

Abstract
While asthma is a chronic inflammatory disorder that is managed with inhaled controller and reliever drugs, there remains a large unmet need at the severe end of the disease spectrum. Here, a novel stratified approach to its treatment is reviewed, based upon identification of causal pathways, with a focus on biologics. A systematic search of the literature was made using Medline, and publications were selected on the basis of their relevance to the topic. Despite strong preclinical data for many of the more recently identified asthma targets, especially those relating to the T-helper 2 allergic pathway, clinical trials with specific biologics in moderate to severe asthma as a group have been disappointing. However, subgroup analyses based upon pathway-specific biomarkers suggest specific endotypes that are responsive. Application of hypothesis-free analytical approaches (the ‘omics’) to well-defined phenotypes is leading to the stratification of asthma along causal pathways. Refinement of this approach is likely to be the future for diagnosing and treating this group of diseases, as well as helping to define new causal pathways. The identification of responders and nonresponders to targeted asthma treatments provides a new way of looking at asthma diagnosis and management, especially with biologics that are costly. The identification of novel biomarkers linked to well-phenotyped patients provides a stratified approach to disease management beyond simple disease severity and involving causal pathways. In order to achieve this effectively, a closer interaction will be required between industry (therapeutic and diagnostic), academia and health workers.
Keywords: asthma, biologics, biomarkers, stratification, treatment
Historical perspective
In 1860, Henry Hyde Salter first identified asthma as a distinct disease entity, separating it from the previously broad use of the Greek term meaning ‘shortness of breath’. His experiences are related in his treatise ‘On Asthma: Its Pathology and Treatment’; he carefully separated asthma from other obstructive diseases of the airways by identifying contraction of smooth muscle as the primary cause of the airway obstruction 1. As a physician practising in London, over many years he was able to collect 50 patients who provided the basis for his observations including, purportedly, having asthma himself. With the recognition of the importance of ‘bronchospasm’ in the symptomatology of the disease, treatment was directed towards bronchodilators. In the 19th century this included Datura stramonium, which was smoked to release anticholinergic alkaloids. Asthma cigarettes were available in the UK up to 1985. Much earlier, the Chinese had identified Ma huang, a herb used as an oral treatment for respiratory disease, from which ephedrine was subsequently identified and synthesized 2. Salter also recognized that black coffee offered some relief to asthma sufferers, but it was not until the 1930s that theophylline, a methylxanthine, was identified as the active agent, along with other xanthines, such as caffeine and theobromine 3. Theophylline and its ethylenediamine salt, aminophylline, were widely adopted as asthma therapies up to the end of the 20th century, administered orally for chronic disease control and intravenously or rectally for acute asthma 3. Theophylline and its analogues are still widely used, especially in low- and middle-income countries, because it is a cheap and efficacious therapy; however, its popularity is waning, largely on account of potentially serious side-effects, especially cardiac arrhythmias, convulsions and diuresis, as well as nausea and vomiting. Slow-release preparations and blood drug level monitoring were introduced to increase the therapeutic index of theophylline, but use of these preparations also is now falling 3.
A major breakthrough in asthma treatment came in 1901 with the identification of adrenaline 4, which was shown to be a powerful bronchodilator and cardiovascular stimulant when administered systemically 5. Over the following 100 years, the potent bronchodilator action of adrenaline was separated from cardiovascular effects by separating β- from α-adrenoceptor activity, leading to the introduction of isoprenaline, followed by subdivision of β-receptors into β1 and β2, the latter carrying the bronchodilator response, leading to the introduction of salbutamol and related β2 agonists, such as terbutaline 5 (Figure 1). Finally, the identification of key molecular characteristics that produce bronchodilatation led to the introduction of salmeterol and formoterol as long-acting β2-agonists (LABAs) 6. Ultra-long-acting bronchodilators are now appearing (e.g. indacaterol) 7, suggesting that molecular manipulation of adrenergic bronchodilators has probably reached the maximum that can be reasonably achieved.
Figure 1.

The evolution of inhaled β-adrenoceptor bronchodilators for the treatment of asthma. The discovery of adrenaline in 1901 and its powerful bronchodilator effect, initially when administered by injection but later as a nebulized aqueous aerosol, was to stimulate a relentless search for improved specificity, potency and duration of action. Application of medicinal chemistry and classical structure–activity pharmacology led to the sequential separation of α- (noradrenaline) from β-receptor activity (isoprenaline), followed by the selection of β2 activity, mediating airway smooth muscle relaxation and anti-bronchoconstriction, from cardiovascular β1 effects (e.g. salbutamol, turbutaline). The next discovery was to incorporate structural properties that extended the duration of action to 12 h (salmeterol, formoterol) and finally to 24 h (indacaterol) following a single inhalation
The rise in asthma mortality in the 1960s and 1970s that was seen in those countries where inhaled β-agonists became available without prescription and in high doses 8 led to an intense series of studies that pointed to β-adrenoceptor tolerance as the most likely cause 9. A second peak of asthma deaths in New Zealand in the late 1980s has been attributed to the introduction of a high-dose formulation of inhaled fenoterol 8. While controversial, these peaks in asthma mortality have driven research into the underlying causes of asthma. The recognition that airway inflammation is a common feature of asthma 10 and the discovery that inhaled corticosteroids can both control asthma and release airway inflammation 11 has been the driving force for using anti-inflammatory drugs to control asthma. Beginning with the discovery of the potent anti-inflammatory activity of cortisone by Phillip Hench in 1947 12 and the discovery of inhaled beclomethasone disproprionate in the early 1970s as a highly active ‘controller’ drug for asthma 13, there have been many attempts to improve potency and safety, with limited success. The main problem has been producing anti-inflammatory drugs free of endocrine activity and, at the same time, maintaining desirable potency, bioavailability and pharmacokinetics 14. The search continues, with the important discovery that corticosteroids produce much of their suppressive effect on inflammation by interfering with transcription factor activation of pro-inflammatory and prosurvival genes in immune and inflammatory cells (transrepression), whereas their endocrine effects result from the corticosteroid–receptor complex activating selective genes (transactivation) 15.
In parallel with these developments, there has been development of inhaler devices, from the hand-held nebulizer through to the pressurized metered-dose aerosol to inhaled dry powder devices and those that time and meter dose administration 16. The vast range of different inhaler devices now available has its problems in causing patient confusion as well as operating difficulties and, along with the social stigma accompanying their use and fear of side-effects, this could be an important factor contributing to lack of adherence to prescribed therapies 17. Accepting this as an ongoing problem in asthma management, corticosteroid refractoriness is increasingly being recognized as an unmet problem in asthma, especially in those patients with more severe disease, and constitutes the majority of patients with ‘difficult-to-treat’ asthma 18. A range of factors have been incriminated in such corticosteroid refractory disease 19, but one that has recently come to the forefront is tobacco smoking 20.
To avoid the inevitable endocrine side-effects of ever-increasing doses of inhaled corticosteroid (ICS) for uncontrolled asthma, combination therapy is advocated, in which a LABA and an ICS are combined in a single inhaler. However, even this approach has ‘responders’ and ‘nonresponders’. It should also be recognized that ICS started at the inception of asthma has no effects on the natural history of the disease despite effectively suppressing airway inflammation 21.
Anti-allergic approaches to asthma
All of the treatments so far referred to act on the secondary consequences of asthma (i.e. bronchospasm and inflammation) rather than treating the underlying mechanistic and aetiological causes of the disease 21. Atopy and allergic mechanisms have long been recognized as contributing to asthma in a high proportion of patients, leading to its classification as an atopic disorder. While allergen exposure is a powerful trigger for early (mast cell-driven) and late (inflammatory cell-driven) airway narrowing in asthma 22, somewhat counterintuitively, attempts at allergen avoidance have had limited success, even with such common allergens as those from dust mites 23. Likewise, while there has been some success with allergen-specific immunotherapy, either systemically by subcutaneous injections or sublingually, this is in large part in those with single-allergen sensitization, e.g. cat or pollen, rather than multiple allergens, which is the situation in most asthmatics 24. Moreover, allergen-reduction strategies in young children genetically at risk of asthma have had little success 25 combined with other interventions (e.g. breast-feeding, environmental tobacco smoke avoidance) 26.
Recognition that mediator release from mast cells is important in the acute asthmatic response with allergen challenge but also that mast cell activation is a feature of more chronic asthma 27 has stimulated interest in agents that can inhibit mast cell activation and mediator release. The first of these was sodium cromoglicate (SCG), derived from the flavonoid khelin extracted from the herb Ammi visnaga by Altounyan in the 1950s. Inhaled sodium cromoglicate was shown to inhibit both allergen-induced early and late asthmatic responses and exercise-induced asthma 28. In vitro, SCG inhibited IgE-dependent mast cell mediator release and, in 1968, was shown to be effective as a treatment for asthma when inhaled regularly 29. Nedocromil sodium is a second-generation drug with similar properties but is more active 30. Even during its early development, it was recognized that only a proportion of patients responded well to SCG, especially children 31. However, subsequent meta-analysis and a Cochrane Review concluded that ‘there was insufficient evidence to be sure about the efficacy of SCG over placebo’ 32 and, as a consequence, it has been withdrawn from the World Health Organization list of drugs, despite some concerns over the methods used and the limited number of trials selected for the analysis 33. For those asthmatics who benefited from SCG as a safe and effective anti-allergic therapy, this was a loss. If only it had been possible at the time of its development to have a clear idea of the asthma phenotype most likely to respond to SCG, then the drug might be available today. Another stumbling block was the lack of an underlying pharmacological mechanism. Although there was some evidence that SCG inhibited the chloride flux associated with mast cell activation 34, the precise ion channel involved eluded discovery. Both SCG and nedocromil sodium have recently been found to be potent and selective inhibitors of the G protein-coupled receptor 35 that recognizes its natural ligand 2-acyl-lysophosphatidic 35, 36. G protein-coupled receptor 35 is one of several lysophosphatidic acid receptors that are involved in mast cell development and activation 37. Identification of this receptor class on mast cells might stimulate a search for compounds more active than SCG and nedocromil sodium because, while these drugs were active at suppressing mast cell activation in vitro and in vivo, they lacked potency and were also subject to tachyphylaxis, so that dose estimation was difficult 38.
Mast cell-derived histamine is a powerful bronchoconstrictor in asthma, acting via the H1-G protein-coupled receptor, which has led to its use as a provocation test to assess ‘nonspecific airway hyperresponsiveness’ (AHR). Given the importance of histamine as a contractile agonist of airway smooth muscle, it is somewhat surprising that H1-antihistamines, particularly the more potent selective inverse agonists, such as cetirizine, loratidine and fexofenadine, are not effective in asthma, in stark contrast to their proven efficacy in allergic rhinoconjunctivitis 39. Although there may be certain types of asthma that are responsive to H1-receptors, especially asthma associated with acute pollenosis 40, this lack of efficacy is puzzling if the mast cell is so important in driving AHR. One possible explanation is that in airways the smooth muscle H1-receptors activate an alternative cellular signalling mechanism from the one utilized by vascular endothelial cells in the nasal mucosa (responsible for much of the sympomatology of allergic rhinitis). This would require inhibitors to bind to a different component of the H1-receptor for effective inhibition, as has recently been proposed for β2-adrenoceptor functions with repeated dosing 41.
The situation of low efficacy is different for inhibitors of a second mast cell mediator class, the cysteinyl leukotrienes (cyst-LTs). The discovery that slow reacting substance of anaphylaxis (SRS-A), first identified by Kellaway and Trethewie in 1940, is a powerful smooth muscle contractile agent released upon allergen challenge that cannot not be inhibited with antihistamines led to a 50 year search for its structure. In 1989, Samuelsson identified SRS-A with a new family of lipid mediators, the cyst-LTs, of which LTC4 was the secreted form 42. Subsequent extracellular processing of LTC4 into LTD4 and eventually LTE4, in which the peptide side-chain was progressively shortened, provided the molecular basis for the biological effects of SRS-A, which has smooth muscle contractile activity almost 1000 times greater than that of histamine 43. The development of cyst-LT receptor antagonists (LTRAs; most notably, montelukast, zafirlukast and pranlukast) has provided the first orally active anti-asthma controller drugs beyond corticosteroids and xanthines. Shortly after their development, the receptor via which LTC4 and LTD4 contracts airway smooth muscle, the cyst-LTR1, was identified 44. With a remarkably good safety record, cyst-LTR1 antagonists are now widely used in asthma treatment, although head-to-head trials with inhaled corticosteroids have generally shown them to be less efficacious, and for most asthma guidelines ICSs are the first-line controller drugs. However, this may be an over simplification, because in head-to-head trials in which patient-related outcome measures have been used the difference in efficacy between ICSs and LTRAs is far less apparent 45. Moreover, in effectiveness studies conducted in the community (as opposed to efficacy studies in highly selected patients), montelukast used as first-line therapy was not different from ICS, and as add-on therapy to ICS, not different from the LABA salmeterol 46. This may be in part because adherence to treatment with once daily oral montelukast is greater than with inhaled drugs 47. In addition, montelukast is more active than ICS in asthmatic patients who smoke 48. It is salutary to know that <4% of asthmatic patients are represented in efficacy trials for drug registration 49. However, this greatly underestimates the spectrum of patients who eventually receive the drug in the ‘real world’, once it is approved 50.
The assessment of responsiveness to LTRAs is also greatly influenced by whether this mediator pathway is dominant in causing airway dysfunction in different patients. A responder analysis of head-to-head comparator trials revealed that the mean response to both drugs masks a remarkable heterogeneity of responsiveness 51 ( Figure 2). For both the ICS arm and the montelukast arm, there are clear differences from placebo in a range of asthma outcome measures. However, within each active arm there were patients who experienced dramatic and consistent improvements, whereas others experienced no change or even deteriorated (Figure 2). In the past, such variability has been brushed aside as being part of the ‘normally expected spectrum of response’ to any drug; however, recent research suggests that there may be very specific reasons why one drug may work well in one patient but not in another. The 5-lipoxygenase pathway responsible for generating cyst-LTs is selectively upregulated in patients with aspirin-intolerant asthma 52, 53; in particular, there is a selective overexpression of the terminal synthetic enzyme, LTC4 synthase, that overrides the suppressive effects of locally generated inhibitory prostaglandin prostaglandin E2 54, 55. Thus, both the 5-lipoxygenase inhibitor zileuton 56 and the cyst LT1R antagonist montelukast 57 are especially efficacious in this subgroup who exhibit increased airway LTC4 and airway LTE4 levels at baseline and following aspirin challenge commensurate with overexpression of this pathway in pathogenesis 58. In a broader range of asthma, the urinary LTE4/exhaled nitric oxide (eNO) ratio predicts a superior response to montelukast compared with the inhaled corticosteroid fluticasone propionate in children with mild to moderate asthma 59. Exhaled NO is generated by epithelial inducible NO synthase, which is upregulated in asthma and is suppressed by corticosteroids 60 and has proved to be a sensitive biomarker of responsiveness to inhaled corticosteroids in mild to moderate disease 61. Exhaled NO, as a biomarker of corticosteroid responsiveness, is much less useful in severe asthma, possibly owing to alternative cellular sources and the fact that most severe asthmatics are already receiving high doses of inhaled corticosteroids 62.
Figure 2.
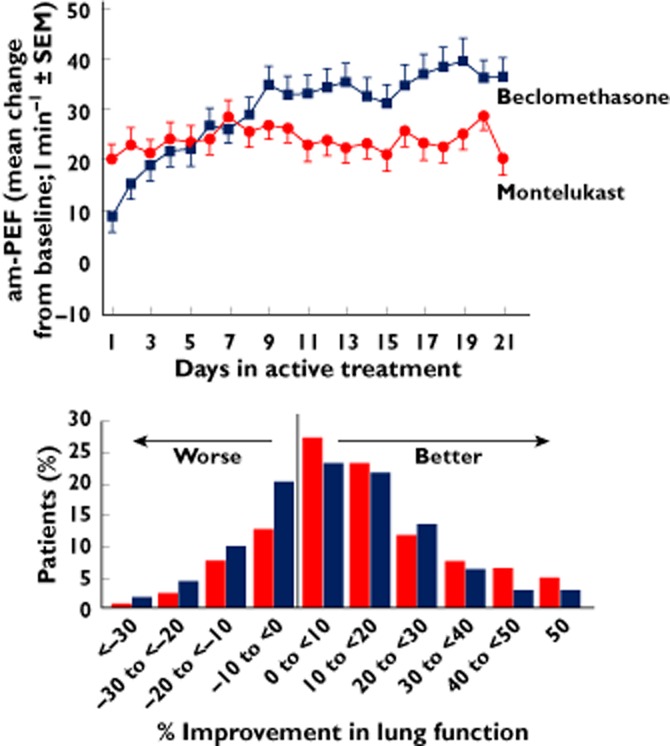
Top panel shows results from a randomized controlled trial of oral montelukast (10 μg twice daily) and inhaled beclomethasone (200 μg twice daily) against the asthma outcome measure of morning peak expiratory flow (am-PEF), over 21 days of treatment of moderate asthma. Improvement in am-PEF was more rapid and initially greater with montelukast compared with beclomethasone, but after day 8, the beclomethasone treatment effect surpassed that of montelukast. Bottom panel shows results from the same clinical trial, but displayed as the percentage of individuals achieving changes in peak expiratory flow at week 12, showing the large range of responders and nonresponders for both drugs (Adapted from 53).  , beclomethasone (n = 246);
, beclomethasone (n = 246);  , montelukast (n = 375)
, montelukast (n = 375)
Mechanism-directed treatment of asthma
Anti-IgE monoclonal antibody (mAb)
Over the last 50 years, there has been an explosion in knowledge about the cells and mediators involved in the allergic tissue response. Prominent in this has been the identification of the T-helper 2 (Th2)-type T cell as the ‘orchestrator’ of allergic responses 63, culminating in the interleukin (IL)-4- and IL-13-dependent generation of IgE by dedicated follicular B cells and plasma cells, and represents the principal trigger of the allergic response. Allergen-specific IgE is the mechanism through which the acute mast cell/basophil-mediated early response is generated, by cross-linking of IgE bound to its high-affinity receptors (FcεR1). When allergen binds to cell-bound IgE, it undergoes a major conformational change 64 to initiate the secretory response, involving the noncytotoxic release of preformed mediators, cytokines, chemokines and growth factors and the generation of newly formed products, including prostaglandin D2 and LTC4, as well as cytokines such as IL-4, IL-5, IL-13, thymic stromal lymphopoietin, tumour necrosis factor-α (TNFα) and a range of chemokines 65.
Although IgE was structurally identified as the fifth immunoglobulin class in 1968 66, it took a further 35 years before a therapeutic agent targeting IgE was developed. Omalizumab, a noncomplement-fixing IgG mAb binds to the FCε3 of IgE, thereby blocking the binding to the α-chain of FcεR1 and to the low-affinity receptor FcεR2 (CD23) 67. The small tri- and hexameric complexes formed are taken up by the reticuloendothelial system and rapidly eliminated. In addition to blocking the IgE-dependent mechanism involved with the acute allergic response, omalizumab blocks IgE-dependent facilitated uptake of allergens by mature myeloid dendritic cells 68 and, through this mechanism, is also able to modify the more chronic allergic response. After administration of intravenous or subcutaneous omalizumab as a once monthly or 2 weekly subcutaneous injection (according to an algorithm calculated from the total serum IgE and bodyweight), free circulating IgE levels fall precipitously, but tissue cell-bound IgE levels decrease more slowly over 12–16 weeks 69. At this time, the allergen-induced early and late allergic response are both almost ablated, and there is a reduction of the influx of eosinophils into the airways 70. Clinical trials in adults and children have confirmed clinical efficacy of omalizumab in moderate to severe allergic asthma, but the responses have not been uniform across patients or asthma-related end-points 71. Over 12 months of treatment in severe allergic asthma, omalizumab exerts a far greater impact on patient-related outcome measures (e.g. asthma control and quality of life) than over lung function (assessed as forced expiratory volume in 1 second or AHR) 72. In mild to moderate asthma, omalizumab efficacy was shown to be accompanied by a dramatic loss of mast cell-associated IgE and FcεR1 and reductions in eosinophils, T cells and B cells 73.
Another feature of omalizumab treatment is its dramatic life-transforming effect in some patients (∼30%), while in others (∼30%) only moderate effects have been observed despite there being no apparent differences in asthma phenotypes 71, 74. For those who response well to omalizumab, the relationship between free serum IgE and asthma outcome measures after beginning and stopping therapy are closely paralleled, whereas for those who do not respond, there is no such relationship during therapy induction or withdrawal 75. There has been much speculation about why such a variable response exists when targeting the principal activation pathway of the allergic response. Although genetic studies conducted by Novartis failed to find any associations of polymorphisms along the IgE–receptor–signalling pathway that could explain even part of the variability, there may be considerable subtlety in the way in which omalizumab is able to interact with different IgE species in relationship to their affinity for binding allergen 76 and the relative importance of IgE signalling vs. other non-IgE Th2-related inflammatory pathways in the disease 77. There is clearly a need to understand more about the immunopathology and physiology of the airways in omalizumab responders vs. nonresponders and the propensity of the mAb to bind and eliminate subtypes of IgE. Another possible explanation for lack of efficacy in certain subjects is lack of potency/tissue penetration; these possibilities will be testable for the more potent form of anti-IgE (QGE031) currently in clinical development (http://clinicaltrials.gov/ct2/show/NCT01451450).
Targeting T cells in asthma
From extensive observational studies, in vitro experiments and animal models, the Th2 cell has been identified as central in driving the allergic response 77. It is entirely reasonable to consider this as an attractive therapeutic target. Experience with immunosuppressants, such as cyclosporine, tacrolimus, methotrexate, azathioprine and cyclophosphamide, has been mixed 78. These drugs are usually given as oral corticosteroid-sparing agents, and some patients respond well, with T-cell reductions in airway biopsies 79, whereas other patients experience either severe side-effects or lack of efficacy that precludes their use.
Given that major histocompatibility antigen class II-dependent allergen presentation to the T-cell receptor (CD3) is critical to propagation of the allergic cascade, more specific immunotherapeutics have been tried. Blockade of CD4 with the mAb keliximab initially looked promising for treatment of severe corticosteroid-refractory disease, but while a small clinical trial revealed significant effects on lung function, other asthma outcomes were either minimally affected or unchanged 80, despite finding that over the three doses used the T-cell proliferation in response to allergen was markedly suppressed 81. More recently, the mAb daclizumab, which is directed at the T-cell activation marker CD25, has been trialled in moderate to severe asthma on the basis of its powerful immunosuppressive effect in organ transplant rejection 82. While some benefit was shown, this was not statistically significant until 6 months of therapy had been given and, again, was restricted to a limited number of end-points 83. Immunosuppressive side-effects were also a problem. As CD25 is an important marker for forkhead box P3 (FOXP3) regulatory T cells, there was concern that its blockade with daclizumab might break any allergen-specific tolerance 84. However, it has now been shown that the apparent decrease in CD25+ regulatory T cells observed with daclizumab therapy reflected lack of detection of the cells, as a result of antibody allosterically blocking the immunoreactive epitopes on the CD25 protein 85. These somewhat disappointing clinical results for anti-T-cell therapies in chronic asthma are in stark contrast to the efficacy that might be expected if T cells were obligatory in driving the asthmatic response in more severe disease. However, the recognition that a myriad of different T-cell subtypes, as well as Th2 cells, are involved in asthma as it becomes more severe (e.g. Th1, Th9, Th17, Th21, γδT, iKT and CD8+ cells) mitigates against specific T-cell therapies unless it is possible to endotype (i.e. defined by a distinct functional or pathobiological mechanism) asthma better, according to specific causative T-cell subsets 86.
An alternative approach has been to target the proliferation and activation of T cells by interfering with dendritic cell–T cell co-stimulation (the ‘second signal’ in the immunological synapse). Two targets have come to the forefront: CD28/CD80/82 and OX40/OX40 ligand, the former being regulated by the negative signalling molecule CTLA-4 on T cells 87, the latter by the positive regulator epithelial and mast cell-derived thymic stromal lymphopoietin 88. The Ig-CTLA-4-Ig fusion protein, abatacept, has proven effective in rheumatoid arthritis (RA) 89, but the response is variable. A clinical trial of CTLA-4-Ig in allergen-induced airway inflammation is in progress (http://clinicaltrials.gov/ct2/show/NCT00784459), while a trial on the use of mAbs to block OX40 ligand has also been completed (http://clinicaltrials.gov/ct2/show/NCT00983658), although the outcome is currently not published.
Cytokines and their receptors as therapeutics
The cluster of cytokines genetically encoded on chromosome 5q31 and secreted by Th2 cells, as well as mast cells and basophils, have been strongly implicated in the causal pathway of the allergic cascade in in vitro, animal and human studies. Indeed, this cytokine gene cluster encoding IL-3, IL-4, IL-5, IL-9, IL-13 and granuloctye macrophage-colony stimulating factor (GM-CSF) has almost become synonymous with asthma 90. It is therefore hardly surprising that each of these cytokines has become the target for new therapeutics, especially biologics.
Interleukin-5
Given that most asthma, whether allergic or non-allergic, is characterized by eosinophilic inflammation of the airways and sputum eosinophilia (which is a sensitive index for assessment of disease control and corticosteroid responsiveness) the factors that influence the influx, maturation and survival of eosinophils are obvious therapeutic targets 91. Eosinophils mature from CD34+ precursors, both in the bone marrow and resident in the airways, and mature under the influence of IL-3, IL-5 and GM-CSF. However, based on strong evidence from gene-manipulated mice and blocking antibodies in rodents and nonhuman primates 92, IL-5 was selected as the optimal target, in part because of its relatively selective actions on eosinophils in promoting their terminal maturation and survival. Interleukin-5 binds to the IL-5 receptor α chain and signals via a common β chain. Blockade of IL-5, using the IgG1 mAb mepolizumab administered intravenously, had a dramatic effect in almost ablating circulatory and sputum eosinophils in asthma but, somewhat surprisingly, had no significant effect on the late allergic response 93 following allergen challenge, nor on any clinical outcomes in moderate to severe asthma 94. Subsequent studies showed that mepolizumab decreased tissue 95 and bone marrow eosinophils 96 by ∼50%, possibly because some eosinophils lose their IL-5α chain as they migrate into the airways. Partial depletion of airway eosinophils was argued to be responsible for the unexpected lack of efficacy of mepolizumab in asthma, with mepolizumab-resistant eosinophils being dependent upon other factors for their survival, e.g. GM-CSF.
Recently, two small clinical trials have shown that mepolizumab markedly reduced exacerbations of asthma in patients with severe disease who exhibited persistent sputum eosinophilia and elevated eNO despite moderate- to high-dose oral corticosteroid treatment (representing ∼1% of the asthmatic population) 97, 98. Thus, sputum eosinophilia in refractory asthma could be used as a biomarker for determining anti-IL-5 responsive patients. Similar results have been obtained with a second mAb, reslizumab (CTx55700), which initially showed no overall response in moderate to severe asthma apart from a possible beneficial trend in baseline lung function 99. In a larger trial of eosinophilic asthma poorly controlled by high-dose ICS, when compared with placebo those receiving reslizumab exhibited improved lung function and a trend towards improved asthma control in parallel with a reduction in sputum eosinophilia 100. The beneficial response to reslizumab was especially noted in those patients with concomitant nasal polyposis. Indeed, in a separate randomized controlled trial, reslizumab significantly suppressed corticosteroid-resistant polyps in proportion to IL-5 levels in nasal lavage 101. Given that eosinophil infiltration is common to both asthma and nasal polyposis, a recent small placebo-controlled trial with mepolizumab has likewise shown efficacy against nasal polyposis 102.
As with anti-IgE, a further possible explanation for variable efficacy is incomplete tissue penetration and removal of bioavailable IL-5. The development of an antibody-dependent cell-cytotoxic mAb against the IL-5 receptor α chain by the removal of fucose from the Fc portion of IgG 103 (MEDI 563, Immunex) will definitively test the role of IL-5 signalling and eosinophils in asthma. An initial proof-of-concept and safety study of MEDI 563 administered as single, escalating, intravenous doses (0.0003-3 mg/kg) to patients with mild asthma has demonstrated a dose-dependent reduction in blood eosinophils, with total ablation lasting for 8–12 weeks occurring at the highest dose 104. At lower doses, circulating eosinophils were reduced by of those remaining; their ability to secrete cytokines and mediators ex vivo was markedly reduced.
Interleukin-4 and interleukin-13
Interleukin-4 and IL-13 are of particular interest in asthma based upon the ability of these two cytokines to drive Th2 (IL-4) cell differentiation and activation, to enhance inflammatory responses by upregulating Vascular cell adhesion protein 1 (in concert with TNFα) involved in eosinophil and basophil recruitment (IL-4 and IL-13) 105 and to prime inflammatory cells for secretion of mediators (IL-4 and IL-13) 106. Interleukin-4 and especially IL-13 also drive aspects of airway remodelling, including mucous metaplasia, fibroblast activation and smooth muscle development 107. Animal models (especially involving mice) of acute and chronic Th2-type lung inflammation have reinforced the importance of these two cytokines as candidates involved in the pathophysiology of both airway inflammatory and remodelling responses 107. There is accumulating genetic evidence incriminating these two cytokines in human asthma 108. Interleukin-4 and IL-13 signal through a complex set of receptor subunits, some of which are shared by the two cytokines (Figure 3). Interleukin-4 binds to IL-4 receptor alpha (IL-4Rα), leading to phosphorylation of signal transducer and activator of transcription factor-6 (STAT-6) by the second subunit, γC (common to the interferon receptors) to activate Janus kinase (JAK) 3. Alternatively, IL-4 can bind to IL-4Rα associated with the IL-13Rα1 rather than the γC chain to activate JAK2 and tyrosine kinsae (TYK) 2 as a way of phosphorylating STAT-6 109. Finally, a second IL-13 subunit, IL-13α2, has a higher affinity for IL-13 than the IL-13Rα1 and serves as a decoy, while the greatly shortened cytoplasmic tail of IL-13α2 may interfere with the association or activation of signalling molecules, such as JAK1, on IL-4Rα to provide an inhibitory feedback mechanism 110. There is also evidence that in certain conditions soluble IL-4Rα has the potential to stabilize binding of IL-13 to its receptor to augment IL-13-mediated responses 111. Thus, the cellular disposition of IL-4Rα and the IL-13α2 subunit is able to regulate IL-13 agonist signalling activity tightly.
Figure 3.
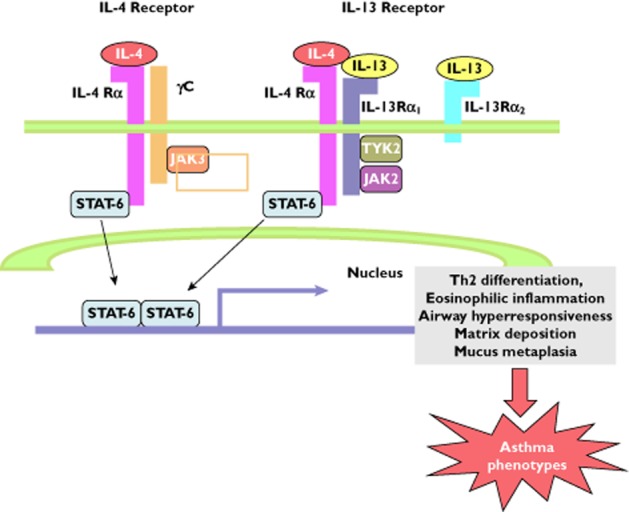
Schematic diagram of the interleukin (IL)-4/IL-13 signal transducer and activator of transcription factor (STAT)-6 signalling pathways linked to T-helper 2 (Th2)-type inflammation. Interleukin-4 and IL-13 are recognized by IL-4Rα, a component of the IL-4 type I (IL-4Rα and γC) and type II receptors (IL-4Rα and IL-13Rα1). Interleukin-4 signals through both type I and type II receptor pathways, whereas IL-13 signals only through the type II IL-4R. Interleukin-13 also binds to the IL-13Rα2 chain with greater affinity, lacking a transmembrane-signalling domain, but functions to interfere with janus kinase (JAK) 2 activation in the IL-4Rα/ IL-13Rα1 complex as well as functioning as a decoy receptor to down-regulate IL-13 signalling. γC activates JAK3, while IL-13Rα1 activates tyrosine kinase 2 (TYK2) and JAK2. Activated JAKs phosphorylate STAT-6 which, upon dimerization, translocates to the nucleus, where it binds to the promoters of the IL-4- and IL-13-responsive genes associated with Th2 cell differentiation, airway inflammation, airway hyperresponsiveness, fibrosis and epithelial mucous metaplasia (Adapted from reference 105; reproduced with permission of Trends in Immunology)
Initial studies using an inhaled formulation of an IL-4R Ig fusion protein (Nuvance) showed initial promise in two small trials 112, 113; however, in a large phase II study, efficacy in moderate to severe asthma could not be confirmed (http://www.clinicaltrials.gov/ct2/show/NCT00001909). These initial disappointing results might be due to rapid breakdown of Nuvance by proteolysis in the asthmatic airway, although, following inhalation, circulating levels of Nuvance could be detected, as well as activity in induced sputum. Attention, therefore, has moved to systemic administration of anti-IL-4 and anti-IL-13 biologics. The first of these to reach clinical trial was an IL-4Rα double mutein (pitrakinra) involved in both IL-4 and IL-13 signalling 114. When administered intravenously (and also by inhalation), pitrakinra attenuated the allergen-induced late allergic reaction, as well as reducing sputum and circulating eosinophilia 115. Trials are now in progress in clinical asthma. It is worth noting, however, that the late allergic reaction was only partially attenuated and there was no effect on allergen-induced AHR. Nevertheless, there were encouraging results validating the IL-4/IL-13 target in human asthma.
Most of the subsequent attempts to interrupt the STAT-6 pathway have been through mAbs targeting the IL-4Rα (AMG317, Amgen), IL-13 itself (QAX-5676; Novartis, CAT-354; Astra Zeneca/Immunex, IMA-638 and IMA-026; Pfizer/Wyeth, lebrikizumab; Roche, TNX-650; Tanox) and IL-13Rα (Merck) or by using mAb fragments (IL-13, DOM 100P; IL-4/IL-13 DOM-0910; both Zenyth Therapeutics and UCB) 116. As with pitrakinra, the anti-IL-13 mAb IMA-638 (Pfizer) attenuated the allergen-provoked late allergic reaction, but interestingly, a related mAb, IMA-026, directed to the same target but a different epitope, does not 117. A search for the reasons for this difference has been informative with regard to how the IL-4/IL-13 receptor complex functions. IMA-638 binds IL-13 in such a way that it still allows it to bind to both the IL-13Rα1 and IL-13Rα2 subunits but inhibits the docking of the IL-4Rα to the IL-13/IL-13Rα1 complex 118. In contrast, IMA-026 binds to IL-13 at a point that blocks its interaction with IL-13Rα1 and IL-13Rα2. Thus, when compared with IMA-026, the efficacy of IMA-638 in depleting IL-13 indicates that IL-13Rα2 on the cell surface is important for the removal of IL-13. This clearly has implications for the design of any future IL-13 inhibitors.
AMG-317 (Amgen), directed to IL-4Rα, was the first mAb targeting this pathway to enter clinical trial in moderate to severe asthma 119. Three doses vs. placebo were assessed, with the Juniper Asthma Control Questionnaire used as the primary actions measure. No significant change in this or any other asthma end-point was observed, although at the highest tertile of disease severity there was a trend towards improvement in a number of end-points, as well as ∼50% reduction in serum total IgE. Based upon the strong preclinical data supporting the STAT-6 pathway as a therapeutic target, this was a disappointing result. The second mAb for which there are reports is CAT-354 (MedImmune, Astra Zeneca), an IgG4 mAb directed to IL-13 120, 121, again showing no overall benefit in either the Juniper Asthma Control Questionnaire or lung function over a range of three doses 122. However, in a small subset of patients who had elevated sputum IL-13 levels measured at baseline, CAT-354 did show efficacy against both end-points, suggesting that the sputum level of IL-13 might be a good biomarker for anti-IL-13 responsiveness.
Identification of biomarkers for Th2 responsiveness
Although there is mounting evidence that eosinophils and cytokines in sputum, as well as eNO, could serve as biomarkers for allergic-type disease of the airways, as well as responses to corticosteroid anti-IgE and LTRA treatment, what are needed are more precise biomarkers of the different types of inflammatory response to help direct treatment to causative pathways. In the past, subphenotyping of asthma has largely been in relationship to disease severity, although classification has included some causal associations, e.g. allergic asthma, aspirin-induced asthma, occupational asthma and reactive airways disease 123. More recently, nonhierarchical statistical approaches, such as cluster analyses, have been applied to subdivide asthma. Up to six ‘endotypes’ of adult asthma and four endotypes of childhood asthma have now been identified, but to date none of these has been linked directly to causal pathways, although allergen sensitization, eosinophils and elevated NO predominate in some but not others 124–128.
Given that asthma is primarily an airway disease driven through the epithelium, this structure has provided some of the first insights into disease causality and responsiveness to specific treatments. For example, elevation of the FK506 binding protein (FKBP51) gene in epithelial cells has proved to be a highly sensitive marker of ICS sensitivity 129, 130, an observation also confirmed for this target in peripheral blood mononuclear cells when seeking a biomarker for oral corticosteroid responsiveness 131.
Applying this approach to a wider set of Th2-responsive genes, Woodruff and colleagues examined IL-13-responsive genes in epithelial cells obtained from bronchial brushings of asthmatic and normal airways 130, 132. After examining a wide range of genes, they focused on the following three: RSNT (periostin), encoding an epithelial secreted matrix protein; CLCAI, encoding a chloride channel involved in mucus secretion; and SERPINB2, encoding a plasminogen activator inhibitor type II, on the basis of showing marked IL-13 upregulation and consistency of expression over time 132. There was also a broad expression of these three genes across the asthmatic population. The asthmatic patients were subdivided into two groups designated Th2high (IL-13+ response) and Th2low (little or no change in the expression level of the 13 responsive genes). Th2high asthmatics had a greater number of circulating eosinophils and bronchoalveolar lavage eosinophilia and, in bronchial biopsies, had increased expression of IL-5, IL-13 and tryptase+ mast cells 132, 133. The Th2high phenotype had great AHR, serum levels of total IgE, thickening of the epithelial basement membrane lamina reticularis and airway mucin (MUC5AC) gene expression. In separate studies, POSTN gene expression has been shown to correlate closely with thickening of the lamina reticularis 134, and in monolayer epithelial cultures, periostin protein was secreted into the basal medial in response to IL-13 134, 135. Periostin activates transforming growth factor-β to drive the secretion of ‘repair’ collagens, such as type I, by underlying myofibroblasts, as well as cross-linking collagen fibrils, which causes matrix stiffening 134. Subepithelial matrix deposition is a characteristic feature of asthma as a possible marker of airway wall remodelling in this disease 136. A proof-of-concept randomized controlled trial of the anti-IL-13 mAb lebrikizumab (Roche) revealed a small but significant improvement in baseline lung function over the 12 weeks of treatment, but it was of considerable interest that this was almost entirely restricted to those patients with elevated serum periostin levels 137. However, in this trial other asthma-related end-points were not affected, including patient-related outcome measures.
Recently, sputum cells have been used as a source of transcriptomics. In moderate to severe asthma, three gene profiles have been described, one almost identical to the Th2high endotype and two with characteristics of the Th2low endotype, one being dominated by neutrophils and the other macrophages 138. The neutrophil-dominant endotype had increased expression of IL-1-, TNFα- and nuclear factor-κB-associated genes, indicating activation of oxidant and inflammazone pathways 138, and was associated with greater systemic inflammation, as revealed by elevated circulating C-reactive protein and IL-6 and increased sputum IL-8 and neutrophil elastase and CXCL-8 gene expression 139.
The identification of TNFα as being overexpressed at both gene and protein levels in severe corticosteroid-refractory asthma in which neutrophils are prominent has led to anti-TNF strategies as potential therapies 140. While several small trials with the TNF-R1-Ig fusion protein etanercept (Wyeth/Pfizer) looked promising 140–143), a phase II trial with etanercept in patients with rather less severe asthma on high-dose ICS showed no overall benefit 144. A further trial in moderate to severe asthma with the anti-TNF mAb golimumab (Centecor) also showed no overall effect over 6 months of treatment, although substratification into those displaying rhinosinusitis and >12% bronchodilator reversibility did identify a dose-dependent responsive subgroup 145. However, concerns over increased infection have halted further development of this mAb for asthma, even though it is highly efficacious in RA.
Conclusions
Asthma can no longer be regarded as a homogeneous disorder, with increasing evidence for multiple endotypes now emerging. Beginning with Th2high and Th2low asthma subtypes, it is increasingly clear that different causative pathways will become linked to different disease endotypes. The identification of such novel pathways will provide the opportunity to develop novel animal models beyond the allergen sensitization/challenge (Th2) model 145 and will form the basis for the stratified treatment of this disease, hopefully attacking those pathways high up the causal cascade. What is now required to achieve this is a close collaboration between academia, clinicians and industry to enable careful mapping of these causative pathways onto the distinct clinical, physiological and laboratory phenotypes that occur in humans.
Acknowledgments
The author is an MRC Clinical Professor and programme grant holder. Thanks are given to the National Institute for Health Research (NIHR) Respiratory Biomedical Research Unit in Southampton. The author wishes to acknowledge the help of Mrs Christine Vincent in the preparation of this manuscript.
Competing Interests
As sole author, I have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf and declare no support from any organization for the submitted work; I have listed on the form any potential organizations that might have an interest in the submitted work in the previous 3 years. No other relationships exist.
References
- 1.Sakula A. Henry Hyde Salter (1823–71): a biographical sketch. Thorax. 1985;40:887–888. doi: 10.1136/thx.40.12.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee MR. The history of Ephedra (ma-huang) J R Coll Physicians Edinb. 2011;41:78–84. doi: 10.4997/JRCPE.2011.116. [DOI] [PubMed] [Google Scholar]
- 3.Tilley SL. Methylxanthines in asthma. Handb Exp Pharmacol. 2011;200:439–456. doi: 10.1007/978-3-642-13443-2_17. [DOI] [PubMed] [Google Scholar]
- 4.Parascandola JAbel. Takamine, and the isolation of epinephrine. J Allergy Clin Immunol. 2010;125:514–517. doi: 10.1016/j.jaci.2009.11.044. [DOI] [PubMed] [Google Scholar]
- 5.Rau JL. Inhaled adrenergic bronchodilators: historical development and clinical application. Respir Care. 2000;45:854–862. [PubMed] [Google Scholar]
- 6.Crompton G. A brief history of inhaled asthma therapy over the last fifty years. Prim Care Respir J. 2006;1:326–331. doi: 10.1016/j.pcrj.2006.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beeh KM, Beier J. Indacaterol: a new once daily long-acting beta(2) adrenoceptor agonist. Core Evid. 2010;4:37–41. doi: 10.2147/ce.s6013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pearce N, Crane J, Burgess C, Beasley R, Jackson R. Beta-agonists and death from asthma. N Engl J Med. 1992;327:355–357. [PubMed] [Google Scholar]
- 9.Holgate ST, Baldwin CJ, Tattersfield AE. Beta-adrenergic agonist resistance in normal human airways. Lancet. 1977;2:375–377. doi: 10.1016/s0140-6736(77)90304-x. [DOI] [PubMed] [Google Scholar]
- 10.Beasley R, Roche W, Holgate ST. Inflammatory processes in bronchial asthma. Drugs. 1989;37(Suppl. 1):117–122. doi: 10.2165/00003495-198900371-00021. [DOI] [PubMed] [Google Scholar]
- 11.Djukanović R, Wilson JW, Britten KM, Wilson SJ, Walls AF, Roche WR, Howarth PH, Holgate ST. Effect of an inhaled corticosteroid on airway inflammation and symptoms in asthma. Am Rev Respir Dis. 1992;145:669–674. doi: 10.1164/ajrccm/145.3.669. [DOI] [PubMed] [Google Scholar]
- 12.Shampo MA, Kyle RA, Philip S. Hench – 1950 Nobel laureate. Mayo Clin Proc. 2001;76:1073. doi: 10.4065/76.11.1073. [DOI] [PubMed] [Google Scholar]
- 13.Brown HM, Storey G, George WH. Beclomethasone dipropionate: a new steroid aerosol for the treatment of allergic asthma. Br Med J. 1972;1:585–590. doi: 10.1136/bmj.1.5800.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cerasoli F., Jr Developing the ideal inhaled corticosteroid. Chest. 2006;130(1 Suppl):54S–64S. doi: 10.1378/chest.130.1_suppl.54S. [DOI] [PubMed] [Google Scholar]
- 15.Barnes PJ. Biochemical basis of asthma therapy. J Biol Chem. 2011;286:32899–32905. doi: 10.1074/jbc.R110.206466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sanders M. Inhalation therapy: an historical review. Prim Care Respir J. 2007;16:71–81. doi: 10.3132/pcrj.2007.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haughney J, Price D, Kaplan A, Chrystyn H, Horne R, May N, Moffat M, Versnel J, Shanahan ER, Hillyer EV, Tunsäter A, Bjermer L. Achieving asthma control in practice: understanding the reasons for poor control. Respir Med. 2008;102:1681–1693. doi: 10.1016/j.rmed.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 18.Currie GP, Douglas JG, Heaney LG. Difficult to treat asthma in adults. BMJ. 2009;338:b494. doi: 10.1136/bmj.b494. [DOI] [PubMed] [Google Scholar]
- 19.Ito K, Chung KF, Adcock IM. Update on glucocorticoid action and resistance. J Allergy Clin Immunol. 2006;117:522–543. doi: 10.1016/j.jaci.2006.01.032. [DOI] [PubMed] [Google Scholar]
- 20.Livingston E, Thomson NC, Chalmers GW. Impact of smoking on asthma therapy: a critical review of clinical evidence. Drugs. 2005;65:1521–1536. doi: 10.2165/00003495-200565110-00005. [DOI] [PubMed] [Google Scholar]
- 21.Holgate ST. Asthma: a simple concept but in reality a complex disease. Eur J Clin Invest. 2011;41:1339–1352. doi: 10.1111/j.1365-2362.2011.02534.x. [DOI] [PubMed] [Google Scholar]
- 22.Bloemen K, Verstraelen S, Van Den Heuvel R, Witters H, Nelissen I, Schoeters G. The allergic cascade: review of the most important molecules in the asthmatic lung. Immunol Lett. 2007;113:6–18. doi: 10.1016/j.imlet.2007.07.010. [DOI] [PubMed] [Google Scholar]
- 23.O'Connor GT. Allergen avoidance in asthma: what do we do now? J Allergy Clin Immunol. 2005;116:26–30. doi: 10.1016/j.jaci.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 24.Barnes PJ. Is there a role for immunotherapy in the treatment of asthma? No. Am J Respir Crit Care Med. 1996;154:1227–1228. doi: 10.1164/ajrccm.154.5.8912730. [DOI] [PubMed] [Google Scholar]
- 25.Passalacqua G, Canonica GW. Specific immunotherapy in asthma: efficacy and safety. Clin Exp Allergy. 2011;41:1247–1255. doi: 10.1111/j.1365-2222.2010.03688.x. [DOI] [PubMed] [Google Scholar]
- 26.Arshad SH, Bateman B, Sadeghnejad A, Gant C, Matthews SM. Prevention of allergic disease during childhood by allergen avoidance: the Isle of Wight prevention study. J Allergy Clin Immunol. 2007;119:307–313. doi: 10.1016/j.jaci.2006.12.621. [DOI] [PubMed] [Google Scholar]
- 27.Robinson C, Holgate ST. Mast cell-dependent inflammatory mediators and their putative role in bronchial asthma. Clin Sci (Lond) 1985;68:103–112. doi: 10.1042/cs0680103. [DOI] [PubMed] [Google Scholar]
- 28.Edwards AM. The discovery of cromolyn sodium and its effect on research and practice in allergy and immunology. J Allergy Clin Immunol. 2005;115:885–888. doi: 10.1016/j.jaci.2005.01.063. [DOI] [PubMed] [Google Scholar]
- 29.Howell JB, Altounyan RE. A double-blind trial of disodium cromoglycate in the treatment of allergic bronchial asthma. Lancet. 1967;2:539–542. doi: 10.1016/s0140-6736(67)90499-0. [DOI] [PubMed] [Google Scholar]
- 30.Holgate ST. Clinical evaluation of nedocromil sodium in asthma. Eur J Respir Dis Suppl. 1986;147:149–159. [PubMed] [Google Scholar]
- 31.Edwards A, Stevens M, Holgate S, Iikura Y, Aberg N, König P, Reinhardt D, Stenius-Aarniala B, Warner J, Weinberg E, Callaghan B, Howell J. Inhaled sodium cromoglycate in children with asthma. Thorax. 2002;57:282. doi: 10.1136/thorax.57.3.282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van der Wouden JC, Uijen JH, Bernsen RM, Tasche MJ, de Jongste JC, Ducharme F. Inhaled sodium cromoglycate for asthma in children. Cochrane Database Syst Rev. 2008;(4) doi: 10.1002/14651858.CD002173.pub2. CD002173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stevens MT, Edwards AM, Howell JB. Sodium cromoglicate: an ineffective drug or meta-analysis misused? Pharm Stat. 2007;6:123–137. doi: 10.1002/pst.279. [DOI] [PubMed] [Google Scholar]
- 34.Alton EW, Norris AA. Chloride transport and the actions of nedocromil sodium and cromolyn sodium in asthma. J Allergy Clin Immunol. 1996;98:S102–105. [PubMed] [Google Scholar]
- 35.Oka S, Lu JY, Wu X, Summer S, Whoriskey J, Saris C. Reagan JD GPR35 is a novel lysophosphatidic acid receptor. Biochem Biophys Res Commun. 2010;395:232–237. doi: 10.1016/j.bbrc.2010.03.169. [DOI] [PubMed] [Google Scholar]
- 36.Yang Y, Lu JY, Wu X, Summer S, Whoriskey J, Saris C, Reagan JD. G-protein-coupled receptor 35 is a target of the asthma drugs cromolyn disodium and nedocromil sodium. Pharmacology. 2010;86:1–5. doi: 10.1159/000314164. [DOI] [PubMed] [Google Scholar]
- 37.Bagga S, Price KS, Lin DA, Friend DS, Austen KF, Boyce JA. Lysophosphatidic acid accelerates the development of human mast cells. Blood. 2004;104:4080–4087. doi: 10.1182/blood-2004-03-1166. [DOI] [PubMed] [Google Scholar]
- 38.Church MK, Hiroi J. Inhibition of IgE-dependent histamine release from human dispersed lung mast cells by anti-allergic drugs and salbutamol. Br J Pharmacol. 1987;90:421–429. doi: 10.1111/j.1476-5381.1987.tb08972.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Simons FE, Simons KJ. Histamine and H1-antihistamines: celebrating a century of progress. J Allergy Clin Immunol. 2011;128:1139–1150. doi: 10.1016/j.jaci.2011.09.005. [DOI] [PubMed] [Google Scholar]
- 40.Lordan JL, Holgate ST. H1-antihistamines in asthma. Clin Allergy Immunol. 2002;17:221–248. [PubMed] [Google Scholar]
- 41.Hill SJ, Baker JG. The ups and downs of Gs- to Gi-protein switching. Br J Pharmacol. 2003;138:1188–1189. doi: 10.1038/sj.bjp.0705192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Samuelsson B. Prostaglandins, thromboxanes, and leukotrienes: formation and biological roles. Harvey Lect. 1979;75:1–40. –1980. [PubMed] [Google Scholar]
- 43.Drazen JM, Austen KF. Leukotrienes and airway responses. Am Rev Respir Dis. 1987;136:985–998. doi: 10.1164/ajrccm/136.4.985. [DOI] [PubMed] [Google Scholar]
- 44.Lynch KR, O'Neill GP, Liu Q, Im DS, Sawyer N, Metters KM, Coulombe N, Abramovitz M, Figueroa DJ, Zeng Z, Connolly BM, Bai C, Austin CP, Chateauneuf A, Stocco R, Greig GM, Kargman S, Hooks SB, Hosfield E, Williams DL, Jr, Ford-Hutchinson AW, Caskey CT, Evans JF. Characterization of the human cysteinyl leukotriene CysLT1 receptor. Nature. 1999;399:789–793. doi: 10.1038/21658. [DOI] [PubMed] [Google Scholar]
- 45.Bjermer L, Bisgaard H, Bousquet J, Fabbri LM, Greening AP, Haahtela T, Holgate ST, Picado C, Menten J, Dass SB, Leff JA, Polos PG. Montelukast and fluticasone compared with Salmeterol and fluticasone in protecting against asthma exacerbation in adults: one year, double blind, randomised, comparative trial. BMJ. 2003;327:891. doi: 10.1136/bmj.327.7420.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Price D, Musgrave SD, Shepstone L, Hillyer EV, Sims EJ, Gilbert RF, Juniper EF, Ayres JG, Kemp L, Blyth A, Wilson EC, Wolfe S, Freeman D, Mugford HM, Murdoch J, Harvey I. Leukotriene antagonists as first-line or add-on asthma-controller therapy. N Engl J Med. 2011;364:1695–1707. doi: 10.1056/NEJMoa1010846. [DOI] [PubMed] [Google Scholar]
- 47.Rand C, Bilderback A, Schiller K, Edelman JM, Hustad CM, Zeiger RS MIAMI Study Research Group. Adherence with montelukast or fluticasone in a long-term clinical trial: results from the mild asthma montelukast versus inhaled corticosteroid trial. J Allergy Clin Immunol. 2007;119:916–923. doi: 10.1016/j.jaci.2006.12.664. [DOI] [PubMed] [Google Scholar]
- 48.Lazarus SC, Chinchilli VM, Rollings NJ, Boushey HA, Cherniack R, Craig TJ, Deykin A, DiMango E, Fish JE, Ford JG, Israel E, Kiley J, Kraft M, Lemanske RF, Jr, Leone FT, Martin RJ, Pesola GR, Peters SP, Sorkness CA, Szefler SJ, Wechsler ME, Fahy JV National Heart Lung and Blood Institute's Asthma Clinical Research Network. Smoking affects response to inhaled corticosteroids or leukotriene receptor antagonists in asthma. Am J Respir Crit Care Med. 2007;175:783–790. doi: 10.1164/rccm.200511-1746OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Herland K, Akselsen JP, Skjønsberg OH, Bjermer L. How representative are clinical study patients with asthma or COPD for a larger ‘real life’ population of patients with obstructive lung disease? Respir Med. 2005;99:11–19. doi: 10.1016/j.rmed.2004.03.026. [DOI] [PubMed] [Google Scholar]
- 50.Price D, Chisholm A, van der Molen T, Roche N, Hillyer EV, Bousquet J. Reassessing the evidence hierarchy in asthma: evaluating comparative effectiveness. Curr Allergy Asthma Rep. 2011;11:526–538. doi: 10.1007/s11882-011-0222-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Szefler SJ, Martin RJ. Lessons learned from variation in response to therapy in clinical trials. J Allergy Clin Immunol. 2010;125:285–292. doi: 10.1016/j.jaci.2009.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cowburn AS, Sladek K, Soja J, Adamek L, Nizankowska E, Szczeklik A, Lam BK, Penrose JF, Austen FK, Holgate ST, Sampson AP. Overexpression of leukotriene C4 synthase in bronchial biopsies from patients with aspirin-intolerant asthma. J Clin Invest. 1998;101:834–846. doi: 10.1172/JCI620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Malmström K, Rodriguez-Gomez G, Guerra J, Villaran C, Piñeiro A, Wei LX, Seidenberg BC, Reiss TF. Oral montelukast, inhaled beclomethasone, and placebo for chronic asthma. A randomized, controlled trial. Montelukast/beclomethasone study group. Ann Intern Med. 1999;130:487–495. doi: 10.7326/0003-4819-130-6-199903160-00005. [DOI] [PubMed] [Google Scholar]
- 54.Mastalerz L, Sanak M, Gawlewicz-Mroczka A, Gielicz A, Cmiel A, Szczeklik A. Prostaglandin E2 systemic production in patients with asthma with and without aspirin hypersensitivity. Thorax. 2008;63:27–34. doi: 10.1136/thx.2007.080903. [DOI] [PubMed] [Google Scholar]
- 55.Pierzchalska M, Soja J, Woś M, Szabó Z, Nizankowska-Mogielnicka E, Sanak M, Szczeklik A. Deficiency of cyclooxygenases transcripts in cultured primary bronchial epithelial cells of aspirin-sensitive asthmatics. J Physiol Pharmacol. 2007;58:207–218. [PubMed] [Google Scholar]
- 56.Dahlén B, Nizankowska E, Szczeklik A, Zetterström O, Bochenek G, Kumlin M, Mastalerz L, Pinis G, Swanson LJ, Boodhoo TI, Wright S, Dubé LM, Dahlén SE. Benefits from adding the 5-lipoxygenase inhibitor zileuton to conventional therapy in aspirin-intolerant asthmatics. Am J Respir Crit Care Med. 1998;157:1187–1194. doi: 10.1164/ajrccm.157.4.9707089. [DOI] [PubMed] [Google Scholar]
- 57.Dahlén SE, Malmström K, Nizankowska E, Dahlén B, Kuna P, Kowalski M, Lumry WR, Picado C, Stevenson DD, Bousquet J, Pauwels R, Holgate ST, Shahane A, Zhang J, Reiss TF, Szczeklik A. Improvement of aspirin-intolerant asthma by montelukast, a leukotriene antagonist: a randomized, double-blind, placebo-controlled trial. Am J Respir Crit Care Med. 2002;165:9–14. doi: 10.1164/ajrccm.165.1.2010080. [DOI] [PubMed] [Google Scholar]
- 58.Daffern PJ, Muilenburg D, Hugli TE, Stevenson DD. Association of urinary leukotriene E4 excretion during aspirin challenges with severity of respiratory responses. J Allergy Clin Immunol. 1999;104:559–564. doi: 10.1016/s0091-6749(99)70324-6. [DOI] [PubMed] [Google Scholar]
- 59.Rabinovitch N, Graber NJ, Chinchilli VM, Sorkness CA, Zeiger RS, Strunk RC, Bacharier LB, Martinez FD, Szefler SJ Childhood Asthma Research and Education Network of the National Heart, Lung, and Blood Institute. Urinary leukotriene E4/exhaled nitric oxide ratio and montelukast response in childhood asthma. J Allergy Clin Immunol. 2010;126:545–551. doi: 10.1016/j.jaci.2010.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Redington AE, Meng QH, Springall DR, Evans TJ, Créminon C, Maclouf J, Holgate ST, Howarth PH, Polak JM. Increased expression of inducible nitric oxide synthase and cyclo-oxygenase-2 in the airway epithelium of asthmatic subjects and regulation by corticosteroid treatment. Thorax. 2001;56:351–357. doi: 10.1136/thorax.56.5.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zeiger RS, Schatz M, Zhang F, Crawford WW, Kaplan MS, Roth RM, Chen W. Elevated exhaled nitric oxide is a clinical indicator of future uncontrolled asthma in asthmatic patients on inhaled corticosteroids. J Allergy Clin Immunol. 2011;128:412–414. doi: 10.1016/j.jaci.2011.06.008. [DOI] [PubMed] [Google Scholar]
- 62.Moore WC, Bleecker ER, Curran-Everett D, Erzurum SC, Ameredes BT, Bacharier L, Calhoun WJ, Castro M, Chung KF, Clark MP, Dweik RA, Fitzpatrick AM, Gaston B, Hew M, Hussain I, Jarjour NN, Israel E, Levy BD, Murphy JR, Peters SP, Teague WG, Meyers DA, Busse WW, Wenzel SE National Heart, Lung, Blood Institute's Severe Asthma Research Program. Characterization of the severe asthma phenotype by the National Heart, Lung, and Blood Institute's Severe Asthma Research Program. J Allergy Clin Immunol. 2007;119:405–413. doi: 10.1016/j.jaci.2006.11.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bosnjak B, Stelzmueller B, Erb KJ, Epstein MM. Treatment of allergic asthma: modulation of Th2 cells and their responses. Respir Res. 2011;12:114. doi: 10.1186/1465-9921-12-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Holdom MD, Davies AM, Nettleship JE, Bagby SC, Dhaliwal B, Girardi E, Hunt J, Gould HJ, Beavil AJ, McDonnell JM, Owens RJ, Sutton BJ. Conformational changes in IgE contribute to its uniquely slow dissociation rate from receptor FcεRI. Nat Struct Mol Biol. 2011;18:571–576. doi: 10.1038/nsmb.2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Amin K. The role of mast cells in allergic inflammation. Respir Med. 2012;106:9–14. doi: 10.1016/j.rmed.2011.09.007. [DOI] [PubMed] [Google Scholar]
- 66.Bennich HH, Ishizaka K, Johansson SG, Rowe DS, Stanworth DR, Terry WD. Immunoglobulin E: a new class of human immunoglobulin. Immunology. 1968;15:323–324. [PMC free article] [PubMed] [Google Scholar]
- 67.Di Domenico M, Bisogno A, Polverino M, De Rosa C, Ricci V, Capasso A. Xolair in asthma therapy: an overview. Inflamm Allergy Drug Targets. 2011;10:2–12. doi: 10.2174/187152811794352042. [DOI] [PubMed] [Google Scholar]
- 68.Schroeder JT, Bieneman AP, Chichester KL, Hamilton RG, Xiao H, Saini SS, Liu MC. Decreases in human dendritic cell-dependent T(H)2-like responses after acute in vivo IgE neutralization. J Allergy Clin Immunol. 2010;125:896–901. doi: 10.1016/j.jaci.2009.10.021. [DOI] [PubMed] [Google Scholar]
- 69.Lowe PJ, Tannenbaum S, Gautier A, Jimenez P. Relationship between omalizumab pharmacokinetics, IgE pharmacodynamics and symptoms in patients with severe persistent allergic (IgE-mediated) asthma. Br J Clin Pharmacol. 2009;68:61–76. doi: 10.1111/j.1365-2125.2009.03401.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.van Rensen EL, Evertse CE, van Schadewijk WA, van Wijngaarden S, Ayre G, Mauad T, Hiemstra PS, Sterk PJ, Rabe KF. Eosinophils in bronchial mucosa of asthmatics after allergen challenge: effect of anti-IgE treatment. Allergy. 2009;64:72–80. doi: 10.1111/j.1398-9995.2008.01881.x. [DOI] [PubMed] [Google Scholar]
- 71.Bousquet J, Rabe K, Humbert M, Chung KF, Berger W, Fox H, Ayre G, Chen H, Thomas K, Blogg M, Holgate S. Predicting and evaluating response to omalizumab in patients with severe allergic asthma. Respir Med. 2007;101:1483–1492. doi: 10.1016/j.rmed.2007.01.011. [DOI] [PubMed] [Google Scholar]
- 72.Harris JM, Wong DA, Kapp AV. Development of the Asthma Control Composite outcome measure to predict omalizumab response. Ann Allergy Asthma Immunol. 2011;107:273–280. doi: 10.1016/j.anai.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 73.Djukanović R, Wilson SJ, Kraft M, Jarjour NN, Steel M, Chung KF, Bao W, Fowler-Taylor A, Matthews J, Busse WW, Holgate ST, Fahy JV. Effects of treatment with anti-immunoglobulin E antibody omalizumab on airway inflammation in allergic asthma. Am J Respir Crit Care Med. 2004;170:583–593. doi: 10.1164/rccm.200312-1651OC. [DOI] [PubMed] [Google Scholar]
- 74.Humbert M, Boulet LP, Niven RM, Panahloo Z, Blogg M, Ayre G. Omalizumab therapy: patients who achieve greatest benefit for their asthma experience greatest benefit for rhinitis. Allergy. 2009;64:81–84. doi: 10.1111/j.1398-9995.2008.01846.x. [DOI] [PubMed] [Google Scholar]
- 75.Slavin RG, Ferioli C, Tannenbaum SJ, Martin C, Blogg M, Lowe PJ. Asthma symptom re-emergence after omalizumab withdrawal correlates well with increasing IgE and decreasing pharmacokinetic concentrations. J Allergy Clin Immunol. 2009;123:107–113. doi: 10.1016/j.jaci.2008.09.050. [DOI] [PubMed] [Google Scholar]
- 76.Xiong H, Dolpady J, Wabl M, Curotto de Lafaille MA, Lafaille JJ. Sequential class switching is required for the generation of high affinity IgE antibodies. J Exp Med. 2012;209:353–364. doi: 10.1084/jem.20111941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kay AB. The role of T lymphocytes in asthma. Chem Immunol Allergy. 2006;91:59–75. doi: 10.1159/000090230. [DOI] [PubMed] [Google Scholar]
- 78.Polosa R, Benfatto GT. Managing patients with chronic severe asthma: rise to the challenge. Eur J Intern Med. 2009;20:114–124. doi: 10.1016/j.ejim.2008.06.010. [DOI] [PubMed] [Google Scholar]
- 79.Vrugt B, Wilson S, Bron A, Shute J, Holgate ST, Djukanovic R, Aalbers R. Low-dose methotrexate treatment in severe glucocorticoid-dependent asthma: effect on mucosal inflammation and in vitro sensitivity to glucocorticoids of mitogen-induced T-cell proliferation. Eur Respir J. 2000;15:478–485. doi: 10.1034/j.1399-3003.2000.15.09.x. [DOI] [PubMed] [Google Scholar]
- 80.Kon OM, Sihra BS, Compton CH, Leonard TB, Kay AB, Barnes NC. Randomised, dose-ranging, placebo-controlled study of chimeric antibody to CD4 (keliximab) in chronic severe asthma. Lancet. 1998;352:1109–1113. doi: 10.1016/S0140-6736(97)12261-9. [DOI] [PubMed] [Google Scholar]
- 81.Kon OM, Sihra BS, Loh LC, Barkans J, Compton CH, Barnes NC, Larché M, Kay AB. The effects of an anti-CD4 monoclonal antibody, keliximab, on peripheral blood CD4+ T-cells in asthma. Eur Respir J. 2001;18:45–52. doi: 10.1183/09031936.01.00064101. [DOI] [PubMed] [Google Scholar]
- 82.van den Hoogen MW, Hilbrands LB. Use of monoclonal antibodies in renal transplantation. Immunotherapy. 2011;3:871–880. doi: 10.2217/imt.11.72. [DOI] [PubMed] [Google Scholar]
- 83.Busse WW, Israel E, Nelson HS, Baker JW, Charous BL, Young DY, Vexler V, Shames RS Daclizumab Asthma Study Group. Daclizumab improves asthma control in patients with moderate to severe persistent asthma: a randomized, controlled trial. Am J Respir Crit Care Med. 2008;178:1002–1008. doi: 10.1164/rccm.200708-1200OC. [DOI] [PubMed] [Google Scholar]
- 84.Oh U, Blevins G, Griffith C, Richert N, Maric D, Lee CR, McFarland H, Jacobson S. Regulatory T cells are reduced during anti-CD25 antibody treatment of multiple sclerosis. Arch Neurol. 2009;66:471–479. doi: 10.1001/archneurol.2009.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hire K, Ngo DK, Stewart-Maynard KM, Hering B, Bansal-Pakala P. FoxP3+, and not CD25+, T cells increase post-transplant in islet allotransplant recipients following anti-CD25+ rATG immunotherapy. Cell Immunol. 2012;274:83–88. doi: 10.1016/j.cellimm.2012.01.008. [DOI] [PubMed] [Google Scholar]
- 86.Holgate ST. Innate and adapotive immune responses in asthma. Nat Med. 2012;18:1–11. doi: 10.1038/nm.2731. [DOI] [PubMed] [Google Scholar]
- 87.Bellou A, Finn PW. Costimulation: critical pathways in the immunologic regulation of asthma. Curr Allergy Asthma Rep. 2005;5:149–154. doi: 10.1007/s11882-005-0089-6. [DOI] [PubMed] [Google Scholar]
- 88.Jani M, Hyrich KL. Abatacept in the long-term treatment of rheumatoid arthritis. Expert Rev Clin Immunol. 2012;8:231–234. doi: 10.1586/eci.11.98. [DOI] [PubMed] [Google Scholar]
- 89.Lavender P, Cousins D, Lee T. Regulation of Th2 cytokine gene transcription. Chem Immunol. 2000;78:16–29. doi: 10.1159/000058813. [DOI] [PubMed] [Google Scholar]
- 90.Molfino NA, Gossage D, Kolbeck R, Parker JM, Geba GP. Molecular and clinical rationale for therapeutic targeting of interleukin-5 and its receptor. Clin Exp Allergy. 2012;42:712–737. doi: 10.1111/j.1365-2222.2011.03854.x. [DOI] [PubMed] [Google Scholar]
- 91.Foster PS, Hogan SP, Yang M, Mattes J, Young IG, Matthaei KI, Kumar RK, Mahalingam S, Webb DC. Interleukin-5 and eosinophils as therapeutic targets for asthma. Trends Mol Med. 2002;8:162–167. doi: 10.1016/s1471-4914(02)02302-x. [DOI] [PubMed] [Google Scholar]
- 92.Hart TK, Cook RM, Zia-Amirhosseini P, Minthorn E, Sellers TS, Maleeff BE, Eustis S, Schwartz LW, Tsui P, Appelbaum ER, Martin EC, Bugelski PJ, Herzyk DJ. Preclinical efficacy and safety of mepolizumab (SB-240563), a humanized monoclonal antibody to IL-5, in cynomolgus monkeys. J Allergy Clin Immunol. 2001;108:250–257. doi: 10.1067/mai.2001.116576. [DOI] [PubMed] [Google Scholar]
- 93.Leckie MJ, Brinke A, Khan J, Diamant Z, O'Connor BJ, Walls CM, Mathur AK, Cowley HC, Chung KF, Djukanovic R, Hansel TT, Holgate ST, Sterk PJ, Barnes PJ. Effects of an interleukin-5 blocking monoclonal antibody on eosinophils, airway hyper-responsiveness, and the late asthmatic response. Lancet. 2000;356:2144–2148. doi: 10.1016/s0140-6736(00)03496-6. [DOI] [PubMed] [Google Scholar]
- 94.Flood-Page P, Swenson C, Faiferman I, Matthews J, Williams M, Brannick L, Robinson D, Wenzel S, Busse W, Hansel TT, Barnes NC. International Mepolizumab Study Group. A study to evaluate safety and efficacy of mepolizumab in patients with moderate persistent asthma. Am J Respir Crit Care Med. 2007;176:1062–1107. doi: 10.1164/rccm.200701-085OC. [DOI] [PubMed] [Google Scholar]
- 95.Kay AB, Menzies-Gow A. Eosinophils and interleukin-5: the debate continues. Am J Respir Crit Care Med. 2003;167:1586–1587. doi: 10.1164/rccm.2304001. [DOI] [PubMed] [Google Scholar]
- 96.Menzies-Gow AN, Flood-Page PT, Robinson DS, Kay AB. Anti-IL-5 (mepolizumab) therapy induces bone marrow eosinophil maturational arrest and decreases eosinophil progenitors in the bronchial mucosa of atopic asthmatics. J Allergy Clin Immunol. 2003;111:714–719. doi: 10.1067/mai.2003.1382. [DOI] [PubMed] [Google Scholar]
- 97.Haldar P, Brightling CE, Hargadon B, Gupta S, Monteiro W, Sousa A, Marshall RP, Bradding P, Green RH, Wardlaw AJ, Pavord ID. Mepolizumab and exacerbations of refractory eosinophilic asthma. N Engl J Med. 2009;360:973–984. doi: 10.1056/NEJMoa0808991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Nair P, Pizzichini MM, Kjarsgaard M, Inman MD, Efthimiadis A, Pizzichini E, Hargreave FE, O'Byrne PM. Mepolizumab for prednisone-dependent asthma with sputum eosinophilia. N Engl J Med. 2009;360:985–993. doi: 10.1056/NEJMoa0805435. [DOI] [PubMed] [Google Scholar]
- 99.Kips JC, O'Connor BJ, Langley SJ, Woodcock A, Kerstjens HA, Postma DS, Danzig M, Cuss F, Pauwels RA. Effect of SCH55700, a humanized anti-human interleukin-5 antibody, in severe persistent asthma: a pilot study. Am J Respir Crit Care Med. 2003;167:1655–1659. doi: 10.1164/rccm.200206-525OC. [DOI] [PubMed] [Google Scholar]
- 100.Castro M, Mathur S, Hargreave F, Boulet LP, Xie F, Young J, Wilkins HJ, Henkel T, Nair P Res-5-0010 Study Group. Reslizumab for poorly controlled, eosinophilic asthma: a randomized, placebo-controlled study. Am J Respir Crit Care Med. 2011;184:1125–1132. doi: 10.1164/rccm.201103-0396OC. [DOI] [PubMed] [Google Scholar]
- 101.Gevaert P, Lang-Loidolt D, Lackner A, Stammberger H, Staudinger H, Van Zele T, Holtappels G, Tavernier J, van Cauwenberge P, Bachert C. Nasal IL-5 levels determine the response to anti-IL-5 treatment in patients with nasal polyps. J Allergy Clin Immunol. 2006;118:1133–1141. doi: 10.1016/j.jaci.2006.05.031. [DOI] [PubMed] [Google Scholar]
- 102.Gevaert P, Van Bruaene N, Cattaert T, Van Steen K, Van Zele T, Acke F, De Ruyck N, Blomme K, Sousa AR, Marshall RP, Bachert C. Mepolizumab, a humanized anti-IL-5 mAb, as a treatment option for severe nasal polyposis. J Allergy Clin Immunol. 2011;128:989–995. doi: 10.1016/j.jaci.2011.07.056. [DOI] [PubMed] [Google Scholar]
- 103.Kolbeck R, Kozhich A, Koike M, Peng L, Andersson CK, Damschroder MM, Reed JL, Woods R, Dall'acqua WW, Stephens GL, Erjefalt JS, Bjermer L, Humbles AA, Gossage D, Wu H, Kiener PA, Spitalny GL, Mackay CR, Molfino NA, Coyle AJ. MEDI-563, a humanized anti-IL-5 receptor alpha mAb with enhanced antibody-dependent cell-mediated cytotoxicity function. J Allergy Clin Immunol. 2010;125:1344–1353. doi: 10.1016/j.jaci.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 104.Busse WW, Katial R, Gossage D, Sari S, Wang B, Kolbeck R, Coyle AJ, Koike M, Spitalny GL, Kiener PA, Geba GP, Molfino NA. Safety profile, pharmacokinetics, and biologic activity of MEDI-563, an anti-IL-5 receptor alpha antibody, in a phase I study of subjects with mild asthma. J Allergy Clin Immunol. 2010;125:1237–1244. doi: 10.1016/j.jaci.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 105.Oh CK, Geba GP, Molfino N. Investigational therapeutics targeting the IL-4/IL-13/STAT-6 pathway for the treatment of asthma. Eur Respir Rev. 2010;19:46–54. doi: 10.1183/09059180.00007609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Saggini A, Maccauro G, Tripodi D, De Lutiis MA, Conti F, Felaco P, Fulcheri M, Galzio R, Caraffa A, Antinolfi P, Felaco M, Pandolfi F, Sabatino G, Neri G, Shaik-Dasthagirisaheb YB. Allergic inflammation: role of cytokines with special emphasis on IL-4. Int J Immunopathol Pharmacol. 2011;24:305–311. doi: 10.1177/039463201102400204. [DOI] [PubMed] [Google Scholar]
- 107.Carter RJ, Bradding P. The role of mast cells in the structural alterations of the airways as a potential mechanism in the pathogenesis of severe asthma. Curr Pharm Des. 2011;17:685–698. doi: 10.2174/138161211795428975. [DOI] [PubMed] [Google Scholar]
- 108.Howard TD, Koppelman GH, Xu J, Zheng SL, Postma DS, Meyers DA, Bleecker ER. Gene-gene interaction in asthma: IL4RA and IL13 in a Dutch population with asthma. Am J Hum Genet. 2002;70:230–236. doi: 10.1086/338242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Townley RG, Sapkota M, Sapkota K. IL-13 and its genetic variants: effect on current asthma treatments. Discov Med. 2011;12:513–523. [PubMed] [Google Scholar]
- 110.Andrews AL, Nordgren IK, Kirby I, Holloway JW, Holgate ST, Davies DE, Tavassoli A. Cytoplasmic tail of IL-13Ralpha2 regulates IL-4 signal transduction. Biochem Soc Trans. 2009;37:873–876. doi: 10.1042/BST0370873. [DOI] [PubMed] [Google Scholar]
- 111.Andrews AL, Holloway JW, Holgate ST, Davies DE. IL-4 receptor alpha is an important modulator of IL-4 and IL-13 receptor binding: implications for the development of therapeutic targets. J Immunol. 2006;176:7456–7461. doi: 10.4049/jimmunol.176.12.7456. [DOI] [PubMed] [Google Scholar]
- 112.Borish LC, Nelson HS, Lanz MJ, Claussen L, Whitmore JB, Agosti JM, Garrison L. Interleukin-4 receptor in moderate atopic asthma. A phase I/II randomized, placebo-controlled trial. Am J Respir Crit Care Med. 1999;160:1816–1823. doi: 10.1164/ajrccm.160.6.9808146. [DOI] [PubMed] [Google Scholar]
- 113.Borish LC, Nelson HS, Corren J, Bensch G, Busse WW, Whitmore JB, Agosti JM IL-4R Asthma Study Group. Efficacy of soluble IL-4 receptor for the treatment of adults with asthma. J Allergy Clin Immunol. 2001;107:963–970. doi: 10.1067/mai.2001.115624. [DOI] [PubMed] [Google Scholar]
- 114.Burmeister Getz E, Fisher DM, Fuller R. Human pharmacokinetics/pharmacodynamics of an interleukin-4 and interleukin-13 dual antagonist in asthma. J Clin Pharmacol. 2009;49:1025–1036. doi: 10.1177/0091270009341183. [DOI] [PubMed] [Google Scholar]
- 115.Wenzel S, Wilbraham D, Fuller R, Getz EB, Longphre M. Effect of an interleukin-4 variant on late phase asthmatic response to allergen challenge in asthmatic patients: results of two phase 2a studies. Lancet. 2007;370:1422–1431. doi: 10.1016/S0140-6736(07)61600-6. [DOI] [PubMed] [Google Scholar]
- 116.Holgate ST. Trials and tribulations in identifying new biologic treatments for asthma. Trends Immunol. 2012;33:238–246. doi: 10.1016/j.it.2012.02.003. [DOI] [PubMed] [Google Scholar]
- 117.Gauvreau GM, Boulet LP, Cockcroft DW, Fitzgerald JM, Carlsten C, Davis BE, Deschesnes F, Duong M, Durn BL, Howie KJ, Hui L, Kasaian MT, Killian KJ, Strinich TX, Watson RM, Y N, Zhou S, Raible D, O'Byrne PM. Effects of interleukin-13 blockade on allergen-induced airway responses in mild atopic asthma. Am J Respir Crit Care Med. 2011;183:1007–1014. doi: 10.1164/rccm.201008-1210OC. [DOI] [PubMed] [Google Scholar]
- 118.Kasaian MT, Raible D, Marquette K, Cook TA, Zhou S, Tan XY, Tchistiakova L. IL-13 antibodies influence IL-13 clearance in humans by modulating scavenger activity of IL-13Rα2. J Immunol. 2011;187:561–569. doi: 10.4049/jimmunol.1100467. [DOI] [PubMed] [Google Scholar]
- 119.Corren J, Busse W, Meltzer EO, Mansfield L, Bensch G, Fahrenholz J, Wenzel SE, Chon Y, Dunn M, Weng HH, Lin SL. A randomized, controlled, phase 2 study of AMG 317, an IL-4Ralpha antagonist, in patients with asthma. Am J Respir Crit Care Med. 2010;181:788–796. doi: 10.1164/rccm.200909-1448OC. [DOI] [PubMed] [Google Scholar]
- 120.Kakkar T, Sung C, Gibiansky L, Vu T, Narayanan A, Lin SL, Vincent M, Banfield C, Colbert A, Hoofring S, Starcevic M, Ma P. Population PK and IgE pharmacodynamic analysis of a fully human monoclonal antibody against IL4 receptor. Pharm Res. 2011;28:2530–2542. doi: 10.1007/s11095-011-0481-y. [DOI] [PubMed] [Google Scholar]
- 121.Thom G, Minter R. Optimization of CAT-354, a therapeutic antibody directed against interleukin-13, using ribosome display. Methods Mol Biol. 2012;805:393–401. doi: 10.1007/978-1-61779-379-0_22. [DOI] [PubMed] [Google Scholar]
- 122.May R, Monk P, Cohen E, Manuel D, Dempsey F, Davis N, Dodd A, Corkill D, Woods J, Joberty-Candotti C, Conroy L, Koentgen F, Martin E, Wilson R, Brennan N, Powell J, Anderson IK. Preclinical development of CAT-354, an IL-13-neutralising antibody, for the treatment of severe uncontrolled asthma. Br J Pharmacol. 2012;166:177–193. doi: 10.1111/j.1476-5381.2011.01659.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wenzel SE. Asthma: defining of the persistent adult phenotypes. Lancet. 2006;368:804–813. doi: 10.1016/S0140-6736(06)69290-8. [DOI] [PubMed] [Google Scholar]
- 124.Haldar P, Pavord ID, Shaw DE, Berry MA, Thomas M, Brightling CE, Wardlaw AJ, Green RH. Cluster analysis and clinical asthma phenotypes. Am J Respir Crit Care Med. 2008;178:218–224. doi: 10.1164/rccm.200711-1754OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Weatherall M, Travers J, Shirtcliffe PM, Marsh SE, Williams MV, Nowitz MR, Aldington S, Beasley R. Distinct clinical phenotypes of airways disease defined by cluster analysis. Eur Respir J. 2009;34:812–818. doi: 10.1183/09031936.00174408. [DOI] [PubMed] [Google Scholar]
- 126.Moore WC, Meyers DA, Wenzel SE, Teague WG, Li H, Li X, D'Agostino R, Jr, Castro M, Curran-Everett D, Fitzpatrick AM, Gaston B, Jarjour NN, Sorkness R, Calhoun WJ, Chung KF, Comhair SA, Dweik RA, Israel E, Peters SP, Busse WW, Erzurum SC, Bleecker ER National Heart, Lung, and Blood Institute's Severe Asthma Research Program. Identification of asthma phenotypes using cluster analysis in the Severe Asthma Research Program. Am J Respir Crit Care Med. 2010;181:315–323. doi: 10.1164/rccm.200906-0896OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Siroux V, Basagaña X, Boudier A, Pin I, Garcia-Aymerich J, Vesin A, Slama R, Jarvis D, Anto JM, Kauffmann F, Sunyer J. Identifying adult asthma phenotypes using a clustering approach. Eur Respir J. 2011;38:310–317. doi: 10.1183/09031936.00120810. [DOI] [PubMed] [Google Scholar]
- 128.Fitzpatrick AM, Teague WG. Severe asthma in children: insights from the national heart, lung, and blood institute's severe asthma research program. Pediatr Allergy Immunol Pulmonol. 2010;23:131–138. doi: 10.1089/ped.2010.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Vermeer H, Hendriks-Stegeman BI, Verrijn Stuart AA, van Buul-Offers SC, Jansen M. A comparison of in vitro bioassays to determine cellular glucocorticoid sensitivity. Eur J Endocrinol. 2004;150:41–47. doi: 10.1530/eje.0.1500041. [DOI] [PubMed] [Google Scholar]
- 130.Woodruff PG, Boushey HA, Dolganov GM, Barker CS, Yang YH, Donnelly S, Ellwanger A, Sidhu SS, Dao-Pick TP, Pantoja C, Erle DJ, Yamamoto KR, Fahy JV. Genome-wide profiling identifies epithelial cell genes associated with asthma and with treatment response to corticosteroids. Proc Natl Acad Sci U S A. 2007;104:15858–15863. doi: 10.1073/pnas.0707413104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Chun E, Lee HS, Bang BR, Kim TW, Lee SH, Kim JH, Cho SH, Min KU, Kim YY, Park HW. Dexamethasone-induced FKBP51 expression in peripheral blood mononuclear cells could play a role in predicting the response of asthmatics to treatment with corticosteroids. J Clin Immunol. 2011;31:122–127. doi: 10.1007/s10875-010-9463-9. [DOI] [PubMed] [Google Scholar]
- 132.Woodruff PG, Modrek B, Choy DF, Jia G, Abbas AR, Ellwanger A, Koth LL, Arron JR, Fahy JV. T-helper type 2-driven inflammation defines major subphenotypes of asthma. Am J Respir Crit Care Med. 2009;180:388–395. doi: 10.1164/rccm.200903-0392OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Dougherty RH, Sidhu SS, Raman K, Solon M, Solberg OD, Caughey GH, Woodruff PG, Fahy JV. Accumulation of intraepithelial mast cells with a unique protease phenotype in T(H)2-high asthma. J Allergy Clin Immunol. 2010;125:1046–1053. doi: 10.1016/j.jaci.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sidhu SS, Yuan S, Innes AL, Kerr S, Woodruff PG, Hou L, Muller SJ, Fahy JV. Roles of epithelial cell-derived periostin in TGF-beta activation, collagen production, and collagen gel elasticity in asthma. Proc Natl Acad Sci U S A. 2010;107:14170–14175. doi: 10.1073/pnas.1009426107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Takayama G, Arima K, Kanaji T, Toda S, Tanaka H, Shoji S, McKenzie AN, Nagai H, Hotokebuchi T, Izuhara K. Periostin: a novel component of subepithelial fibrosis of bronchial asthma downstream of IL-4 and IL-13 signals. J Allergy Clin Immunol. 2006;118:98–104. doi: 10.1016/j.jaci.2006.02.046. [DOI] [PubMed] [Google Scholar]
- 136.Holgate ST, Holloway J, Wilson S, Bucchieri F, Puddicombe S, Davies DE. Epithelial-mesenchymal communication in the pathogenesis of chronic asthma. Proc Am Thorac Soc. 2004;1:93–98. doi: 10.1513/pats.2306034. [DOI] [PubMed] [Google Scholar]
- 137.Corren J, Lemanske RF, Hanania NA, Korenblat PE, Parsey MV, Arron JR, Harris JM, Scheerens H, Wu LC, Su Z, Mosesova S, Eisner MD, Bohen SP, Matthews JG. Lebrikizumab treatment in adults with asthma. N Engl J Med. 2011;365:1088–1098. doi: 10.1056/NEJMoa1106469. [DOI] [PubMed] [Google Scholar]
- 138.Baines KJ, Simpson JL, Wood LG, Scott RJ, Gibson PG. Transcriptional phenotypes of asthma defined by gene expression profiling of induced sputum samples. J Allergy Clin Immunol. 2011;127:153–160. doi: 10.1016/j.jaci.2010.10.024. [DOI] [PubMed] [Google Scholar]
- 139.Wood LG, Baines KJ, Fu J, Scott HA, Gibson PG. The neutrophilic inflammatory phenotype is associated with systemic inflammation in asthma. Chest. 2012;142:86–93. doi: 10.1378/chest.11-1838. [DOI] [PubMed] [Google Scholar]
- 140.Howarth PH, Babu KS, Arshad HS, Lau L, Buckley M, McConnell W, Beckett P, Al Ali M, Chauhan A, Wilson SJ, Reynolds A, Davies DE, Holgate ST. Tumour necrosis factor (TNFalpha) as a novel therapeutic target in symptomatic corticosteroid dependent asthma. Thorax. 2005;60:1012–1018. doi: 10.1136/thx.2005.045260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Stoll ML, Solomon DH, Batra KL, Simard JF, Karlson EW, Dellaripa PF, Weinblatt ME, Glass R, Shadick NA. TNFalpha inhibitors may improve asthma symptoms: a case series of 12 patients with rheumatoid arthritis and asthma. J Clin Rheumatol. 2009;15:198–200. doi: 10.1097/RHU.0b013e3181a7ace9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Berry MA, Hargadon B, Shelley M, Parker D, Shaw DE, Green RH, Bradding P, Brightling CE, Wardlaw AJ, Pavord ID. Evidence of a role of tumor necrosis factor alpha in refractory asthma. N Engl J Med. 2006;354:697–708. doi: 10.1056/NEJMoa050580. [DOI] [PubMed] [Google Scholar]
- 143.Morjaria JB, Chauhan AJ, Babu KS, Polosa R, Davies DE, Holgate ST. The role of a soluble TNFalpha receptor fusion protein (etanercept) in corticosteroid refractory asthma: a double blind, randomised, placebo controlled trial. Thorax. 2008;63:584–591. doi: 10.1136/thx.2007.086314. [DOI] [PubMed] [Google Scholar]
- 144.Holgate ST, Noonan M, Chanez P, Busse W, Dupont L, Pavord I, Hakulinen A, Paolozzi L, Wajdula J, Zang C, Nelson H, Raible D. Efficacy and safety of etanercept in moderate-to-severe asthma: a randomised, controlled trial. Eur Respir J. 2011;37:1352–1359. doi: 10.1183/09031936.00063510. [DOI] [PubMed] [Google Scholar]
- 145.Wenzel SE, Barnes PJ, Bleecker ER, Bousquet J, Busse W, Dahlén SE, Holgate ST, Meyers DA, Rabe KF, Antczak A, Baker J, Horvath I, Mark Z, Bernstein D, Kerwin E, Schlenker-Herceg R, Lo KH, Watt R, Barnathan ES, Chanez P T03 Asthma Investigators. A randomized, double-blind, placebo-controlled study of tumor necrosis factor-alpha blockade in severe persistent asthma. Am J Respir Crit Care Med. 2009;179:549–558. doi: 10.1164/rccm.200809-1512OC. [DOI] [PubMed] [Google Scholar]