation of copper and occurs as a result of a functional deficit in the ATP7B gene.27 As already discussed, ATP7B also plays a key role in the cellular export of platinum compounds, the overexpression of which is associated with chemo-resistance in cancer cell lines.19 As ATP7B is normally highly expressed in the liver, it would be reasonable to expect that it would also be relatively protected from the platinum toxicity.
Polymorphisms in the gene ATP7B are relatively common in the general population. One study that examined genomic DNA isolated from the peripheral leukocytes of 203 cancer patients found that 61 patients had a polymorphism in ATP7B.28 This study made no attempt to correlate these polymorphisms with chemotherapy side-effects, however, and thus the functional consequence of such polymorphisms is unknown.
Although it is entirely logical to believe there is a direct correlation between the number of cycles of chemotherapy administered to patients and the development of SOS, the evidence for this is not particularly clear. In a multivariate analysis performed in a series of 90 patients undergoing resection of CLM following preoperative chemotherapy, Nakano et al. identified the receipt of more than six cycles of oxaliplatin-based chemotherapy as an independent risk factor for the development of SOS.29 By contrast, a separate study of 196 patients, conducted by Tamandl et al., failed to identify any association between the number of chemotherapy cycles and SOS development.2 One possible explanation for these divergent observations is that pharmacogenomics and, in particular, polymorphisms affecting ATP7B function may play a key role in determining individual susceptibility to the toxic effects of oxaliplatin-based chemotherapy.
The concept of modifying a chemotherapy strategy based on the presence of genetic polymorphisms is not new. Irinotecan is an alternative chemotherapeutic widely applied in the management of patients with CLM, the use of which can be associated with several significant complications, including febrile neutropenia and severe diarrhoea. The active metabolite of irinotecan, SN38, undergoes metabolism itself utilizing a group of enzymes known as UDP-glucuronosyltransferase (UGT) 1A. Single-nucleotide polymorphisms (SNPs) in the genes encoding members of this family are common and, when present, particularly in the gene UGT1A1, have been associated with an increased incidence of irinotecan toxicity. When these SNPs are detected, it is usual practice to reduce the administered dose of irinotecan.30,31
The identification of a cohort of individuals at high risk for developing SOS following oxaliplatin-based chemotherapy would be extremely useful in establishing which patients will be best served by the provision of alternative and equally effective regimens in place of prolonged and detrimental chemotherapy. Screening for SNPs is an attractive way of doing this as genomic DNA can be easily isolated from peripheral blood leukocytes for analysis.
It should be noted that the current study is intended to generate hypotheses rather than to provide a direct causative link between ATP7B expression and the occurrence of oxaliplatin-induced SOS. Nonetheless, despite these limitations, it does appear to offer justification for human studies to explore whether SNPs in ATP7B could be used to identify a cohort of patients at high risk for oxaliplatin-induced SOS.
Acknowledgments
SMR was supported by a Wellcome Trust Clinical Research Training Fellowship (WT090974MA).
Conflicts of interest
None declared.
Supporting information
Additional supporting information may be found in the online version of this article.
Table S1. Primer sequences for quantitative reverse transcription polymerase chain reaction (qRT-PCR).
References
- 1.Vauthey JN, Pawlik TM, Ribero D, Wu TT, Zorzi D, Hoff PM, et al. Chemotherapy regimen predicts steatohepatitis and an increase in 90-day mortality after surgery for hepatic colorectal metastases. J Clin Oncol. 2006;24:2065–2072. doi: 10.1200/JCO.2005.05.3074. [DOI] [PubMed] [Google Scholar]
- 2.Tamandl D, Klinger M, Eipeldauer S, Herberger B, Kaczirek K, Gruenberger B, et al. Sinusoidal obstruction syndrome impairs longterm outcome of colorectal liver metastases treated with resection after neoadjuvant chemotherapy. Ann Surg Oncol. 2011;18:421–430. doi: 10.1245/s10434-010-1317-4. [DOI] [PubMed] [Google Scholar]
- 3.Aloysius MM, Zaitoun AM, Beckingham IJ, Neal KR, Aithal GP, Bessell EM, et al. The pathological response to neoadjuvant chemotherapy with FOLFOX-4 for colorectal liver metastases: a comparative study. Virchows Arch. 2007;451:943–948. doi: 10.1007/s00428-007-0497-1. [DOI] [PubMed] [Google Scholar]
- 4.Hubert C, Fervaille C, Sempoux C, Horsmans Y, Humblet Y, Machiels JP, et al. Prevalence and clinical relevance of pathological hepatic changes occurring after neoadjuvant chemotherapy for colorectal liver metastases. Surgery. 2010;147:185–194. doi: 10.1016/j.surg.2009.01.004. [DOI] [PubMed] [Google Scholar]
- 5.Nordlinger B, Sorbye H, Glimelius B, Poston GJ, Schlag PM, Rougier P, et al. Perioperative chemotherapy with FOLFOX4 and surgery versus surgery alone for resectable liver metastases from colorectal cancer (EORTC Intergroup trial 40983): a randomized controlled trial. Lancet. 2008;371:1007–1016. doi: 10.1016/S0140-6736(08)60455-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Keizman D, Maimon N, Ish-Shalom M, Buchbut D, Inbar M, Klein B, et al. An animal model for chemotherapy-associated steatohepatitis and its prevention by the oral administration of fatty acid bile acid conjugate. Cancer. 2010;116:251–255. doi: 10.1002/cncr.24710. [DOI] [PubMed] [Google Scholar]
- 7.Rickenbacher A, DeOliveira ML, Tian Y, Jang JH, Riener MO, Graf R, et al. Arguments against toxic effects of chemotherapy on liver injury and regeneration in an experimental model of partial hepatectomy. Liver Int. 2011;31:313–321. doi: 10.1111/j.1478-3231.2010.02446.x. [DOI] [PubMed] [Google Scholar]
- 8.Boughattas NA, Levi F, Fournier C, Lemaigre G, Roulon A, Hecquet B, et al. Circadian rhythm in toxicities and tissue uptake of 1,2-diamminocyclohexane(trans-1)oxalatoplatinum(II) in mice. Cancer Res. 1989;49:3362–3368. [PubMed] [Google Scholar]
- 9.Kweekel DM, Gelderblom H, Guchelaar HJ. Pharmacology of oxaliplatin and the use of pharmacogenomics to individualize therapy. Cancer Treat Rev. 2005;31:90–105. doi: 10.1016/j.ctrv.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 10.Katano K, Kondo A, Safaei R, Holzer A, Samimi G, Mishima M, et al. Acquisition of resistance to cisplatin is accompanied by changes in the cellular pharmacology of copper. Cancer Res. 2002;62:6559–6565. [PubMed] [Google Scholar]
- 11.Safaei R, Katano K, Samimi G, Naerdemann W, Stevenson JL, Rochdi M, et al. Cross-resistance to cisplatin in cells with acquired resistance to copper. Cancer Chemother Pharmacol. 2004;53:239–246. doi: 10.1007/s00280-003-0736-3. [DOI] [PubMed] [Google Scholar]
- 12.Holzer AK, Manorek GH, Howell SB. Contribution of the major copper influx transporter CTR1 to the cellular accumulation of cisplatin, carboplatin, and oxaliplatin. Mol Pharmacol. 2006;70:1390–1394. doi: 10.1124/mol.106.022624. [DOI] [PubMed] [Google Scholar]
- 13.Larson CA, Blair BG, Safaei R, Howell SB. The role of the mammalian copper transporter 1 in the cellular accumulation of platinum-based drugs. Mol Pharmacol. 2009;75:324–330. doi: 10.1124/mol.108.052381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu JJ, Jamieson SM, Subramaniam J, Ip V, Jong NN, Mercer JF, et al. Neuronal expression of copper transporter 1 in rat dorsal root ganglia: association with platinum neurotoxicity. Cancer Chemother Pharmacol. 2009;64:847–856. doi: 10.1007/s00280-009-1017-6. [DOI] [PubMed] [Google Scholar]
- 15.Lee YY, Choi CH, Do IG, Song SY, Lee W, Park HS, et al. Prognostic value of the copper transporters, CTR1 and CTR2, in patients with ovarian carcinoma receiving platinum-based chemotherapy. Gynecol Oncol. 2011;122:361–365. doi: 10.1016/j.ygyno.2011.04.025. [DOI] [PubMed] [Google Scholar]
- 16.Blair BG, Larson CA, Adams PL, Abada PB, Safaei R, Howell SB. Regulation of copper transporter 2 expression by copper and cisplatin in human ovarian carcinoma cells. Mol Pharmacol. 2010;77:912–921. doi: 10.1124/mol.109.062836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blair BG, Larson CA, Safaei R, Howell SB. Copper transporter 2 regulates the cellular accumulation and cytotoxicity of cisplatin and carboplatin. Clin Cancer Res. 2009;15:4312–4321. doi: 10.1158/1078-0432.CCR-09-0311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Samimi G, Safaei R, Katano K, Holzer AK, Rochdi M, Tomioka M, et al. Increased expression of the copper efflux transporter ATP7A mediates resistance to cisplatin, carboplatin, and oxaliplatin in ovarian cancer cells. Clin Cancer Res. 2004;10:4661–4669. doi: 10.1158/1078-0432.CCR-04-0137. [DOI] [PubMed] [Google Scholar]
- 19.Komatsu M, Sumizawa T, Mutoh M, Chen ZS, Terada K, Furukawa T, et al. Copper-transporting P-type adenosine triphosphatase (ATP7B) is associated with cisplatin resistance. Cancer Res. 2000;60:1312–1316. [PubMed] [Google Scholar]
- 20.Martinez-Balibrea E, Martinez-Cardus A, Musulen E, Gines A, Manzano JL, Aranda E, et al. Increased levels of copper efflux transporter ATP7B are associated with poor outcome in colorectal cancer patients receiving oxaliplatin-based chemotherapy. Int J Cancer. 2009;124:2905–2910. doi: 10.1002/ijc.24273. [DOI] [PubMed] [Google Scholar]
- 21.Robinson S, Manas DM, Pedley I, Mann D, White SA. Systemic chemotherapy and its implications for resection of colorectal liver metastasis. Surg Oncol. 2011;20:57–72. doi: 10.1016/j.suronc.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 22.Longley DB, Harkin DP, Johnston PG. 5-fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer. 2003;3:330–338. doi: 10.1038/nrc1074. [DOI] [PubMed] [Google Scholar]
- 23.Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O, et al. Molecular mechanisms of cisplatin resistance. Oncogene. 2012;31:1869–1883. doi: 10.1038/onc.2011.384. [DOI] [PubMed] [Google Scholar]
- 24.Martin LP, Hamilton TC, Schilder RJ. Platinum resistance: the role of DNA repair pathways. Clin Cancer Res. 2008;14:1291–1295. doi: 10.1158/1078-0432.CCR-07-2238. [DOI] [PubMed] [Google Scholar]
- 25.Pilgrim CH, Brettingham-Moore K, Pham A, Murray W, Link E, Smith M, et al. mRNA gene expression correlates with histologically diagnosed chemotherapy-induced hepatic injury. HPB. 2011;13:811–816. doi: 10.1111/j.1477-2574.2011.00365.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lutsenko S, Barnes NL, Bartee MY, Dmitriev OY. Function and regulation of human copper-transporting ATPases. Physiol Rev. 2007;87:1011–1046. doi: 10.1152/physrev.00004.2006. [DOI] [PubMed] [Google Scholar]
- 27.Huster D. Wilson disease. Best Pract Res Clin Gastroenterol. 2010;24:531–539. doi: 10.1016/j.bpg.2010.07.014. [DOI] [PubMed] [Google Scholar]
- 28.Fukushima-Uesaka H, Saito Y, Maekawa K, Kurose K, Sugiyama E, Katori N, et al. Genetic polymorphisms of copper and platinum drug-efflux transporters ATP7A and ATP7B in Japanese cancer patients. Drug Metab Pharmacokinet. 2009;24:565–574. doi: 10.2133/dmpk.24.565. [DOI] [PubMed] [Google Scholar]
- 29.Nakano H, Oussoultzoglou E, Rosso E, Casnedi S, Chenard-Neu MP, Dufour P, et al. Sinusoidal injury increases morbidity after major hepatectomy in patients with colorectal liver metastases receiving preoperative chemotherapy. Ann Surg. 2008;247:118–124. doi: 10.1097/SLA.0b013e31815774de. [DOI] [PubMed] [Google Scholar]
- 30.Kweekel D, Guchelaar HJ, Gelderblom H. Clinical and pharmacogenetic factors associated with irinotecan toxicity. Cancer Treat Rev. 2008;34:656–669. doi: 10.1016/j.ctrv.2008.05.002. [DOI] [PubMed] [Google Scholar]
- 31.Marsh S, Hoskins JM. Irinotecan pharmacogenomics. Pharmacogenomics. 2010;11:1003–1010. doi: 10.2217/pgs.10.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.