

Abstract
T cells are a multifaceted family pivotal in the operations of the immune system and many of its associated diseases. The pathway to understanding T cells has been marked by several pharmacological advances including the discoveries of ciclosporin, tacrolimus and the mTOR inhibitors which revolutionized transplant therapy along with providing relief for severe eczema, asthma and other immunological disorders towards the end of the last century. This article will revisit the current understanding and new developments in T cell pharmacology 10 years on from the TeGenero (TGN 1412) debacle and look at more recent successes with ex vivo antigen presenting cell incubation technologies; T cell receptor (TCR) engineering and adoptive T cell therapy both with chimaeric antibodies and also with modified T cell receptors themselves. Features of T cell biology will be explored and processes often highly unique to humans will be used to highlight what many are beginning to see as an exciting new monoclonal (T cell) frontier for drug development.
Keywords: adoptive cell therapy, bi-specific, cancer, T cells
Introduction to T cells
The human lymphocyte antigen (HLA) system is present on almost all human cells (Figure 1). Exogenous antigens (foreign antigens, e.g. viruses, micro-organisms, mutations, toxins or drugs, etc.) are processed by antigen presenting cells and married with HLA destined for the self-cell surface.
Figure 1.
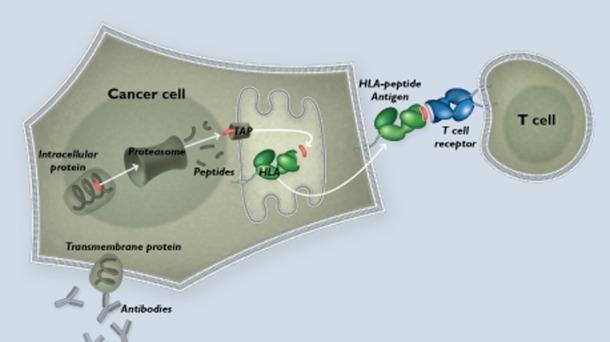
External and internal cell membranes work to envelop and process proteins that are operating within the cell interior and extracellular environment. Merging internalised proteins with lysosomes results in the degradation of internalizes protiens to smaller pepide fragments. Further subsequent merging of the the phagolysosome with HLA emerging from the endoplasmic reticulum allows digested peptides to be married up with their cognate MHC protein and thereafter presented on the surface of the cell. Through this mechanism, viruses, bacteria, cellular debris and internal cellular proteins (mutated and normal) can all be presented to passing T cells
The HLA protein complex attaches to proteins that are being processed internally by cells and shuttles them to the cell surface to display peptide fragments of 8–11 amino acids in length. Through the HLA display system passing T cells bearing complimentary (cognate) T cell receptors can then recognize non-self and mutated proteins that happen to be occupying and may be corrupting indigenous cell hosts. The HLA system does not respect the origin of the proteins that are being processed and therefore it is as likely to display a constitutive housekeeping protein fragment as it is an occupying viral protein fragment or a cancerous mutation in an oncogene. In truth the HLA system is not perfectly ‘agnostic’ to the origin of a protein but in fact has evolved to favour presentation of foreign or abnormal antigens at a higher frequency. However this is not the critical determining factor in determining if a T cell thinks a protein is ‘safe’ or not ‘safe’. The critical determining factor is that the cognate T cell receptor is more likely to bind with high affinity those unusual proteins that it did not encounter when present in the embryo. A low affinity interaction is much less likely to result in attack. Embryonic thymic selection processes have, in earlier life, removed T cell receptors that bind to self-protein fragments with high affinity leaving by subtraction only those high affinity TCRs that recognize foreign peptide sequences. These amino acid sequences are unique in many cases and act as the substrate for what is often an exquisitely specific line of defence against cancer and infestation. T cells carrying the requisite cognate T cell receptor to the HLA-peptide immunologically synapse with the cell and orchestrate its destruction or even, in the case of a regulatory T cell, its defence. A process of cell-induced killing is triggered by T cell secretion of the cytotoxins granulysin, granzyme and perforin. A further refinement of this system is that not only do the majority of the body's native cells constitutively present HLA peptide fragments on a dynamic turnover basis but there is also a specialist set of antigen presenting cells that do this on a more comprehensive footing. These antigen presenting cells (APCs) have specialized protein degradation units known as immuno-proteosomes where the proteins are digested. APCs also phagocytize external debris and process and present any proteinaceous contents therein. Antigen presenting cells can be found as a natural part of most tissues and are more frequently encountered at ‘sentry’ points in organs that sit at the interface with the ‘outside world’ like gut lymphoid tissue, liver Kupfer cells or skin Langerhan's cells. Fully functioning T cell receptors have the potential to overcome one limiting factor that antibodies suffer from – T cell receptors can target intra-cellular proteins as well as extra-cellular or surface proteins. A further difference between antibodies and T cell receptor systems is that the binding dynamics between a T cell receptor and an HLA complex appear to be at least two or three orders of magnitude more sensitive than those that exist between an antibody and its target protein. Proprietary soluble TCRs work in the femto to picomolar range whilst most antibodies appear more commonly to engage around the nanomolar range 1
Calcineurin and mTOR inhibitors
Ciclosporin, pimecrolimus, tacrolimus, temsirolimus, sirolimus and everolimus are all molecules derived from soil-based Streptomyces species except for ciclosporin which comes from the soil-based fungus Tolypocladium inflatum (Beauveria nivea). These agents profoundly influence T cell function through inhibition of two significant pathways (Table 1) 2 The calcineurin pathway is inhibited by ciclosporin, tacrolimus and pimecrolimus and downstream pathway consequences are associated with potent suppression of T cell-mediated inflammatory activity. Everolimus, temsirolimus and sirolimus abrogate T cell activity as well but through inhibition of the mTOR1 molecule complex. For completeness there are two different types of mTOR complexes, though the mTOR 2 complex appears less related to T cell function and has initially proved more of interest to metabolic biologists because of its role in carbohydrate metabolism. The mTOR 1 agents are pivotal in the cascade of molecular interactions that are initiated by growth factor receptor activation all the way down to resultant nuclear DNA transcription which initiates growth and proliferation effects central to many inflammation/organ rejection episodes, graft vs. host-disease and cancer growth. What the development of all these agents has taught us is that the number of degrees of freedom within the immune system is inordinately large (Figure 2). The effects of these molecules were discovered in laboratories using high throughput screening assays and then transferred over into animal models and eventually into successful transplant trials in the clinic. Defining the subtle effects on balances of T cell populations, for example CD4:CD8 ratios, T regulatory cell growth and down regulation, mast cell and antigen presenting cell modulation, and then fitting these observations to the emergent phenomena seen in the clinic has been and continues to be an immense challenge to biologists and pharmacologists alike. The outputs from this large body of work have informed and cautioned the work of newly emerging T cell therapies going forward 2–4.
Table 1.
Small molecule suppressors of T cell activity influence outcomes in cancer, transplantation and autoimmune diseases predominantly through two closely associated pathways. Ciclosporin, tacrolimus and pimecrolimus all work with an intracellular binding partner (cyclophilins or FKBPs) to negatively influence calcineurin and the NFAT nuclear transcription regulator whilst everolimus, sirolimus and temsirolimus bind directly to the mTOR intracellular target and negatively regulate growth factor signalling
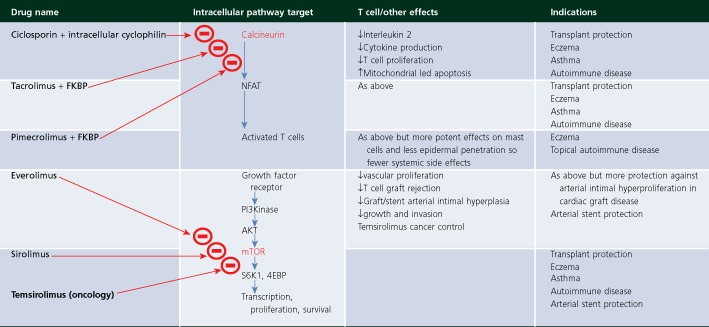 |
Figure 2.
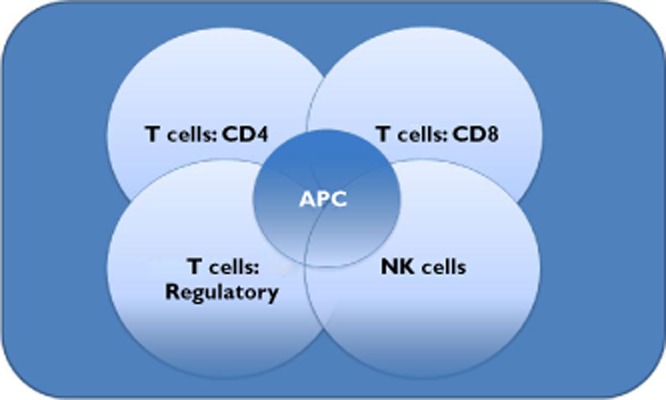
A simplified diagram showing the number of degrees of freedom operating between antigen presenting cells (APC), CD4 T helper cells, CD8 cytotoxic T cells, suppressive regulatory T cells and Natural Killer cells. Depending on the relative balance of these interacting cells and the cytokine milieu in which they operate outcomes could involve self-tolerance, organ rejection, auto-immunity and clearance or persistence of a given pathogen
Emerging T cell treatment paradigms
New T cell treatments can be divided for simplicity into those that modify the patient's T cells in vivo and those that modify the cells ex vivo directly with a view to subsequent therapeutic reinstatement in the patient 5. The intention with both approaches is to educate and provoke the indigenous immune system into reacting against dysfunctional cells whether they are cancer or virally occupied cells. The first ‘biotechnology’ live cell vaccination for infectious disease (smallpox in 1796) predates the first successful small molecule treatment of an infectious disease (penicillin in the 1930s) by over a century. The last century, arguably was dominated by small molecules and injected attenuated vaccines 6–8. Antibodies occupied many at the turn of the century and may prove to have been forerunners of a wider and more diverse armamentarium of immunologic therapies. The advent of gene therapy and the successful solubilization of the TCR 1 now offer an opportunity to further the field of T cell oriented therapeutics. This article will examine a sample of the pharmacologic interventions that are being developed across the T cell field and look at how new approaches may enhance understanding of the complex immune system.
Vaccines
Humans are born with a respectable repertoire of T cells which are already selected for their ability to recognize peptides that are not found normally within the host. During early child or adult life a T cell may come into contact with its pre-determined HLA peptide antigen and this contact brings about a T cell mediated attack as well as invoking the critical step of memory formation. The memory is manifested by a T cell response subsequently becoming stronger and more rapid to invoke. This ability of T cells to form memory cells is the fundamental basis of vaccination and poses one of the greatest prospects and challenges to a T cell oriented drug developer. If a therapeutic strategy cannot only utilize T cell killing function but also can do so in a way that induces long term memory formation, then the patient can be made therapeutically competent perhaps even for a lifetime. Furthermore T cells can, through a process known as antigenic spread, gain new mechanisms for attacking diverse aspects of a target. This is also possible for B cell mediated therapies but it more classically is the domain of T cell activity. The potential for a therapy that co-evolves with its target and that can in part at least continue to work even though this initial target, through processes of mutation or down regulation, may long since have been rendered irrelevant offers up an alluring prospect for drug development – life-long efficacy (and toxicity) from a limited course of intervention. As a preventative strategy for patients with operable cancer or other immediate high risk features in any disease, even pre-diabetes, this prospect is significant.
It is notable that most current therapeutic strategies dichotomize therapies into either those that are ‘preventative’ or those that are ‘therapeutic’. T cell therapy offers the real prospect of an intervention that can be immediately efficacious whilst also simultaneously offering potential for long term vaccination/engraftment that might result in more definitive cure or prevention of relapse. The complexity and evolution of T cells mean that the rules and paradigms of the immunoglobulin-schooled pharmacologist can be successfully applied to the T cell domain but only once certain nuances are appreciated. The single biggest challenge is perhaps to move from thinking about individual human beings to populations of human beings, the best example of which is the phenomenon of Herd immunity 9. This phenomenon means that spread of an infectious agent within a community can effectively be suppressed below a given threshold rate of viability such that no further spread is possible and the infection dies out. This threshold rate of viability is critically dependent upon a certain majority of individuals being immune to that agent. However not all individuals need to be immune and those that are not immunized are protected by the ‘quenching’ herd effect of the others who effectively act like levies against the oncoming epidemic surge. This means that intervention (exposure) with a drug in one individual can result in powerful efficacy (in absence of exposure or side effects) in another connected but separate individual. Models of exposure and efficacy can still be constructed but require more stochastic elements and have to be applied to groups of exposed individuals not just one subject (see Table 2 for a comparison of similarities and differences between T cell drug development and other more conventional modalities).
Table 2.
A table comparing and contrasting approaches with traditional small molecule pharmacology and those observed with T cell based technologies
| Conventional pharmacology | Vaccines and T cell products |
|---|---|
| Drug is absorbed through an epithelial surface or injected | Outcomes may be as much a product of the process as the ‘drug’ and some products involve nothing more than education of a cell or system |
| Efficacy is predominantly and largely related to exposure – i.e. how much drug gets into the relevant compartment | Efficacy is related to exposure though threshold systems more commonly operate – the system typically will need to ‘catch’ i.e. it either takes after three inoculations or it does not |
| No small molecule or biotech product ever eradicated a disease – prevalence can even increase as a result of successful intervention | Vaccine eradicated smallpox (possibly polio by the end of the decade) |
| The drug will undergo metabolism, commonly by the liver and then excretion, commonly by the kidney | Metabolism (especially through opsonization) is typically how the vaccine generates efficacy and activation. |
| Efficacy is related to modulation of a large enzyme by a small molecule interaction | Efficacy is related to the learning of the immune system and persists as memory long after the initial intervention has been removed |
| Habituation and tachyphylaxis are more frequent problems with small molecules. Sensitization can be seen with hepatotoxic or dermatotoxic substances | Through memory formation patient/population may show sensitization. Sensitizsation with small or large molecules often implies immune system involvement |
| Toxicity is mediated by effects that relate to parent drug or metabolites binding to and interacting with non-target molecules | Toxicity may be related to on target activation in the wrong organ as is seen with graft vs. host disease or may be related to the miseducation or misappropriation of signal in the form of alloreactivity after bone marrow transplant whereby HLA mismatching causes off target toxicity due to host T cell receptors binding graft HLA |
| Pharmacodynamic effects will typically track with decay of parent compound which is predominantly determined by the distribution and excretion of product | Efficacy and toxicity may persist for years after all administered product is excreted. Memory cells are very hard to detect though repeat stimulus with antigen could show a retained ‘memory’ in the absence of initial vaccine. |
| Most commercial and ethical arguments will drive the pharmacologist towards hoping to achieve once daily dosing | Infrequent or one-time administrations are possible and once daily dosing would probably mean commercial non-viability |
| Compliance introduces a large variability and tends to be on an individual basis, with the exception perhaps of directly observed dosing regimes for tuberculosis, HIV, malaria etc | Compliance is on a population basis and may even be mandated in governmental policy e.g. MMR vaccination or cervical cancer vaccines |
| Cost of goods is important though rarely dominant in price discussions on a small molecule. There are therefore also often relatively large amounts of drug available for multiple phase 1 activities | Cost of goods/administration can eclipse all other discussions. Approaches can fail in phase 1 if efficacy is not immediately perceptible |
Some early T cell pionnering technology
TGN 1412
T cells recognize protein sequences that are not indigenous and a subset of T cells (regulatory cells) provides suppressive and dampening signals in this context. It is hypothesized that the balance between stimulatory and suppressive cytokine and TCR contact signals is integrated and orchestrated by the T cell itself and the result of this signal then invokes either the appropriate attack of ‘foreign peptide’ bearing cells or the signal decision could result in the defence of innocent native cell types 5. When this decision process is corrupted an overly aggressive and inappropriate attack of host tissue can occur giving way to potentially devastating autoimmune diseases such as multiple sclerosis or ulcerative colitis 10.
TeGenero's antibody TGN 1412 is an antibody raised, by design, as a T cell agonist. TGN 1412 is more potent than any endogenous known ligand and as such the name ‘super agonist’ was applied. As shown in Figure 2, given the relative balance of stimuli acting upon different suppressive and stimulatory components of the T cell community, it is possible to explain any given drug effect through this matrix. Accordingly a stimulant signal can supress (provided that it stimulates suppressive cells more than effector cells) and a suppressive signal can stimulate providing that it suppresses the suppressive regulatory cells more than the effector cells. Given this set of interactions potential explanations can be derived as to why a CD 28 super agonist could suppress the immune system in rheumatoid arthritis and simultaneously re-invigorate bone marrow after chemotherapy induced aplasia during the treatment of B cell leukaemia. TGN 1412 is a monoclonal antibody directed with sizeable activity towards the CD 28 ligand on T cells. Antigen presenting cells upregulate CD 80 on their surface when activated and this, in combination with their constitutively expressed CD86 molecule, binds to and activates CD 28 which is present constantly on T cells. T cells mostly require the CD28 molecule to be activated as a contingency for T cell stimulation under normal physiological circumstances and usually the activation of the CD28 molecule in unison with stimulation of the T cell's own T cell receptor will produce various stimulatory cytokines such as interleukins-2 and 6. This contingency for co-stimulation of both the CD 28 molecule and TCR is furthermore usually in the context of other stimuli such that the T cell can be seen to be subject to a community of competing and complimentary stimuli. The caveat with CD28 is that when activation of the CD 28 molecule is above a given physiological threshold the contingency for co-activation no longer operates. This is true of super agonist TGN 1412 which can activate T cells non-specifically and in the absence of TCR binding events 11, 12.
On 26 March 2006 eight healthy volunteers were randomly dosed in a 3:1 compound : placebo ratio with 0.1 mg kg−1 of candidate drug/placebo. The six actively dosed subjects suffered overwhelming immune activation (‘cytokine storm’) and all were subsequently hospitalized, some for many weeks with serious multiple organ complications 11–15. The press became involved principally because the victims were healthy volunteers and the near simultaneous dosing of all six volunteers created a perfect storm as the final patient in the cohort was dosed before there was any real sign of a major problem in the first patient. A governmental enquiry showed that the correct regulatory procedures had been followed and that all pre-clinical testing was, even with hindsight, valid and in line with prevailing clinical practice. Not only had pre-clinical testing failed to show the overwhelming clinical agonistic activity but it was not clear in the aftermath how anything could have been done better other than avoiding parallel dosing and perhaps even avoiding the use of healthy volunteers when doing first-in-man studies with immunotherapies 16.
Animal tests and spiking of human blood have subsequently revealed some of the underlying protagonistic conditions 12. The human immune system, and very specifically the T cell system, is critically dependent on prevailing context 17–19. This means that unless T cells and their activities are studied in realistic conditions, that is to say in humans or very humanized culture conditions, results will be and indeed were misleading. Subsequent, well-validated work has shown that TGN 1412, when presented with both peripheral blood monocytes and a human epithelial monolayer releases sentinel supra-physiological amounts of cytokine 12. Everything was ostensibly tested correctly in the preclinical manner that was prescribed by both the regulatory authorities and the company's pharmacology advisors and therefore it has to be assumed that the error was not a mistake. Receptor occupancy (RO) models 16 have been applied in criticism of the preclinical work up for TeGenero. However these RO models are predicated on the findings regarding TGN 1412 activity that were discovered after the trial had failed. ‘Epithelialized’ models would not be ones that would normally be commissioned as part of a pre-clinical work up and it was these models that informed the RO models. TGN 1412 provides evidence on two levels of a dangerous and sometimes prevalent activity in clinical science: Both the use of complex T cell interactions to explain the potentially opposing therapeutic modalities and the use of complex post hoc clinical findings to criticize pre-clinical testing are examples of teleology or the art of making up explanations after observation has been made 20. Whilst the complexity of the immune system demands profound respect and caution science is best practised when applied to predicting the future and not the past. Any contemporary therapy allying itself with T cell activity (stimulation or suppression) should be subject to a high degree of regulatory scrutiny, and hopefully regulatory permissiveness, in the context of well thought out and adequately prosecuted research. The challenge in moving T cell pharmacology forward is balancing the immense therapeutic power of the T cell with the equally destructive power that lies therein.
Sipuleucel-T (APC8015, trade name Provenge™)
Sipuleucel T is a novel dendritic cell-based vaccine, also described as an autologous cell based immunotherapy. Sipuleucel T is a treatment for advanced metastatic prostate cancer that is no longer responding to testosterone depriving therapies and might otherwise be subject to palliative chemotherapy strategies 21. The sipuleucel T processes involved in production of the therapeutic product and also those processes pertaining to the clinical trials that putatively demonstrated efficacy provide lessons in trial interpretation and governance. The production process can be broken up into three phases, extraction, modulation and re-introduction (see Figure 3). Sipuleucel-T is a cellular immunotherapy consisting of activated autologous peripheral-blood mononuclear cells (PBMCs) which have been removed from the patient at a previous appointment. These activated PBMCs include antigen-presenting cells (APCs) that are para-vaccinated ex vivo by being pulsed with recombinant fusion protein (PA2024). The concept is that the fed cells process antigen, break it up into sentinel peptides and these are then presented on the antigen-presenting cells surface HLA complexes such that when these cells are returned to the patient they stimulate endogenous T cell reactivity. Upon reinstatement the cells engraft in the patient's immune system and thereafter are redirected to attack the patient's other antigen-bearing cells, their prostate cancer cells. The PA2024 construct consists of a prostate antigen-prostatic acid phosphatase that is fused to granulocyte–macrophage colony stimulating factor, another immune-cell activator.
Figure 3.
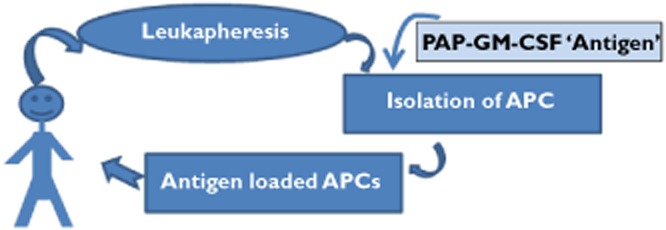
Diagrammatic representation of the procedure that patients undergo in order to receive sipuleucel T therapy
To show efficacy this cycle (remove, modulate and return) in clinical trials was repeated three times over approximately 5 weeks in patients with late stage prostate cancer who were then observed for a number of years (Table 3). In 2010 Kantoff et al. 22 reported a statistically significant overall survival benefit against placebo for the sipuleucel T arm (see Table 3). The reported benefit in the Kantoff trial and other later phase trials is probably ‘real’ though the magnitude of this benefit and in particular its clinical significance has not been robustly established 23–26. This lack of robust evidence is true even in the face of some evidence of overall survival benefit. It remains to be seen if the cost of sipuleucel T (which for the full three courses currently costs ∼$93000) is counter balanced by weighted evidence of efficacy of sufficient magnitude 24. The role of placebo in this trial demonstrates principles that are relevant for many controlled clinical trials. The patients were not noted to show any tumour shrinkage or demonstrable benefit in progression free survival in this study and benefits were only demonstrated in overall survival differences. Taking the well demonstrated correlation between tumour progression and overall survival generally, it can be inferred that this would have to mean that if the patients' tumours were not shrinking then at least patients' tumours were growing less quickly in the intervention arm. The criticism remains that patients randomized to the placebo arm were unfairly denuded of their white cells over the period of the trial as not all their cells were returned after apheresis whereas the sipuleucel T patients got their full complement of white cells returned along with the activated vaccine treatment. Storage of placebo patients' white cells was undertaken namely to preserve some cells for rescue therapy at a later time point though this ultimately did not happen in a significant proportion of patients and therefore an imbalance in the amount of white cells returned between the two trial arms was introduced. This process may have led to a depression in overall survival in the placebo arm as patients may have been suffering relative immunoparesis owing to their lack of white cells 25, 26. The hazard ratio is a dimensionless statistic and as such overall survival hazard ratios merely report relative differences between two arms. The hazard ratio does not differentiate between a benefit applied to one arm vs. a dis-benefit applied to the other. Interestingly for clinical trialists this kind of discussion does not involve the more typical concerns around type 1 or type 2 statistical errors but an altogether more fundamental question about the integrity of the control. Where the process of therapy and the therapeutic product become inextricably combined adequate care must be undertaken to ensure that the control arm and the intervention arm are treated exactly the same except for only one critical difference, the presence of intervention in the active treatment arm. Currently the sipuleucel T technology requires dedicated facilities both for the extraction and manufacturing of drug. Future developments in this area will potentially deliver a less cumbersome approach.
Table 3.
The efficacy outcomes of three large scale studies conducted with sipuleucel-T in prostate cancer. Efficacy outcomes for study D9901 and study D9902A are shown both individually and combined. The striking observation from an oncological point of view is that the normal relationship between disease progression, which is traditionally easier to demonstrate and overall survival, which is traditionally more difficult to demonstrate, is inverted.
| Parameter | D9901 | D9902A | Integrated analysis D9901 and D9902A | IMPACT study D9902B | ||||
|---|---|---|---|---|---|---|---|---|
| Sipuleucel-T (n = 82) | Placebo (n = 45) | Sipuleucel-T (n = 65) | Placebo (n = 33) | Sipuleucel-T (n = 147) | Placebo (n = 78) | Sipuleucel-T (n = 341) | Placebo (n = 171) | |
| Median survival (months) | 25.9 | 21 | 19 | 15.7 | 23.2 | 18.9 | 25.8 | 21.7 |
| Hazard ratio (CI)‡ | 1.71 (1.13, 2.58) | 1.27 (0.78, 2.07) | 1.50 (1.10, 2.05) | 0.775† (0.614, 0.979) | ||||
| Overall survival, P value§ | P = 0.010 | P = 0.331 | P = 0.011 | P = 0.032† | ||||
| Median time to disease progression (weeks) | 11 | 9.1 | 10.9 | 9.9 | 11.1 | 9 | 14.6 | 14.4 |
| Hazard ratio (CI) | 1.45 (0.99, 2.11) | 1.09 (0.69, 1.70) | 1.26 (0.95, 1.68) | 0.92 (0.75, 1.12) | ||||
| Overall time to disease progression, P value | P = 0.052 | P = 0.719 | P = 0.111 | P = 0.40 | ||||
The hazard ratio and P value are based on the Cox model, adjusted for prostate-specific antigen (ln) and lactic dehydrogenase (ln) and stratified by bisphosphonate use, number of bone metastases and primary Gleason grade.
The hazard ratio is based on the unadjusted Cox model (not prespecified).
The P value is based on a log-rank test (not pre-specified).
Ipilimumab
Ipilimumab, MDX 010, MDX 101 or Yervoy™ is a fully human antibody that binds to and blocks cytotoxic T lymphocyte-associated antigen 4 (CTLA-4). It is indicated for the treatment of late stage melanoma though its mode of action is generally not very specific with respect to the type of cancer indicated and further efficacy signals are being pursued in prostate and both small cell and non-small cell lung cancer. CTLA-4 27 operates as a braking agent reducing the activity of both helper (CD4+) and killer (CD8+) T cells of the immune system (see Figures 4 and 5).
Figure 4.
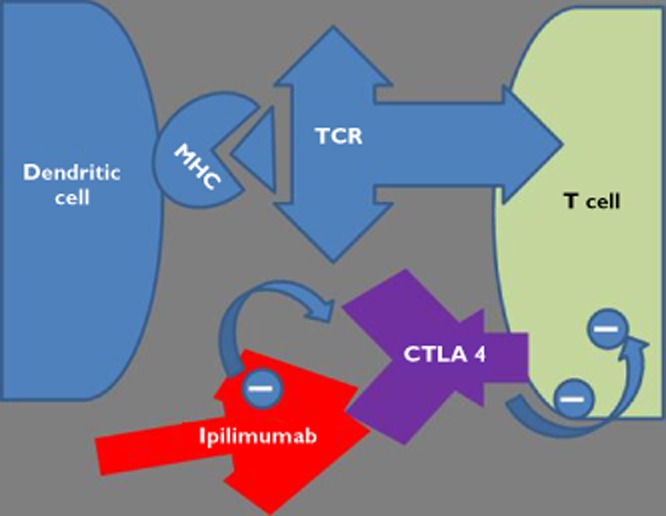
Schematic diagram showing ipilimumab antibody ‘de-repressing the T cell’ by binding to the CTLA 4 molecule and inhibiting the CTLA 4 molecules repression of T cell activity
Figure 5.
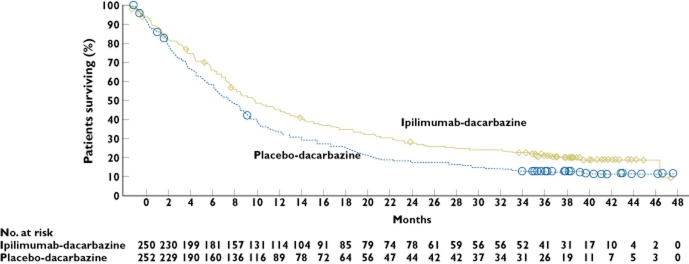
Kaplan–Meier curves for overall survival and progression-free survival in the intention-to-treat population. The median follow-up for overall survival in the ipilimumab (Ipi)-plus-glycop, 8.5 to 11.5); in the ipilimumab-alone group, the median follow-up was 27.8 months, and the median overall survival, 10.1 months (95% CI, 8.0, 13.8); and in the gp100-alone group, the median follow-up was 17.2 months, and the median overall survival, 6.4 months (95% CI, 5.5, 8.7).  , censored;
, censored;  , censored
, censored
Cancer is thought to subvert the immune system by invoking the help of various signalling mechanisms which suppress cancer-attacking B and T cells (CD4+ and CD8+) 27–34. By blocking the breaking action of CTLA 4 on helper and killer T cells these attack cells become de-repressed. The theoretical impact of activating the immune system has been played out in clinical trials where tumour inflammation, flare and regression along with excess immune-related toxicity to self (hepatitis, skin rash, mucositis, thyroiditis, diarrhoea, etc.) has also been seen (Figure 6 and Table 4) 35, 36.
Figure 6.
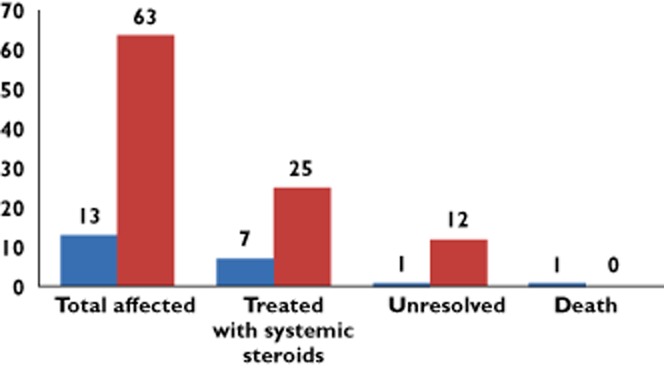
Patients treatment pathways and outcomes associated with ipilimumab toxicity.  , severe dermatitis;
, severe dermatitis;  , moderate dermatitis
, moderate dermatitis
Table 4.
A sample of the variously graded adverse events associated with ipilimumab treatment
| % of patientsa Yervoy + gp100 3 mg kg−1 + gp100 n = 380 | ||||||
|---|---|---|---|---|---|---|
| Selected adverse reactions from ipilimumab prescribing information | Yervoy 3 mg kg−1 n = 131 | gp100 alone n = 132 | ||||
| System organ class/ preferred term | Any grade | Grade 3–5 | Any grade | Grade 3–5 | Any grade | |
| Gastrointestinal disorders | Diarrhoea | 32 | 5 | 37 | 4 | 20 |
| Colitis | 8 | 5 | 5 | 3 | 2 | |
| Skin and subcutaneous tissue disorders | Pruritus | 31 | 0 | 21 | <1 | 11 |
| Rash | 29 | 2 | 25 | 2 | 8 | |
| General disorders and administration site conditions | Fatigue | 41 | 7 | 34 | 5 | 31 |
These rare but severe toxicities are the subject of a black box warning as serious adverse events including death have been observed 37. Withdrawal of drug should by simple reasoning result in senescence of side effects. However this is only partially true. A proportion of ipilimumab immunological toxicities persist indefinitely after the drug has cleared (Figure 5). The immune basis of this toxicity is hinted at by evidence that high dose steroids help the majority of, but not all patients. It would appear that ipilimumab has encouraged self-reactivity to the point of enduring auto-immunization in a small proportion of patients. Releasing an immunological checkpoint appears to be giving rise to a drug-induced T cell vs. host syndrome or in other words drug related autoimmunity. Thyroiditis, rash, hepatitis, colitis and even haemophilia have been ascribed to ipilimumab's immune deviating effects. Once the T cells have started reacting against the patient's own non-cancerous tissues it seems plausible that they secrete further cytokines that encourage further recruitment of T cells to the attack. The immune system's ability to learn and generate memory cells for autotoxicity is the basis for an overall survival benefit with ipilimumab but also the basis for a toxic and occasionally lethal loss of control in a small proportion of patients.
Preliminary results suggest that observed tumour responses with ipilimumab may, in part, be later in onset, may be more durable than with traditional chemotherapy and may even occur after an intervening period of cancer progression 38. The toxicity and efficacy seen with ipilimumab may be represented as a complex phenomenon of the immune system slightly reinventing itself in that ipilimumab has caused the immune system to become less controlled and reactivity against self is becoming embedded in the patients T cell repertoire. New rules are being written for ipilimumab where it appears that the traditional rules for causality are not observed: the usual requirement for temporal association, the extent to which side effects arise in temporal association with starting treatment and also the extent to which side effect subsidence coincides with stopping treatment is, at least in part, not upheld with some of the toxicity and efficacy profiles for ipilimumab. There are many other approaches designed to decrease inherent immune suppression in and around the cancer micro-environment which are also being investigated. These novel approaches may also violate the conventional rules of causality. Examples of other approaches that deregulate the immune sstem include Ontak (an approved drug using an anti IL-2-diptheria toxin conjugate) and PD 1 manipulation (PD 1 sits on regulatory T cells that supress immune mediated cancer killing and there are a number of inhibitors currently in clinical development) 39.
Forthcoming T cell therapies
When T cells are activated they become both potent and unrelenting cytodestructive agents. This can be demonstrated in the setting of acute or even hyperacute rejection of transplantation where the potent and otherwise highly toxic drug cyclophosphamide provides a life-saving counter balance to the T cells trying to reject the foreign transplant. The powerful armamentarium of the activated T cell is being refined and the striking efficacy and, by equal measure, toxicity potential of this system deserves further technological exploitation. Two examples of such approaches are chimaeric antigen receptors (CARs) and Bites.
Chimeric antigen receptors and Bites
Investigators are turning to the T cell using more traditional antibody components as a bridge with the attraction being that the T cell might perhaps be more potent than an equivalently targeted antibody acting alone. Both the CAR 40, 41 technology and the bispecifics are T cell-engaging antibodies that harness the recognized specificity of an antibody directed towards target (in the referenced case it is actually the B cell itself that is being attacked as it bears the specific CD19 molecule). The proposed mechanism of action is to bind the cancer target with an antibody component previously raised against CD19 and ally this with activated T cells. Bites use another fused antibody component (another scFv fragment) which specifically binds the CD3 molecule on T cells (Figure 7). The CD3 molecule is a T cell surface molecule found on all mature T cells and that functions as part of a molecular surface cluster in unison with the T cell receptor α and β complex. The CAR technology is similar in that an antibody is used to recognize target but the whole T cell is ‘attached’ – by virtue of the CAR technology being genetically embedded within the T cell. CAR cells are manufactured ex vivo by taking a patient's harvested T cells and enhancing them with activating CD3 and CD137 moieties spliced to the targeting anti-CD19 antibody (Figure 8). CD137 like CD3 is a co-stimulatory molecule found on the surface of many immune system cells including most T cells. The fundamental proposition with the CAR approach is that of infusion of readily excitable (CD3 and CD137 loaded) T cells that are targeted (antibody construct to CD19) against B cell malignancies. The technology bearing T cells have also been selected in the expansion/selection process based upon their own natural expression of CD28, another activating moiety. The combined effect of CD3 and CD137 expression along with CD28 selection is to create a cell infusion that engrafts in patients whilst not showing too much off-target excitability. Previous CAR approaches failed to engraft and this was attributed in part to cells not expressing as much co-stimulatory apparatus.
Figure 7.
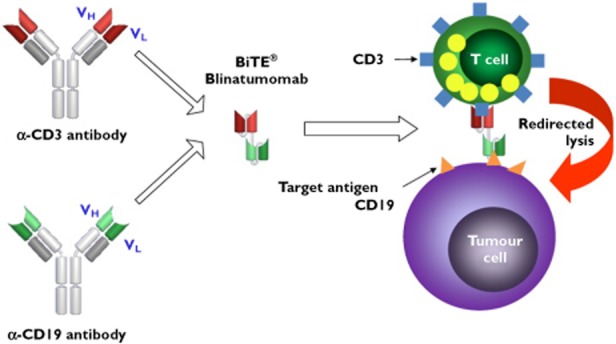
BiTE antibodies. The variable region (ScFv) of an antibody recognizes cell surface epitopes. The short chain component of any given epitope specific antibody can be replaced with another ScFv fragment that recognizes and activates CD3 on T cells. The product is a ‘Siamese’ antibody that specifically and in sequence can recognize two separate entities, e.g. CD 19 and CD 3 hence the term ‘bispecific T cell engaging: BiTE’. The antibody is thought to function as an introduction agent allowing T cells to bind to and become activated around cancer targets such as the cell surface molecule CD19 present on some B cell neoplasms. The subsequent degranulation of the T cell results in release of T cell substances such as perforin and granzyme that are cytotoxic to the target cancer
Figure 8.
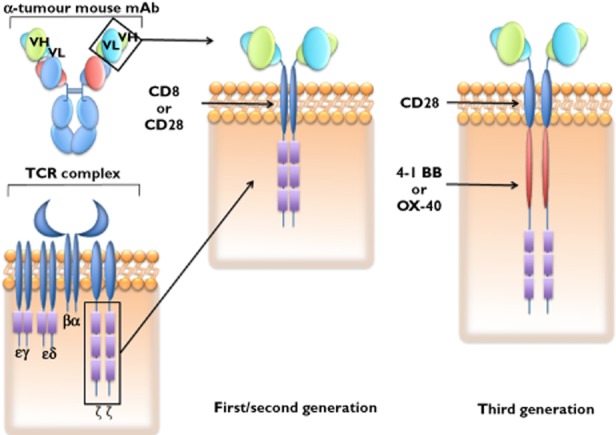
Schematic representation of the evolution of chimeric antigen receptor technology. DNA coding the variable components of both heavy and light chains of an antibody binding domain with a known affinity for a given tumour target is placed, using viral insertion techniques, within the DNA repertoire of T cells. This antibody DNA is incorporated contiguously with additional DNA coding either the CD8 or CD28 chain and DNA coding the CD ζ component of the native T cell receptor complex. Additional code in the latest generation of CAR technology includes the 4-1BB co-stimulatory domain as well. The addition of more than cone co-stimulatory feature appears to be both necessary and sufficient for T cell persistence and memory formation in the clinic
Bites
Blinatumomab is an antibody raised like the CAR technology against CD19. The tail end of this antibody is fused with an anti CD3 binding antibody (effectively a double headed antibody) that engages T cells. In a phase 1 trial 12 out of 13 patients with non-Hodgkins lymphoma were reported to have responded at the two highest doses tested of blinatumomab 42. Significant, though not insurmountable, levels of central nervous system toxicity were also noted. Though these CNS events were characterized as rescinding within days of stopping the otherwise continuously-infused drug (half-life ∼2 h) it remains to be seen if this toxicity will be reproducibly reversible in larger phase trials.
Chimeric antigen receptors technology
Infusing billions of T cells is a departure from the traditional pharmacologic approach but exposure can still be measured and half-lives, pharmacodynamics and pharmacokinetics still apply. A salient difference with CAR cells is the feature of engraftment where a product that is initially ostensibly decaying with a half-life of a number of days is suddenly seen to engraft and develop a half-life that in some cases becomes either effectively indefinite or even reversed as the product starts to proliferate. Such dynamics might even one day require a doubling-time to describe them though such proliferation dynamics have yet to be fully elaborated. A feature of stem cell technology and lymphocyte engraftment is the ability to define a point when the graft has become endemic. The active agent is genetically introduced ex vivo to the host T cells using a lentiviral vector before return to the patient. The CAR methodology includes the co-splicing of genetic code for CD137 (a co-stimulatory receptor in T cells [4-1BB]) and CD3-zeta (an activating signal transduction component of the T cell antigen receptor). Providing both a CD3 and CD137 stimulus might be thought to be excessive though it should be recognized that the CAR technology discussed here is third generation (Figure 7) 40. Previous generations of CAR cells included fewer growth stimuli and it is thought as a consequence they failed to get the T cells to persist in vivo beyond the initial passive transfer stage. The CAR technology in still in early phase trials but either complete, durable or partial, durable tumour clearance has been seen with all of the first three patients treated. The transfused T cells were demonstrated to have actively replicated and probably undertook ‘serial killing’ based on both the pre-clinical laboratory observation of serial killing by individual T cells in culture and the clinical observation that the number of T cells returned to the patient was a couple of orders of magnitude less than the overall (estimated) number of cancer cells that were eradicated 40, 41. The reintroduced T cell constructs enter the patient's system, replicate up to 1000-fold, engraft themselves and proceed to orchestrate mass tumoricide. Once a T cell has undergone a salient interaction with a target antigen then residual memory cell formation is thought to occur. In short the therapy takes on a life of its own. The magnitude of efficacy (and toxicity) merits caution as it was reported that one patient suffered a tumour-lysis syndrome. Tumour lysis syndrome is a phenomenon whereby host organs become overburdened with the by-products of mass leukaemic cell death. The syndrome can manifest in a number of ways including acute gout, hyperkalaemia and/or renal damage. Once a T cell has engrafted so will any ensuing efficacy or toxicity engraft also and, as was seen with steroid treatment of ipilimumab toxicities, reversing this with steroids or other antidotes is of variable success. Only when trials are conducted in larger numbers of patients or in the post-marketing field will the true breadth and depth of toxicity become manifest. However in the short term it appears that these forecast risks will follow and hopefully not outweigh the currently manifest efficacy results.
Engineered T cell receptors
The two previous examples both redirected the T cell effector function, either by splicing on a CD3 targeting antibody fragment placing antibody genes inside a T cell. An alternative approach is to combine CD3 function with its more natural partner the T cell receptor 1. The previous two examples (CARs and Bites) used an antibody/T cell combination thereby establishing early proof for the hypothesis that the T cell is a potentially very effective anti-cancer agent in the oncology context. This insight might have been predicted from the undeniable excess of cancers that patients suffer from when placed on long term immunosuppression in settings such as post-transplantation.
Antibodies are part of a network of immune surveillance but the act of recognition is more of a 1:1 antigen/antibody binding event. With TCRs both the receptor and the HLA molecule have co-evolved around the peptide sequence and therefore the specificity and sensitivity of this recognition event is bolstered by this evolutionary equivalent to the opposable thumb. The sine qua non of the HLA complex is that it helps introduce, specify and engender the T cell peptide recognition step. As a consequence of working with a system which nature itself has invested with ornate functionality it must be noted that very careful engineering of this system is required to avoid unintended dysfunction. An example of this is the observation from our own protein engineering platform that the majority of engineered HLA-TCR binding events remain similar or even degraded in their specificity when mutated from the natural receptor and only very rarely can one introduce new mutations that are both better in their affinity for the HLA-peptide pairing whilst also retaining the critical specificity that is required if they are not to bind to most HLA molecules bearing non-specific peptides i.e. the absence of off-target binding is also retained. Immunoglobulin engineering allows many more sites where mutations can be safely introduced as the diversity at the antibody-antigen interface is itself greater 43.
Of concern in oncology is the fact that many cancers display abnormally low numbers of HLA peptides. Cancers generally down regulate proteins that are targets of attack, not just HLA molecules 28. Such scarcity of HLA is probably selected for if an evolutionary model of heterogeneous cancer cell biology is adopted and T cell surveillance is considered a selection pressure 29–33. Engineering T cell receptors to recognize and kill cells bearing unusually low HLA peptide concentrations represents a potentially novel and wide-ranging paradigm for overcoming this observation and it is likely that such engineering will obtain sensitivities one or two magnitudes better than the equivalent antibody targets because of the intrinsic sensitivity of the HLA system. Currently there are several modified T cell receptor approaches in the clinic 44, 45. In brief T cell receptors are taken from donors and screened for their ability to recognize a candidate antigen. The peptide specific T cell receptor is then engineered towards the two ostensibly dissonant goals of both generating higher binding to the specific HLA-cancer-peptide complex and retaining little or no binding to other non-specific HLA peptide/normal tissue complexes.
Cancer testis antigens (CTAs) are antigens that arise as a result of tumour cells regressing to embryonic type 46, 47. These developmentally-expressed and ‘exclusively’ embryonic antigens become repressed in adult life. Subsequently these CTAs become de-repressed in cancer cells and after proteosomal processing/degradation some can be detected using mass spectroscopy as part of the HLA-peptide expression system on the surface of cancer cells. Targeting these expressed peptide signatures with high affinity T cell receptors offers the possibility that normal tissues will not be subject to attack whilst cancerous tissue will. One such target, NY-ESO, is currently being pursued in the phase 1 setting with TCRs directed against it.
New T cell paradigms for toxicity testing
HLA molecules are so specific that they cause rejection of organs when transplanted from non-matching individuals of the same species 43. Testing of HLA specific molecules requires use of T cell receptors that have specifically been engineered with one chosen HLA type in mind. Binding to other HLA complexes is a reason to reject a candidate TCR in the engineering process because it is an almost certain sign of lack of peptide specificity. The HLA component of T cell activity is so specific for example that in the current phase 1 clinical trial only patients with HLA A2 sub-types can be included. The drug is not intended for, nor is it likely to show any effect in, any other HLA-type of individual. Pre-clinical screening with certain HLA sub-types in blood spiking assays revealed this predicted feature where reactivity was only ever seen in the appropriate HLA matched subjects and non-matched HLA spiking experiments were anodine. For these reasons only humans of a certain HLA restriction are considered valid efficacy and toxicity models for HLA restricted-engineered T cell receptors. Though perhaps obvious this is an important observation because it follows from this that no animal model would ever work. Animal HLA systems are completely different and both on and off target toxicity would likely be completely irrelevant and most likely absent altogether as the animal has no cognate HLA. On this point the MHRA has taken the considered and pioneering step of letting TCRs go into the clinic with animal toxicology at a streamlined minimum.
Adoptive T cell therapy with engineered T cell receptors
The National Institute of Health in North America and some small stage biotechnology companies are working with T cell receptors transducing them into T cells using viral technology not dissimilar to the CAR approach (Figure 9). Transduced T cells bear T cell receptors engineered towards a similar, albeit lower affinity, goal to that of the soluble TCRs. Early clinical results including some complete responses induced on the back of a chemotherapy conditioning regime with an NY-ESO targeting approach in sarcoma and melanoma look exciting 48. One pharmacological challenge with adoptive T cell therapy involves trying to estimate how many cells should be transduced into the patient. Simple logic might embrace the ‘as many as possible’ approach. T cells, however, need to engraft in the patient and this has been achieved historically with typical doses of T cells in the order of 1–10 billion cells. Doses with as few as 10 million cells have been associated with successful engraftment and tumour killing 41. Transducing and transfusing fewer cells offers both theoretical and immediate advantages: Growing cell volumes ex vivo can take weeks and is fraught with danger regarding infection and even the remote potential of oncogenesis (from the lentivirus insertion process). If 10 million cells were no less clinically effective than 10 billion a large amount of time and effort spent between initial cell donation and return of the therapeutic agent could immediately be saved in a context where the patient's time is already painfully limited. From a theoretical perspective it seems innately fair to think that the longer a cell lineage is out of the body and the more replications it undergoes the more removed from safety and efficacy it is likely to be become and the less likely it is to indigenize/engraft upon return. There are theoretical arguments also such that 10 million transfused cells could result in less tumour lysis syndrome than 10 billion cells (if the initial hit on tumour load is drawn out over a predictably less concentrated period of time) and also there is hope that the extra T cell replication that is required in vivo when small doses are infused is more conducive to memory cell formation. Development of these arguments in further detail is beyond the scope of this article but in brief the discussion revolves around the intriguing prospect that white cell levels may be homeostatically controlled within the body and therefore chemotherapy depletion may provide a stronger growth stimulus to incoming cells in circumstances where there are fewer infused cells 48, 49. T cell replication in turn is thought to predispose a patient to the installation of long term immune memory formation. For these reasons the concept in onco-pharmacology that there might be an optimum biological dose significantly below that of the maximum tolerated dose is once again back on the agenda after having initially undergone debate at the beginning of the targeted therapy era 50.
Figure 9.
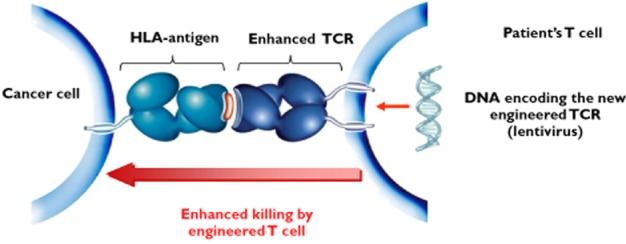
Viruses can be used to introduce the coding material for supra-physiologically high affinity T cell receptors. The National Institute of Health have done this with coding material for a T cell receptor that binds to the cancer specific antigen NY-ESO with higher affinity than operates in natural conditions. Responses have been seen in several sarcoma patients with this approach. Synovial sarcoma specifically is known to express NY-eso in about 75% of cases
Conclusions
The prospect of T cell receptor pharmacology is both exciting and challenging. Initial successes including complete remissions in leukaemia have been achieved at not-inconsequential expenses, such as tumour lysis syndrome. Future therapies look to unlock further the secrets of cancer and will hopefully do so in more a targeted, efficient and cost-considered manner. Animal models have proven helpful in elucidating mechanisms of immune suppression in the natural history of onco-pathogenesis. However these same models can be misleading or irrelevant with respect to disease treatments that are intrinsically human in nature. Investigators are encouraged to proceed with both optimism and caution.
Competing Interests
The author has completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declares DPS had support from Immunocore Ltd for the submitted work, has been within the full time employ of Immunocore Ltd for the last 1 year and has no other relationships or activities that could appear to have influenced the submitted work
References
- 1.Liddy N, Bossi G, Adams KJ, Lissina A, Mahon TM, Hassan NJ, Gavarret J, Bianchi FC, Pumphrey NJ, Ladell K, Gostick E, Sewell AK, Lissin NM, Harwood NE, Molloy PE, Li Y, Cameron BJ, Sami M, Baston EE, Todorov PT, Paston SJ, Dennis RE, Harper JV, Dunn SM, Ashfield R, Johnson A, McGrath Y, Plesa G, June CH, Kalos M, Price DA, Vuidepot A, Williams DD, Sutton DH, Jakobsen BK. Monoclonal TCR-redirected tumor cell killing. Nat Med. 2012;18:980–987. doi: 10.1038/nm.2764. [DOI] [PubMed] [Google Scholar]
- 2.Halloran PF. Immunosuppressive drugs for kidney transplantation. N Engl J Med. 2004;351:2715–2729. doi: 10.1056/NEJMra033540. [DOI] [PubMed] [Google Scholar]
- 3.Schuurman HJ, Cottens S, Fuchs S, Joergensen J, Meerloo T, Sedrani R, Tanner M, Zenke G, Schuler W. SDZ RAD, a new rapamycin derivative: synergism with cyclosporine. Transplantation. 1997;64:32–35. doi: 10.1097/00007890-199707150-00007. [DOI] [PubMed] [Google Scholar]
- 4.Stepkowski SM, Tian L, Napoli KL, Ghobrial R, Wang ME, Chou TC, Kahan BD. Synergistic mechanisms by which sirolimus and cyclosporin inhibit rat heart and kidney allograft rejection. Clin Exp Immunol. 1997;108:63–68. doi: 10.1046/j.1365-2249.1997.d01-984.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.June CH. Adoptive T cell therapy for cancer in the clinic. J Clin Invest. 2007;117:1466–1476. doi: 10.1172/JCI32446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hammarsten JF, Tattersall W, Hammarsten JE. Who discovered smallpox vaccination? Edward Jenner or Benjamin Jesty? Trans Am Clin Climatol Assoc. 1979;90:44–55. [PMC free article] [PubMed] [Google Scholar]
- 7.Brown K. Penicillin Man: Alexander Fleming and the Antibiotic Revolution. Stroud: Sutton; 2004. ISBN 0-7509-3152-3. [Google Scholar]
- 8.Waring MJ, Ponder BAJ, editors. The Search for New Anti-Cancer Drugs. Lancaster: Kluwer Academic Publishers; 1991. p. 284. [Google Scholar]
- 9.John TJ, Samuel R. Herd immunity and herd effect: new insights and definitions. Eur J Epidemiol. 2000;16:601–606. doi: 10.1023/a:1007626510002. doi: 10.1023/A:1007626510002. PMID 11078115. [DOI] [PubMed] [Google Scholar]
- 10.Paust S, Cantor H. Regulatory T cells and autoimmune disease. Immunol Rev. 2005;204:195–207. doi: 10.1111/j.0105-2896.2005.00247.x. [DOI] [PubMed] [Google Scholar]
- 11.Nada A, Somberg J. First-in-Man (FIM) clinical trials post-TeGenero: a review of the impact of the TeGenero trial on the design, conduct, and ethics of FIM trials. Am J Ther. 2007;14:594–604. doi: 10.1097/MJT.0b013e31813737dd. [DOI] [PubMed] [Google Scholar]
- 12.Stebbings R, Findlay L, Edwards C, Eastwood D, Bird C, North D, Mistry Y, Dilger P, Liefooghe E, Cludts I, Fox B, Tarrant G, Robinson J, Meager T, Dolman C, Thorpe SJ, Bristow A, Wadhwa M, Thorpe R, Poole S. ‘Cytokine storm’ in the phase I trial of monoclonal antibody TGN1412: better understanding the causes to improve preclinical testing of immunotherapeutics. J Immunol. 2007;179:3325–3331. doi: 10.4049/jimmunol.179.5.3325. [DOI] [PubMed] [Google Scholar]
- 13.UK Ministry of Health and Regulatory Affairs – Independent Scientific Expert Committee. Clinical trial final report. 25 May 2007. Available at http://www.mhra.gov.uk/NewsCentre/Pressreleases/CON2023822 (last accessed 6 November 2012)
- 14.Suntharalingam G, Perry MR, Ward S, Brett SJ, Castello-Cortes A, Brunner MD, Panoskaltsis N. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med. 2006;355:1018–1028. doi: 10.1056/NEJMoa063842. [DOI] [PubMed] [Google Scholar]
- 15.Wadman M. London's disastrous drug trial has serious side effects for research. Nature. 2006;440:388–389. doi: 10.1038/440388a. [DOI] [PubMed] [Google Scholar]
- 16.Muller PY, Brennan FR. Safety assessment and dose selection for first-in-human clinical trials with immunomodulatory monoclonal antibodies. Clin Pharmacol Ther. 2009;85:247–258. doi: 10.1038/clpt.2008.273. [DOI] [PubMed] [Google Scholar]
- 17.Dietert RR. Fractal immunology and immune patterning: potential tools for immune protection and optimization. J Immunotoxicol. 2011;8:101–110. doi: 10.3109/1547691X.2011.559951. [DOI] [PubMed] [Google Scholar]
- 18.Atlan H, Cohen IR. Immune information, self-organization and meaning. Int Immunol. 1998;10:711–717. doi: 10.1093/intimm/10.6.711. [DOI] [PubMed] [Google Scholar]
- 19.Pillai V, Ortega SB, Wang CK, Karandikar NJ. Transient regulatory T-cells: a state attained by all activated human T-cells. Clin Immunol. 2007;123:18–29. doi: 10.1016/j.clim.2006.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tamir P, Zohar A. Anthropomorphism and teleology in reasoning about biological phenomena. Sci Educ. 1991;75:57–67. [Google Scholar]
- 21.FDA – Vaccines, Blood and Biologics Division. Approval Letter – Provenge. Available at http://www.fda.gov/BiologicsBloodVaccines/CellularGeneTherapyProducts/ApprovedProducts/ucm210012.htm (last accessed 29 April 2010)
- 22.Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, Redfern CH, Ferrari AC, Dreicer R, Sims RB, Xu Y, Frohlich MW, Schellhammer PF IMPACT Study Investigators. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. New Engl J Med. 2010;363:411–422. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- 23.Anassi E, Ndefo UA. Sipuleucel-T (Provenge) injection: the first immunotherapy agent (vaccine) for hormone-refractory prostate cancer. Pharm Ther. 2011;36:197–202. [PMC free article] [PubMed] [Google Scholar]
- 24.Chambers JD, Neumann PJ. Listening to Provenge™ what a costly cancer treatment says about future medicare policy. N Engl J Med. 2011;364:1687–1689. doi: 10.1056/NEJMp1103057. [DOI] [PubMed] [Google Scholar]
- 25.DeFrancesco L. Provenge twists again. Nat Biotechnol. 2010;28:882. [Google Scholar]
- 26.Huber ML, Haynes L, Parker C, Iversen P. Interdisciplinary critique of sipuleucel-T as immunotherapy in castration-resistant prostate cancer. J Natl Cancer Inst. 2012;104:273–279. doi: 10.1093/jnci/djr514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McCoy KD, Le Gros G. The role of CTLA-4 in the regulation of T cell immune responses. Immunol Cell Biol. 1999;77:1–10. doi: 10.1046/j.1440-1711.1999.00795.x. [DOI] [PubMed] [Google Scholar]
- 28.Aris M, Barrio M, Mordoh J. Lessons from cancer immunoediting in cutaneous melanoma. Clin Dev Immunol. 2012;2012 doi: 10.1155/2012/192719. 192719. doi: 10.1155/2012/192719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Levey DL, Srivastava PK. Alterations in T-cells of cancer-bearers: whence specificity? Immunol Today. 1996;17:365–368. doi: 10.1016/0167-5699(96)10013-X. [DOI] [PubMed] [Google Scholar]
- 30.Kiessling R, Wasserman K, Horiguchi S, Kono K, Sjoberg J, Pisa P, Petersson M. Tumor-induced immune dysfunction. Cancer Immunol Immunother. 1999;48:353–362. doi: 10.1007/s002620050586. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dye E, North R. T cell-mediated immunosuppression as an obstacle to adoptive immunotherapy of the P815 mastocytoma and its metastases. J Exp Med. 1981;154:1033–1042. doi: 10.1084/jem.154.4.1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kusmartsev SA, Li Y, Chen SH. Gr-1+ myeloid cells derived from tumor-bearing mice inhibit primary T cell activation induced through CD3/CD28 costimulation. J Immunol. 2000;165:779–785. doi: 10.4049/jimmunol.165.2.779. [DOI] [PubMed] [Google Scholar]
- 33.Thornton AM, Shevach EM. CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J Exp Med. 1998;188:287–296. doi: 10.1084/jem.188.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Herber DL, Nagaraj S, Djeu JY, Gabrilovich DI. Mechanism and therapeutic reversal of immune suppression in cancer. Cancer Res. 2007;67:5067–5069. doi: 10.1158/0008-5472.CAN-07-0897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJ, Lutzky J, Lorigan P, Vaubel JM, Linette GP, Hogg D, Ottensmeier CH, Lebbé C, Peschel C, Quirt I, Clark JI, Wolchok JD, Weber JS, Tian J, Yellin MJ, Nichol GM, Hoos A, Urba WJ. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Robert C, Thomas L, Bondarenko I, O'Day S, Weber J, Garbe C, Lebbe C, Baurain JF, Testori A, Grob JJ, Davidson N, Richards J, Maio M, Hauschild A, Miller WH, Jr, Gascon P, Lotem M, Harmankaya K, Ibrahim R, Francis S, Chen TT, Humphrey R, Hoos A, Wolchok JD. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364:2517–2526. doi: 10.1056/NEJMoa1104621. [DOI] [PubMed] [Google Scholar]
- 37.Bakacs T, Mehrishi JN, Moss RW. Ipilimumab (Yervoy) and the TGN1412 catastrophe. Immunobiology. 2012;217:583–589. doi: 10.1016/j.imbio.2011.07.005. [DOI] [PubMed] [Google Scholar]
- 38.Fischkoff SA, Hersh E, Weber J. Durable responses and long-term progression-free survival observed in a phase II study of MDX-010 alone or in combination with dacarbazine (DTIC) in metastatic melanoma. J Clin Oncol. 2005;23:716s. [Google Scholar]
- 39.Urba WJ, Longo DL. Redirecting T cells. N Engl J Med. 2011;365:754–757. doi: 10.1056/NEJMe1106965. [DOI] [PubMed] [Google Scholar]
- 40.Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, June CH. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3:95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor–modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nagorsen D, Zugmaier G, Viardot A, Goebeler M, Noppeney R, Schmidt M, Klappers P, Baeuerle PA, Kufer P, Bargou RC. Confirmation of safety, efficacy and response duration in non-Hodgkin lymphoma patients treated with 60 μg/m2/d of BiTE® antibody blinatumomab. Blood ASH Annual Meeting Abstracts. 2009;114:2723. [Google Scholar]
- 43.Janeway CA, Jr, Travers P, Walport M, Shlomchik MJ. Immunobiology: The Immune System in Health and Disease. 5th edn. New York: Garland Science; 2001. [Google Scholar]
- 44.Boulter JM, Jakobsen BK. Stable, soluble, high-affinity, engineered T cell receptors: novel antibody-like proteins for specific targeting of peptide antigens. Clin Exp Immunol. 2005;142:454–460. doi: 10.1111/j.1365-2249.2005.02929.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lunde E, Løset GÅ, Bogen B, Sandlie I. Stabilizing mutations increase secretion of functional soluble TCR-Ig fusion proteins. BMC Biotechnol. 2010;10:61. doi: 10.1186/1472-6750-10-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Simpson AJ, Caballero OL, Jungbluth A, Chen YT, Old LJ. Cancer/testis antigens, gametogenesis and cancer. Nat Rev Cancer. 2005;5:615–625. doi: 10.1038/nrc1669. [DOI] [PubMed] [Google Scholar]
- 47.Jungbluth AA, Chen Y-T, Stockert E, Busam KJ, Kolb D, Iversen K, Coplan K, Williamson B, Altorki N, Old LJ. Immunohistochemical analysis of NY-ESO-1 antigen expression in normal and malignant human tissues. Int J Cancer. 2001;92:856–860. doi: 10.1002/ijc.1282. [DOI] [PubMed] [Google Scholar]
- 48.Robbins PF, Dudley ME, Wunderlich J, El-Gamil M, Li YF, Zhou J, Huang J, Powell DJ, Jr, Rosenberg SA. Cutting edge: persistence of transferred lymphocyte clonotypes correlates with cancer regression in patients receiving cell transfer therapy. J Immunol. 2004;173:7125–7130. doi: 10.4049/jimmunol.173.12.7125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gattinoni L, Finkelstein SE, Klebanoff CA, Antony PA, Palmer DC, Spiess PJ, Hwang LN, Yu Z, Wrzesinski C, Heimann DM, Surh CD, Rosenberg SA, Restifo NP. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. Exp Med. 2005;202:907–912. doi: 10.1084/jem.20050732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Adjei AA. What is the right dose? The elusive optimal biologic dose in phase I Clinical Trials. J Clin Oncol. 2006;24:4054–4055. doi: 10.1200/JCO.2006.07.4658. [DOI] [PubMed] [Google Scholar]