

Abstract
The mechanism of oxygen sensing in arterial chemoreceptors is unknown but has often been linked to mitochondrial function. A common criticism of this hypothesis is that mitochondrial function is insensitive to physiological levels of hypoxia. Here we investigate the effects of hypoxia (down to 0.5% O2) on mitochondrial function in neonatal rat type-1 cells. The oxygen sensitivity of mitochondrial [NADH] was assessed by monitoring autofluorescence and increased in hypoxia with a P50 of 15 mm Hg (1 mm Hg = 133.3 Pa) in normal Tyrode or 46 mm Hg in Ca2+-free Tyrode. Hypoxia also depolarised mitochondrial membrane potential (ψm, measured using rhodamine 123) with a P50 of 3.1, 3.3 and 2.8 mm Hg in normal Tyrode, Ca2+-free Tyrode and Tyrode containing the Ca2+ channel antagonist Ni2+, respectively. In the presence of oligomycin and low carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP; 75 nm) ψm is maintained by electron transport working against an artificial proton leak. Under these conditions hypoxia depolarised ψm/inhibited electron transport with a P50 of 5.4 mm Hg. The effects of hypoxia upon cytochrome oxidase activity were investigated using rotenone, myxothiazol, antimycin A, oligomycin, ascorbate and the electron donor tetramethyl-p-phenylenediamine. Under these conditions ψm is maintained by complex IV activity alone. Hypoxia inhibited cytochrome oxidase activity (depolarised ψm) with a P50 of 2.6 mm Hg. In contrast hypoxia had little or no effect upon NADH (P50= 0.3 mm Hg), electron transport or cytochrome oxidase activity in sympathetic neurons. In summary, type-1 cell mitochondria display extraordinary oxygen sensitivity commensurate with a role in oxygen sensing. The reasons for this highly unusual behaviour are as yet unexplained.
Key points
Arterial chemoreceptors measure blood oxygen and are involved in the control of both breathing and the cardiovascular system.
Oxygen is mostly used by cells in their mitochondria to generate energy.
In this study we have investigated the effects of oxygen starvation (hypoxia) on the mitochondria of specialised oxygen sensing cells from arterial chemoreceptors.
Our data confirm that the mitochondria of these oxygen sensing cells are unusually sensitive to modest hypoxia. This effect seems to stem from a reduced affinity of the oxygen utilising enzyme cytochrome oxidase for oxygen.
These results are consistent with a functional adaptation of the mitochondria of oxygen sensing cells that may enable them to play a direct role in the oxygen sensing process itself.
Introduction
The ability to sense changes in oxygen plays a vital role in ensuring adequate oxygenation of animal tissues. There are at least two distinct pathways. One involves the control of gene transcription via hypoxia inducible factor, which is regulated by oxygen sensitive prolyl and asparaginyl hydroxylases (Schofield & Ratcliffe, 2004; Kaelin & Ratcliffe, 2008; Semenza, 2012). This pathway responds slowly to sustained hypoxia and is ubiquitous. The other pathway controls cellular activity by regulating ion channels (Weir et al. 2005) and is found primarily in a few specialised tissues. This latter pathway can signal acute changes in oxygen within seconds. The archetype of this form of acute oxygen sensing is found in the type-1 cells of peripheral arterial chemoreceptors which drive both ventilatory and cardiovascular reflexes to hypoxaemia. The identity of the sensor involved in acute oxygen sensing is, however, proving to be elusive. One of the earliest, and most controversial, hypotheses is that oxygen sensing in chemoreceptors is linked to oxidative phosphorylation (the metabolic hypothesis). This proposal is supported by a wealth of data demonstrating that arterial chemoreceptors are powerfully excited by all inhibitors of oxidative phosphorylation (Heymans et al. 1931; Shen & Hauss, 1939; Anichkov & Belen’kii, 1963; Mulligan et al. 1981; Mulligan & Lahiri, 1982; Wilson et al. 1994). In the type-1 cell these metabolic poisons elicit a classic pattern of sensory neuronal excitation comprising modulation of ion channels to generate a receptor potential which then stimulates electrical activity, calcium influx and neurosecretion (Obeso et al. 1989; Buckler & Vaughan-Jones, 1998; Ortega Saenz et al. 2003; Williams & Buckler, 2004; Wyatt & Buckler, 2004; Varas et al. 2007). The existence of a metabolic signalling pathway in these cells therefore seems well established; the contentious issue is whether this pathway is the same as that used for acute oxygen sensing.
The standard criticism of the metabolic hypothesis is that cytochrome oxidase has such a high affinity for oxygen that ‘physiological hypoxia’ should have no effect upon mitochondrial energy metabolism. This assertion has rarely been directly tested, but three key studies have reported mitochondrial function in the carotid body to have an extraordinarily high sensitivity to hypoxia (Mills & Jöbsis, 1970, 1972; Nair et al. 1986; Duchen & Biscoe, 1992a,b). Whilst there have been theoretical criticisms of these studies there have been little data obtained from arterial chemoreceptors to countermand them. In this paper we have reinvestigated, and sought an explanation for, the effects of hypoxia upon mitochondrial function in isolated type-1 cells.
Our data fully confirmed previous observations (Duchen & Biscoe, 1992a,b) that hypoxia increases mitochondrial NADH and depolarises mitochondrial membrane potential (ψm) over a range of 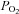 that is both exceptional and commensurate with a role in acute oxygen sensing. We also observed that hypoxia was able to inhibit electron transport and cytochrome oxidase activity at relatively high
that is both exceptional and commensurate with a role in acute oxygen sensing. We also observed that hypoxia was able to inhibit electron transport and cytochrome oxidase activity at relatively high 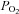 and suggest that this is the most likely cause of changes in NADH and ψm. Possible explanations for the effects of hypoxia on cytochrome oxidase activity and electron transport are considered, but we are as yet unable to give any definitive reason for this unusual behaviour.
and suggest that this is the most likely cause of changes in NADH and ψm. Possible explanations for the effects of hypoxia on cytochrome oxidase activity and electron transport are considered, but we are as yet unable to give any definitive reason for this unusual behaviour.
Methods
Ethical approval
All procedures involving animals were approved by the University of Oxford's Local Ethical Review Process and were conducted in accordance with project and personal licences issued under the UK Animals (Scientific Procedures) Act 1986.
Cell isolation
Carotid artery bifurcations with attached carotid bodies and superior cervical ganglia were excised from neonatal rats aged 12–15 days under terminal anaesthesia (2–4% halothane in oxygen, at the end of the procedure animals were killed by exsanguination or decapitation). Bifurcations were transferred to ice-cold phosphate-buffered saline and the carotid bodies and/or superior cervical ganglia were dissected free. Type-1 cells were isolated from the carotid body using enzymatic digestion with 0.4 mg ml−1 trypsin (Sigma, St Louis, MO, USA) and 0.5 mg ml−1 collagenase (Worthington, Lakewood, NJ, USA) in HAMS F-12 for 25–30 min at 35°C followed by transfer to enzyme-free DMEM/F12 culture media and trituration through fire-polished pipettes. The resultant cell suspension was plated on to poly-l-lysine-coated coverslips and maintained in an incubator at 37°C, 5% CO2 and 11% O2 for between 2 and 8 h before use. Culture media comprised ‘Advanced DMEM/F-12’ (Gibco, Carlsbad, CA, USA) supplemented with 10% heat-inactivated fetal bovine serum, 2 mm l-glutamine and 4 μg ml−1 insulin.
Isolation of sympathetic neurons from the superior cervical ganglia used a similar protocol to that described above but with the following modifications: (i) ganglia were cut into 4–5 pieces before incubation in enzyme media and (ii) enzymatic digestion was for 40–50 min.
Measurement of [Ca2+]i and NADH
Fluorescence measurements were performed using a microspectrofluorimeter based on a Nikon Diaphot 200 (Tokyo, Japan) equipped with a xenon lamp to provide an excitation light source and cooled (−20°C) photomultiplier tubes (PMTs; Thorn EMI, London, UK) to detect emitted fluorescence. [Ca2+]i was measured using Indo-1 loaded into cells by incubation with 2–5 μm of the acetoxymethyl ester derivative in culture media for 1 h at room temperature. Indo-1 was excited at 340 nm and fluorescence intensity measured at 405 ± 16 nm and 495 ± 10 nm. The fluorescence emission ratio (405/495) for Indo-1 was calibrated as previously described (Buckler & Vaughan Jones, 1993).
NADH was excited at 340 nm and fluorescence measured at 450 ± 15 nm. Raw traces showing fluorescence at 450 nm (NADH) are presented without any correction or calibration. The units shown in the ordinate axis in Figs 1–3 represent the output voltage from an I–V converter, with gain 10 nA V-1, connected to the PMT. An offset was applied such that 0 (volts) corresponds to the level of background signal in the absence of cells.
Figure 3. Effects of graded hypoxia on NADH autofluorescence in SCG neuron.
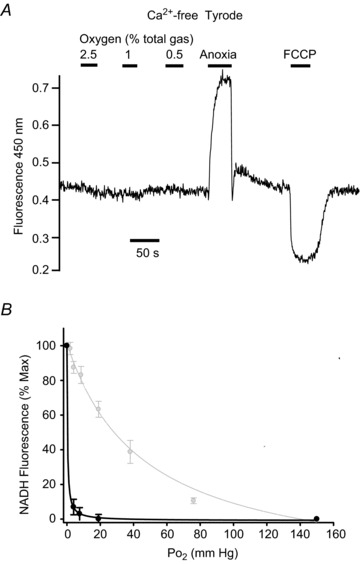
Recording of autofluorescence at 450 nm with 340 nm excitation showing effects of graded levels of hypoxia from 2.5% down to anoxia. A, original recording from a single neuron conducted in Ca2+-free Tyrode containing 100 μm EGTA. B, normalised summary data (mean ± SEM) from 10 neurons with best fit rectangular hyperbola (black line, see Results for details). Also shown in B, in grey, are data and best fit rectangular hyperbola for type-1 cells recorded under identical conditions reproduced from Fig. 2D. Note minimal effects of hypoxia even at 0.5%; only anoxia causes a robust increase in NADH autofluorescence in SCG neurons.
Measurement of mitochondrial membrane potential using rhodamine 123
Mitochondrial membrane potential (ψm) was monitored using rhodamine 123 (Rh123) in the dequench mode. Rh123 is a fluorescent membrane-permeant cation which passively distributes across membranes according to the membrane potential. When cells are incubated in a solution containing Rh123 it is therefore taken up into the cell and then concentrated within the mitochondria, which have a very negative membrane potential. Under suitable loading conditions Rh123 uptake into mitochondria is associated with partial quenching of its fluorescence (Emaus et al. 1986). Once loaded, changes in ψm cause redistribution of Rh123 between mitochondria and cytosol with consequent changes in the degree of fluorescence quenching; for example, mitochondrial depolarisation causes Rh123 efflux from the mitochondrial matrix into the cytosol resulting in decreased quenching and thus an increase in overall Rh123 fluorescence (Emaus et al. 1986; Chen, 1988). Although we cannot directly calibrate Rh123 fluorescence changes in terms of ψm (see Discussion) we have applied some correction for dye bleaching/leakage. Baseline Rh123 fluorescence (measured under specified conditions) and the maximum fluorescence observed in the presence of 1 μm carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP, a mitochondrial uncoupler) were determined before and after experiments with hypoxia. Linear interpolation was then used to estimate baseline and maximum fluorescence during the intervening recording so that data obtained at any time point could be normalised to a 0% (baseline) to 100% (FCCP) scale. The baseline condition varied depending on the experiment (see Results and figure legends). Raw data traces (Figs 4A, B, 5A, B and 6A, B) are shown without any correction; the units shown in the ordinate axis represent the output voltage from the PMT I–V converter (gain 10 nA V-1).
Figure 4. Effects of hypoxia on mitochondrial membrane potential in type-1 cells.
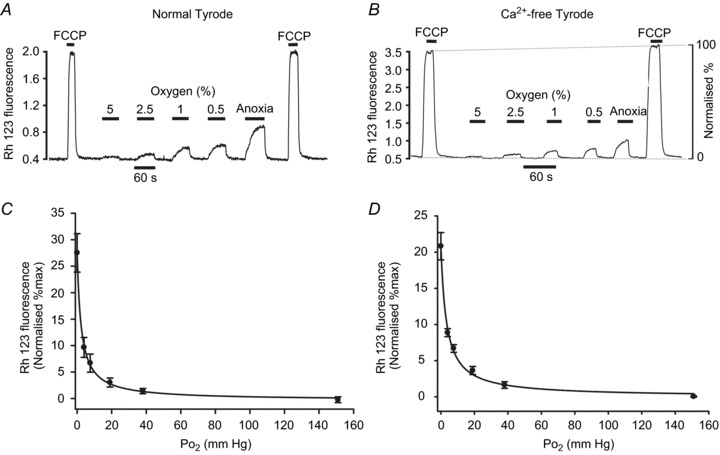
Measurements of mitochondrial membrane potential were performed using rhodamine 123 (see Methods). A, original recording from a small type-1 cell cluster conducted in normal Tyrode. B, similar recording obtained in Ca2+-free Tyrode containing 100 μm EGTA. Hypoxia ranged from 5% O2 down to anoxia. Note application of FCCP (1 μm) causes rapid increase in fluorescence as Rh123 is released from mitochondria due to depolarisation of ψm. Depolarisation is assumed to be complete in 1 μm FCCP. Effects of hypoxia were measured on a 0–100% scale where 0%= interpolated baseline fluorescence in 20% O2 and 100%= interpolated maximum fluorescence in 1 μm FCCP. C and D, normalised summary data (mean ± SEM) from 11 recordings with best fit rectangular hyperbola (see Results for details) for normal Tyrode (C) and Ca-free Tyrode (D).
Figure 5. Effects of hypoxia on electron transport.
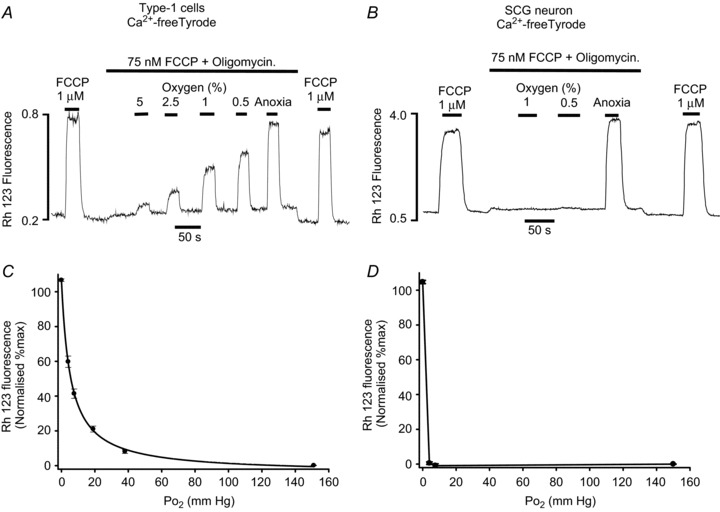
Measurements of mitochondrial membrane potential were performed using rhodamine 123. A and B, original recordings conducted in Ca2+-free Tyrode containing 100 μm EGTA. Type-1 cells (A) or SCG neurons (B) were exposed to a low level of FCCP (75 nm) and oligomycin (2.5 μg ml−1) to inhibit ATP synthase and partially uncouple the mitochondria (see text). The effects of hypoxia were then tested in the presence of FCCP and oligomycin. The effect of hypoxia was assessed on a 0–100% scale where 0%= interpolated baseline fluorescence in 20% O2, 75 μm FCCP + oligomycin and 100%= interpolated maximum fluorescence in 1 μm FCCP (applied as a brief pulse before and after each experiment). C, normalised summary data (mean ± SEM) from 12 recordings obtained in single and small clusters of type-1 cells with best fit rectangular hyperbola (see Results for details). D, normalised summary data (mean ± SEM) from 7 recordings obtained in single SCG neurons. Data points in D are joined by straight lines only.
Figure 6. Effects of hypoxia on cytochrome oxidase activity.
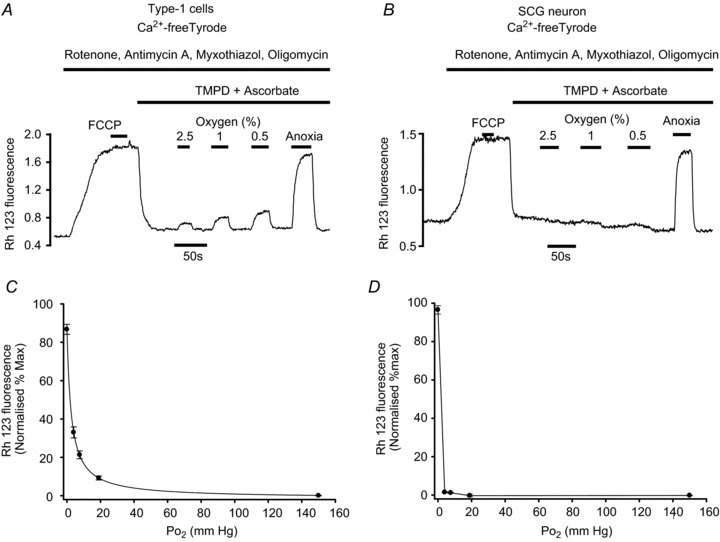
A and B, original recordings of ψm performed using rhodamine 123 conducted in Ca2+-free Tyrode containing 100 μm EGTA. Type-1 cells (A) or SCG neurons (B) were exposed first to a cocktail of rotenone, antimycin A, myxothiazol and oligomycin to block complexes I, III and V. This depolarises ψm, which reaches a steady state within about 50 s. Subsequent addition of 1 μm FCCP has no further effect on the Rh123 signal, suggesting that mitochondria are fully depolarised and electron transport is fully inhibited. Addition of TMPD + ascorbate, to provide a source of electrons to cytochrome c and thus substrate for cytochrome oxidase, causes a brisk repolarisation of ψm. The effects of hypoxia on ψm, which is assumed to be maintained entirely by cytochrome oxidase activity under these conditions, was then tested. The effects of hypoxia were quantified on a 0–100% scale where 0%= interpolated baseline fluorescence in 20% O2+ inhibitors + TMPD/ascorbate and 100%= interpolated maximum fluorescence in 1 μm FCCP (applied as a brief pulse before and after each experiment). C, normalised summary data (mean ± SEM) from 14 recordings obtained in single and small clusters of type-1 cells with best fit rectangular hyperbola (see Results for details). D, normalised summary data (mean ± SEM) from five recordings obtained in single SCG neurons. Data points in D are joined by straight lines only.
Solutions
Standard bicarbonate-buffered Tyrode solutions contained (in mm): NaCl, 117; KCl, 4.5; CaCl2, 2.5; MgCl2, 1; NaHCO3, 23; glucose, 11. In Ca2+-free solutions CaCl2 was omitted and 100 μm EGTA added. Normoxic solutions were equilibrated with 5% CO2 and 95% air, hypoxic solutions were equilibrated with 5% CO2 and 10, 5, 2.5, 1 and 0.5% oxygen (balance nitrogen). Anoxic solutions were produced by equilibration with 5% CO2/95% N2 followed by the addition of 100–200 μm Na2S2O4 (in experiments involving nickel, Na2S2O4 was added immediately before use). The 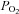 of the control solution was assumed to be 150 mm Hg and that of the anoxic solution to be 0 mm Hg. The
of the control solution was assumed to be 150 mm Hg and that of the anoxic solution to be 0 mm Hg. The 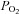 of hypoxic solutions was determined in the recording chamber using a 50 μm fibre optic oxygen sensor (PreSens, Regensburg, Germany) placed close to the bottom of the recording chamber and calibrated using control and anoxic solutions in situ. The
of hypoxic solutions was determined in the recording chamber using a 50 μm fibre optic oxygen sensor (PreSens, Regensburg, Germany) placed close to the bottom of the recording chamber and calibrated using control and anoxic solutions in situ. The 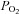 values of the above hypoxic solutions measured 30–60 s after switching from the control perfusate were approximately 76, 38, 19, 8 and 4 mm Hg, respectively. All solutions had a pH of 7.4 at 37°C
values of the above hypoxic solutions measured 30–60 s after switching from the control perfusate were approximately 76, 38, 19, 8 and 4 mm Hg, respectively. All solutions had a pH of 7.4 at 37°C
Acquisition and analysis of data
Data acquisition, calibration of the raw data and measurements taken from the raw data were performed using a 1401 interface and Spike 2 software (Cambridge Electronic Design, Cambridge, UK).
Values are expressed as mean ± standard error of the mean (SEM) unless otherwise specified. Statistical analysis was performed using one of the following: (i) a paired two-tailed t test for simple comparisons (in Excel, Microsoft), (ii) repeated-measures one-way analysis of variance (RM-ANOVA) with post hoc testing against control by the Holm Sidak method (Sigmaplot 12, Sysstat Software Inc, Germany), (iii) RM-ANOVA on ranks with post hoc testing against control by the Dunnet method (Sigmaplot 12). Significance was assumed at P < 0.05. 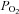 response curves shown in Figs 2–6 are three-parameter hyperbolas of the form y=yo+ (a×b)/(b+
response curves shown in Figs 2–6 are three-parameter hyperbolas of the form y=yo+ (a×b)/(b+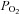 ) where b is the
) where b is the 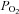 at which a half maximal effect would be observed (P50). Curve fitting was performed using the non-linear regression dynamic fitting routine in Sigmaplot 12.
at which a half maximal effect would be observed (P50). Curve fitting was performed using the non-linear regression dynamic fitting routine in Sigmaplot 12.
Drugs and chemicals
Antimycin A, myxothiazol, rotenone, tetramethyl- p-phenylenediamine (TMPD), FCCP, ascorbate, Rh123 and EGTA were from Sigma; Indo-1-AM was from Invitrogen (Carlsbad, CA, USA); oligomycin was from Calbiochem.
Results
NADH autofluorescence
Recordings of cellular autofluorescence were performed upon small clusters of type-1 cells (approx. 2–10 cells) or on single superior cervical ganglion (SCG) neurons. Signal levels were typically low when working with small clusters (2–3) of type-1 cells but could be easily resolved and recordings lasting ≥ 20 min with less than 25% signal loss were readily achievable. A large part of the autofluorescence at these wavelengths is due to mitochondrial NADH; NAD is non-fluorescent and cytosolic NADH levels are normally low. Mitochondrial NADH can be oxidised to NAD by the uncoupling of mitochondrial respiration. Application of 1 μm FCCP caused a rapid reduction in autofluorescence in all type-1 cells/clusters tested to 74% of control (±2.5%, n= 5, P < 0.005) indicating that approximately 25% of autofluorescence under control conditions was due to mitochondrial NADH (Fig. 1). Just as uncoupling promotes the conversion of mitochondrial NADH to NAD, inhibition of electron transport is expected to promote conversion of mitochondrial NAD into NADH. As expected, application of electron transport inhibitors increased autofluorescence in all type-1 cells tested to 144% (±7.9%, n= 6, P < 0.05) of control with cyanide (1–2 mm) and 136% (± 1.8%, n= 8, P < 0.005) of control with rotenone (1 μm). Similar results have also been obtained with other inhibitors of electron transport, including azide (data not shown) and hydrogen sulphide (Buckler, 2012). Uncouplers and electron transport inhibitors can therefore be used to define a range for the mitochondrial NADH signal between conditions of maximal reduction and maximal oxidation (see Fig. 1A).
Figure 1. NADH autofluorescence in type-1 cells.
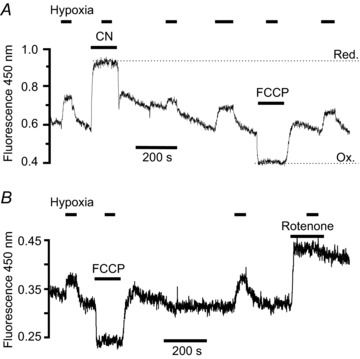
Recording of autofluorescence at 450 nm with 340 nm excitation from two small clusters of type-1 cells. Note increase in fluorescence in response to electron transport inhibition with 2 mm cyanide (A) and 1 μm rotenone (B), and decrease in fluorescence with 1 μm FCCP (A and B). The mitochondrial NAD+/NADH couple is assumed to be maximally oxidised in the presence of FCCP, i.e. all in the form of NAD+ which is non-fluorescent, and any remaining signal under these conditions is therefore assumed to come from some other source. In the presence of cyanide the mitochondrial NAD+/NADH couple is assumed to be maximally reduced, i.e. all in the form of NADH. The difference in fluorescence between the maximally reduced and oxidised states (Red & Ox in A) therefore represents total mitochondrial NADH + NAD+. Note that hypoxia (2.5% oxygen) increases fluorescence under control conditions but not in the presence of CN, rotenone or FCCP, indicating that hypoxia increases mitochondrial NADH but does not affect other sources of cellular fluorescence.
In preliminary experiments we found that superfusing type-1 cells with a moderately hypoxic Tyrode (equilibrated with 2.5% oxygen) also caused a rapid and reversible increase in autofluorescence in all cells/clusters tested (to 123 ± 1.6% of control, n= 7, P < 0.001). This effect of hypoxia was abolished in the presence of cyanide (Fig. 1A, n= 4), rotenone (Fig. 1B, n= 4) and FCCP (Fig. 1, n= 4). These data indicate that the increase in autofluorescence during hypoxia is due to an increase in mitochondrial NADH.
We next sought to define the oxygen sensitivity of changes in mitochondrial NADH in type-1 cells. Experiments were conducted by equilibrating normal Tyrode with a range of different gas mixtures containing 0.5, 1, 2.5 and 5% oxygen (all with 5% CO2). Measurements of 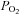 in the recording chamber, made using a 50 μm fibre optic oxygen probe (PreSens), returned values of 3.8, 7.6, 19 and 38 mmHg with each of these gas mixtures, respectively. Anoxic solutions were prepared by equilibrating Tyrode with 5% CO2/96% N2 for > 10 min followed by addition of 100–200 μm Na2S2O4 before use. Exposure of type-1 cells to hypoxic/anoxic Tyrode resulted in an increase in cellular autofluorescence (P < 0.001, n= 9, RM-ANOVA) and this was significant at every level of hypoxia tested (P < 0.001, n= 9, post hoc testing by Holm–Sidak method; see Fig. 2A). The increase in fluorescence at each level of hypoxia was expressed as a percentage of the increase seen in anoxic solution (relative to control) and is plotted in Fig. 2C (data points in anoxia are defined as 100% and those in normal Tyrode (
in the recording chamber, made using a 50 μm fibre optic oxygen probe (PreSens), returned values of 3.8, 7.6, 19 and 38 mmHg with each of these gas mixtures, respectively. Anoxic solutions were prepared by equilibrating Tyrode with 5% CO2/96% N2 for > 10 min followed by addition of 100–200 μm Na2S2O4 before use. Exposure of type-1 cells to hypoxic/anoxic Tyrode resulted in an increase in cellular autofluorescence (P < 0.001, n= 9, RM-ANOVA) and this was significant at every level of hypoxia tested (P < 0.001, n= 9, post hoc testing by Holm–Sidak method; see Fig. 2A). The increase in fluorescence at each level of hypoxia was expressed as a percentage of the increase seen in anoxic solution (relative to control) and is plotted in Fig. 2C (data points in anoxia are defined as 100% and those in normal Tyrode (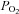 = 150 mm Hg) as 0%). Fitting a rectangular hyperbola (see Methods) to these data returned an R2 value of 0.84 and a P50 (the
= 150 mm Hg) as 0%). Fitting a rectangular hyperbola (see Methods) to these data returned an R2 value of 0.84 and a P50 (the 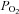 at which a half maximal effect would be observed) of 15 mmHg.
at which a half maximal effect would be observed) of 15 mmHg.
Figure 2. Effects of graded hypoxia on NADH autofluorescence in type-1 cells.
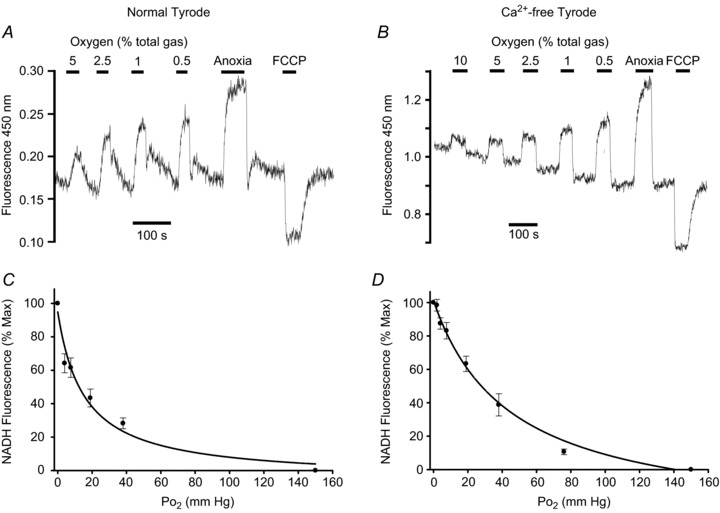
Recordings of autofluorescence at 450 nm with 340 nm excitation showing effects of graded levels of hypoxia from 5/10% down to anoxia. A and B, original recordings. C and D, normalised summary data (mean ± SEM) from 9 (C) and 11–17 (D) recordings with best fit rectangular hyperbola. The recording and graph on the left (A and C) were obtained in a normal Tyrode containing 2.5 mm Ca2+; the recording and graph on the right (B and D) were obtained in a Ca2+-free Tyrode containing 100 μm EGTA. Note rapid effects of hypoxia at all levels.
Hypoxia is known to promote Ca2+ influx in type-1 cells (Buckler & Vaughan Jones, 1994), elevation of [Ca2+]i promotes mitochondrial Ca2+ uptake and elevation of mitochondrial [Ca2+]i activates NADH producing dehydrogenases (see Discussion). The effects of hypoxia upon NADH could therefore be secondary to these events rather that an inherent property of mitochondrial function. We therefore repeated the above experiments under Ca2+-free conditions wherein hypoxia has minimal effect upon cytosolic Ca2+ in rat type-1 cells (Buckler & Vaughan Jones, 1994). We performed 17 recordings of NADH autofluorescence in type-1 cells using the above gas mixtures. In 11 recordings we also included a more severe hypoxia (0 oxygen, 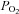 = 2 mm Hg) and in six recordings we included a solution equilibrated with 10% oxygen. Again there was a significant increase in autofluorescence at all levels of hypoxia tested including 10% oxygen (P < 0.05, RM-ANOVA with Holm–Sidak post hoc testing against control; see Fig. 2B). The increase in fluorescence at each level of hypoxia was again expressed as a percentage of the increase seen in anoxic solution (relative to control) and plotted as a function of
= 2 mm Hg) and in six recordings we included a solution equilibrated with 10% oxygen. Again there was a significant increase in autofluorescence at all levels of hypoxia tested including 10% oxygen (P < 0.05, RM-ANOVA with Holm–Sidak post hoc testing against control; see Fig. 2B). The increase in fluorescence at each level of hypoxia was again expressed as a percentage of the increase seen in anoxic solution (relative to control) and plotted as a function of 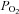 (Fig. 2D). Fitting a rectangular hyperbola to these data returned an R2 value of 0.87 and a P50 of 46 mm Hg.
(Fig. 2D). Fitting a rectangular hyperbola to these data returned an R2 value of 0.87 and a P50 of 46 mm Hg.
As these data suggest an extraordinary degree of oxygen sensitivity in type-1 cells we performed control experiments using another cell type. For this we chose sympathetic neurons from the SCG. These cells were chosen for a number of reasons: (i) the carotid bodies are themselves regarded as part of the sympathetic nervous system (sympathetic paraganglia), (ii) sympathetic neurons are not known to have any oxygen sensing role and (iii) their location in close proximity to the carotid body plus the ability to dissociate neurons from these ganglia using the same enzymatic solutions as those used for dissociating carotid body cells enabled us to ensure that the procedure for preparing isolated SCG neurons was almost identical to that used for type-1 cells (see Methods).
The effects of hypoxia upon autofluorescence in SCG neurons was studied in Ca2+-free media as above. Unlike type-1 cells, hypoxia had very little effect upon autofluorescence in SCG neurons whereas anoxia caused a robust increase in autofluorescence, and FCCP caused a robust decrease in fluorescence (Fig. 3A). Data obtained in the presence of hypoxia was again converted into a percenatge increase relative to that caused by anoxia. Analysis of these data by RM-ANOVA revealed a significant effect of oxygen overall (P < 0.001, n= 10), although post hoc testing revealed that 2.5% oxygen had no significant effect on fluorescence and that whist there was a statistically significant effect of both 0.5 and 1% oxygen (P < 0.05) these effects were very small (7 and 3% of the response to anoxia, respectively). These data are plotted as a function of 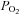 in Fig. 3B. A rectangular hyperbola fitted to these data returned an R2 value of 0.99 and a P50 of 0.3 mmHg, a value 100-fold less than that obtained in type-1 cells under otherwise identical recording conditions. Data from type-1 cells (from Fig. 2D) are reproduced in Fig. 3B (in grey) to facilitate comparison with that obtained from SCG neurons.
in Fig. 3B. A rectangular hyperbola fitted to these data returned an R2 value of 0.99 and a P50 of 0.3 mmHg, a value 100-fold less than that obtained in type-1 cells under otherwise identical recording conditions. Data from type-1 cells (from Fig. 2D) are reproduced in Fig. 3B (in grey) to facilitate comparison with that obtained from SCG neurons.
The significance of these observations is expounded upon further in the Discussion. Suffice to say that our data on type-1 cells are dramatically at variance with conventional wisdom regarding the normal oxygen requirements of mitochondrial respiration but confirm the earlier work of Duchen & Biscoe (1992a) on rabbit type-1 cells. We therefore sought to investigate this phenomenon further.
Mitochondrial membrane potential
Mitochondrial membrane potential was measured using the dye Rh123 in de-quench mode (see Methods). Recordings of ψm were performed under control conditions and in Ca2+-free Tyrode to prevent voltage-gated Ca2+ influx during exposure to either hypoxia or FCCP (Buckler & Vaughan Jones, 1998) as Ca2+ influx has been shown to depolarise type-1 cell mitochondria (Duchen & Biscoe, 1992b). FCCP at 1 μm was applied briefly (for approx. 20 s) both before and after testing the effects of hypoxia upon ψm. Application of FCCP caused a rapid increase in Rh123 fluorescence due to mitochondrial depolarisation and redistribution of Rh123 from mitochondria to cytosol. This response was assumed to represent the maximum (100%) Rh123 fluorescence attainable with complete mitochondrial depolarisation. Measurement of Rh123 fluorescence in the presence of 150 mmHg O2 was taken as the minimum level of Rh123 fluorescence (0%). Responses to hypoxia were then quantified on this relative 0–100% scale (see Fig. 4B). Hypoxia and anoxia caused a significant increase in Rh123 fluorescence both in the presence (P < 0.001, n= 11 RM-ANOVA) and in the absence of external calcium (P < 0.001, n= 11 RM-ANOVA on ranks). This effect was significant (P < 0.05 Holm–Sidak post hoc test) for all levels of hypoxia in the presence of extracellular calcium and 2.5% oxygen and below in the absence of extracellular calcium (Dunnett's post hoc test; note that although data obtained at 5% oxygen failed to reach statistical significance in this test, an increase in Rh123 fluorescence was seen in 10 of the 11 recordings). The increase in fluorescence at each level of hypoxia is plotted as a function of 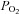 in Fig. 4C and D. Fitting a rectangular hyperbola to these data returned an R2 value of 0.96 and a P50 of 3.1 mmHg in the presence of external calcium and an R2 value of 0.85 and a P50 of 3.3 mmHg in the absence of external calcium. Similar results were also obtained in the presence of extracellular calcium and the calcium channel antagonist Ni2+, and in a subset of data in normal Ca Tyrode representing recordings from single cells only (see supplemental material).
in Fig. 4C and D. Fitting a rectangular hyperbola to these data returned an R2 value of 0.96 and a P50 of 3.1 mmHg in the presence of external calcium and an R2 value of 0.85 and a P50 of 3.3 mmHg in the absence of external calcium. Similar results were also obtained in the presence of extracellular calcium and the calcium channel antagonist Ni2+, and in a subset of data in normal Ca Tyrode representing recordings from single cells only (see supplemental material).
It is notable that the degree of depolarisation (approx. 22%) in complete anoxia was considerably less than that seen with FCCP despite the fact that anoxia should halt electron transport. In part this may simply be due to the fact that ψm did not appear to reach a steady state during the brief exposures to anoxia and so our measurement will underestimate the full effect of anoxia. The failure to rapidly and fully depolarise under anoxic conditions is probably due to reversal of the ATP synthase such that ATP is consumed in order to pump protons out of the inner matrix space and help maintain ψm.
Effects of hypoxia on electron transport
The mitochondrial depolarisation observed in the above experiments could either represent an unusually high degree of oxygen sensitivity in the electron transport chain, or it could be a secondary consequence of increased energy demand leading to increased ATP synthase activity and proton influx into the inner mitochondrial matrix. To evaluate the effects of hypoxia on mitochondrial electron transport alone we sought to isolate ψm and electron transport from changes in energy demand and the activity of ATP synthase by treating cells with the ATP synthase inhibitor oligomycin. We also increased passive proton leak by applying a low level of uncoupler. Under these conditions ψm reaches a steady state when the rate of proton translocation by complexes I, II and IV equals passive proton back flux via the uncoupler. Passive proton leak is dependent upon both ψm and the leak conductance to protons (gH+). If we assume that gH+ is relatively constant then steady-state ψm will be linearly related to proton extrusion. Thus, changes in steady-state ψm will reflect changes in electron transport.
Figure 5A shows the protocol employed in these experiments. Cells, bathed in Ca2+-free Tyrode throughout, were first exposed to 1 μm FCCP for 20 s to calibrate the Rh123. Cells were then exposed to 75 nm FCCP and 2.5 μg ml−1 oligomycin. Oxygen levels in this solution were then reduced, as indicated, in the range 5% to anoxia. Finally, the FCCP/oligomycin solution was removed and a second calibration in 1 μm FCCP performed. Both hypoxia and anoxia caused a brisk and reversible depolarisation in ψm that reached a steady state within 20 s (P < 0.001, n= 12, RM-ANOVA). This effect was significant (P < 0.001) at all levels of hypoxia tested. These data are plotted as a percentage of maximal depolarisation against 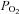 in Fig. 5C (taking fluorescence recorded in the presence of 75 nm FCCP and oligomycin in air equilibrated solutions as the baseline, 0%). Fitting a rectangular hyperbola to these data returned an R2 value of 0.97 and a P50 of 5.4 mmHg.
in Fig. 5C (taking fluorescence recorded in the presence of 75 nm FCCP and oligomycin in air equilibrated solutions as the baseline, 0%). Fitting a rectangular hyperbola to these data returned an R2 value of 0.97 and a P50 of 5.4 mmHg.
The above data indicate that electron transport in type-1 cells would appear to have an unusually high degree of oxygen sensitivity. We therefore repeated the experiment with SCG neurons. The protocol used was identical to that above save for the use of fewer levels of hypoxia (1 and 0.5%). As can be seen from Fig. 5B anoxia caused a rapid depolarisation of ψm to a level comparable to that observed with 1 μm FCCP but little effect was discernible for either of the two hypoxic solutions. The extent of the increase in Rh123 fluorescence as a percentage of that caused by 1 μm FCCP was calculated (as above) and is plotted as a function of 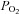 in Fig. 5D. RM-ANOVA showed an effect of oxygen on Rh123 fluorescence (P < 0.001, n= 7) but post hoc testing (Holm–Sidak) revealed that this was only significant for anoxia. In view of this we did not attempt to fit a rectangular hyperbola to the data (symbols in Fig. 5D are joined by straight lines).
in Fig. 5D. RM-ANOVA showed an effect of oxygen on Rh123 fluorescence (P < 0.001, n= 7) but post hoc testing (Holm–Sidak) revealed that this was only significant for anoxia. In view of this we did not attempt to fit a rectangular hyperbola to the data (symbols in Fig. 5D are joined by straight lines).
Oxygen sensitivity of cytochrome oxidase
In their classic paper, Mills & Jöbsis (1972) hypothesised that carotid body cells might contain a cytochrome oxidase with a low oxygen affinity. Such a situation could provide an explanation for the data presented above. We therefore sought to try to assay the oxygen sensitivity of cytochrome oxidase in isolation from the rest of the electron transport chain. To do this we inhibited electron transport complexes I and III and the ATP synthase with a combination of rotenone (1 μm), myxothiazol (0.5 μm), antimycin A (1 μm) and oligomycin (2.5 μg ml−1). This cocktail produced a full depolarisation of mitochondria in both Type-1 cells and SCG neurons, i.e. application of 1 μm FCCP had no further effect upon Rh123 fluorescence (see Fig. 6A and C). Subsequent addition of 5 mm ascorbate plus 40 μg ml−1 TMPD, an artificial electron donor capable of reducing cytochrome c, resulted in a rapid repolarisation of ψm (Fig. 6A and C). Under these conditions ψm is assumed to be maintained by proton pumping through complex IV alone, with electrons being passed exclusively from TMPD, via cytochrome c, through complex IV to oxygen. The effects of reduced oxygen levels on ψm were then tested under these conditions. As can be seen from Fig. 6A, both hypoxia and anoxia caused a depolarisation of ψm in type-1 cells. These effects were quantified as percentage of maximal depolarisation (in 1 μm FCCP) with the baseline (0%) measured in the presence of all inhibitors plus TMPD and ascorbate in air-equilibrated solutions. These data are plotted as a function of 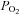 in Fig. 6B for type-1 cells and Fig. 6D for SCG neurons. Reduction of oxygen level caused a significant depolarisation of ψm in type-1 cells (P < 0.001, n= 14, RM-ANOVA). Post hoc testing against control (Holm–Sidak method) revealed a significant depolarisation at every level of hypoxia tested (P < 0.001). Fitting a rectangular hyperbola to these data returned an R2 value of 0.95 and a P50 of 2.6 mmHg. In SCG neurons reducing oxygen also depolarised ψm (P < 0.001, RM-ANOVA, n= 5). Post hoc testing (Holm–Sidak), however, revealed that only anoxia had a significant effect in SCG neurons. In view of this we did not attempt to fit a rectangular hyperbola to data from SCG neurons; the data points shown in Fig. 6D are joined by straight lines only.
in Fig. 6B for type-1 cells and Fig. 6D for SCG neurons. Reduction of oxygen level caused a significant depolarisation of ψm in type-1 cells (P < 0.001, n= 14, RM-ANOVA). Post hoc testing against control (Holm–Sidak method) revealed a significant depolarisation at every level of hypoxia tested (P < 0.001). Fitting a rectangular hyperbola to these data returned an R2 value of 0.95 and a P50 of 2.6 mmHg. In SCG neurons reducing oxygen also depolarised ψm (P < 0.001, RM-ANOVA, n= 5). Post hoc testing (Holm–Sidak), however, revealed that only anoxia had a significant effect in SCG neurons. In view of this we did not attempt to fit a rectangular hyperbola to data from SCG neurons; the data points shown in Fig. 6D are joined by straight lines only.
O2 sensitivity of mitochondrial function vs. chemoreception
One of the objectives of this study was to establish whether mitochondrial respiration in type-1 cells had the requisite oxygen sensitivity to allow it to play a role in acute oxygen sensing. Figure 7 shows measurements of the oxygen sensitivity of calcium signalling in type-1 cells. In this, as in previous studies (Buckler & Vaughan Jones, 1994), hypoxia produced a rapid increase in [Ca2+]i in type-1 cells (P < 0.001, n= 11, RM-ANOVA on ranks). Fitting a rectangular hyperbola to these data returned a value for R2 of 0.52 and P50 of 12.5 mm Hg. An equivalent degree of oxygen sensitivity has also been reported for background potassium channel activity in type-1 cells (Buckler, 1997). The [Ca2+]i data were normalised, relative to baseline [Ca2+]i in air/CO2-equilibrated saline (0%) and maximum [Ca2+]i in anoxia (100%), and are re-plotted in Fig 7C. The oxygen sensitivity of type-1 cell NADH, ψm, electron transport and cytochrome oxidase activity are also plotted in Fig. 7C, for comparison.
Figure 7. Oxygen sensitivity of calcium signalling and mitochondrial function.
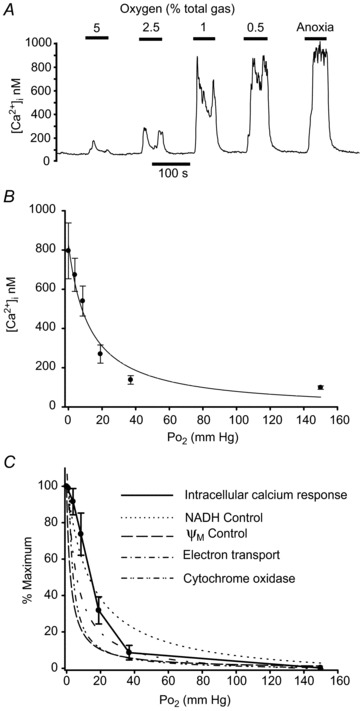
A, original recording of intracellular Ca2+ concentration measured using Indo-1 in a small cluster of type-1 cells bathed in normal Tyrode. Cells are exposed to graded hypoxic stimuli from 5% oxygen down to anoxia. B, summary data (mean ± SEM) from 11 recordings as in A. Curve is a rectangular hyperbola fitted to these data (see text for details). C, effects of hypoxia on a relative scale of 0–100% where 0%= baseline [Ca2+]i recorded in 20% O2 and 100%= maximal [Ca2+]i recorded in anoxia. These data (mean ± SEM) are joined by straight lines only. Curves represent oxygen sensitivity of various measures of mitochondrial function in the form of hyperbolic functions fitted to the original data, including: NADH from Fig 2C, ψm from Fig 4C (both in normal Ca2+ Tyrode), electron transport from Fig. 5C and cytochrome oxidase activity from Fig. 6C. Hyperbolas have been rescaled, where necessary, to a 0 (baseline in 20% O2) to 100% (maximum in anoxia) scale.
Discussion
Our data suggest that modest hypoxia inhibits electron transport in type-1 cells and confirm earlier observations regarding the unusual oxygen sensitivity of mitochondrial function in these cells (Duchen & Biscoe, 1992a,b). Before discussing the significance of these findings we first consider whether changes in ψm or NADH are open to alternative interpretation.
Under normal conditions hypoxia and metabolic inhibitors cause voltage-gated Ca2+ influx in type-1 cells (Buckler & Vaughan Jones, 1994; Wyatt & Buckler, 2004), which could result in Ca2+ uptake by mitochondria (Friel & Tsien, 1994; Werth & Thayer, 1994; Herrington et al. 1996) depolarising ψm and activating Krebs cycle dehydrogenases generating NADH (McCormack & Denton, 1989). Measurements of NADH in Ca-free media and ψm in both Ca-free media and in the presence of the Ca-channel antagonist Ni2+, demonstrate that the effects of hypoxia on NADH and ψm are not a consequence of cytosolic Ca2+ signalling.
Changes in ψm could occur if mitochondria were tightly coupled with low energy demand (state 4) such that ψm reached the limit imposed by the free energy available from NADH oxidation (which is dependent upon oxygen concentration). Alternatively, an increase in energy demand could increase ATP-synthase activity and proton influx (i.e. a switch to state 3 respiration). To address these issues we performed experiments with ATP synthase blocked and mitochondria partially depolarised by a low concentration of uncoupler. Again, however, relatively mild hypoxia decreased ψm in type-1 cells. We assume that with ATP synthase blocked ψm reaches a steady state when proton efflux via complexes I, III and IV matches proton influx via passive leak (presumed to be predominantly via FCCP). Since passive proton influx must decrease as ψm depolarises, a decline in steady-state ψm in hypoxia indicates a corresponding decrease in proton efflux by the electron transport chain. These data therefore confirm that hypoxia can limit electron transport even in partially depolarised mitochondria and independently of ATP synthesis.
Oxygen sensitivity of mitochondrial function in type-1 cells vs. other tissues
Studies of mitochondrial function in other tissues using respirometry indicate a very high affinity for oxygen typically in the range 0.08–0.75 mm Hg (Gnaiger et al. 1998; Scandurra & Gnaiger, 2010). Measurements of cytochrome c redox state, however, show a much greater sensitivity to oxygen with cytochrome c becoming reduced at higher levels of 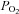 than those required to inhibit oxygen consumption (Wilson et al. 1979a). Indeed this rise in electron transport chain (ETC) intermediates/cytochrome c is thought to partially offset the effects of a fall in oxygen so as to help maintain electron transport rate under conditions of moderate hypoxia (Wilson et al. 1979b). Defining what constitutes the oxygen sensitivity of mitochondrial function is therefore not entirely straightforward as changes in the levels of intermediates may be more sensitive to
than those required to inhibit oxygen consumption (Wilson et al. 1979a). Indeed this rise in electron transport chain (ETC) intermediates/cytochrome c is thought to partially offset the effects of a fall in oxygen so as to help maintain electron transport rate under conditions of moderate hypoxia (Wilson et al. 1979b). Defining what constitutes the oxygen sensitivity of mitochondrial function is therefore not entirely straightforward as changes in the levels of intermediates may be more sensitive to 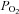 than electron flux. This effect can be seen in our data when comparing the oxygen sensitivity of [NADH] with measurements that reflect electron transport rate (ψm), the former appearing to be more oxygen sensitive than the latter. The most striking feature of our data, however, is that the P50 values for these effects are at least an order of magnitude greater than those reported in other tissues. This leads us to consider whether there could be a major systematic error in our measurements.
than electron flux. This effect can be seen in our data when comparing the oxygen sensitivity of [NADH] with measurements that reflect electron transport rate (ψm), the former appearing to be more oxygen sensitive than the latter. The most striking feature of our data, however, is that the P50 values for these effects are at least an order of magnitude greater than those reported in other tissues. This leads us to consider whether there could be a major systematic error in our measurements.
One potential source of error is the accuracy with which tissue oxygen can be controlled and measured. We have used single cells and small clusters of cells (2–8 cells) dispersed at very low density (<100 cells mm−2). Rat type-1 cells are small (diameter approx. 10–11 μm) so we assume there is no significant diffusion gradient within individual cells or small clusters. Our perfusion system has rapid solution exchange (half time 1–2 s) such that oxygen levels stabilise within 30 s at 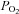 values very close to those expected (see Methods). Significant overestimation of the oxygen levels to which cells were exposed is therefore highly unlikely and measurements of mitochondrial function are made under steady-state conditions with respect to
values very close to those expected (see Methods). Significant overestimation of the oxygen levels to which cells were exposed is therefore highly unlikely and measurements of mitochondrial function are made under steady-state conditions with respect to 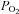 .
.
Another potential source of error is in the calibration of fluorescent signals. NADH autofluorescence bleaches slightly during experiments; as a consequence of this, and applying hypoxic solutions in a decrementing sequence (from high 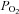 to low
to low 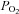 ), there is likely to be systematic overestimation of the effects of higher vs. lower
), there is likely to be systematic overestimation of the effects of higher vs. lower 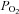 and consequently P50 values will probably also be overestimated. We have calibrated Rh123 fluorescence using a 0–100% scale. A linear relationship between Rh123 fluorescence and ψm has been reported for mitochondria studied in a cuvette (Emaus et al. 1986) but it is by no means certain that the same will apply in a small cell. Indeed if Rh123 distributes between mitochondria and cytoplasm according to the Nernst equation one might expect the relationship between fluorescence and ψm to be non-linear particularly at very negative potentials such that changes in Rh123 fluorescence could underestimate the true change in ψm. It should also be noted that we have implicitly assumed a uniform affinity for O2 (P50) by fitting data to a simple hyperbolic function. This does not allow for the possibility of low- and high-affinity forms of cytochrome oxidase as suggested by Mills & Jöbsis (1972). Our estimation of the oxygen sensitivity (P50) of NADH, ψm, electron transport and cytochrome oxidase can therefore only be considered approximate. These errors are, however, unlikely to account for differences in P50 of 1–2 orders of magnitude. Moreover, none of these errors could account for the robust changes in mitochondrial function we have observed at modest levels of hypoxia. The best test for methodological errors, however, lies in the use of another cell type. Measurements of the effects of hypoxia on NADH in SCG neurons returned a value for P50 that was two orders of magnitude lower than that seen in the carotid body. Similar measurements of the effects of hypoxia on electron transport and cytochrome oxidase activity in SCG neurons were unable to resolve a value for P50 as we were unable to generate
and consequently P50 values will probably also be overestimated. We have calibrated Rh123 fluorescence using a 0–100% scale. A linear relationship between Rh123 fluorescence and ψm has been reported for mitochondria studied in a cuvette (Emaus et al. 1986) but it is by no means certain that the same will apply in a small cell. Indeed if Rh123 distributes between mitochondria and cytoplasm according to the Nernst equation one might expect the relationship between fluorescence and ψm to be non-linear particularly at very negative potentials such that changes in Rh123 fluorescence could underestimate the true change in ψm. It should also be noted that we have implicitly assumed a uniform affinity for O2 (P50) by fitting data to a simple hyperbolic function. This does not allow for the possibility of low- and high-affinity forms of cytochrome oxidase as suggested by Mills & Jöbsis (1972). Our estimation of the oxygen sensitivity (P50) of NADH, ψm, electron transport and cytochrome oxidase can therefore only be considered approximate. These errors are, however, unlikely to account for differences in P50 of 1–2 orders of magnitude. Moreover, none of these errors could account for the robust changes in mitochondrial function we have observed at modest levels of hypoxia. The best test for methodological errors, however, lies in the use of another cell type. Measurements of the effects of hypoxia on NADH in SCG neurons returned a value for P50 that was two orders of magnitude lower than that seen in the carotid body. Similar measurements of the effects of hypoxia on electron transport and cytochrome oxidase activity in SCG neurons were unable to resolve a value for P50 as we were unable to generate 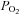 levels low enough to inhibit either electron transport or cytochrome oxidase activity (save for complete anoxia, which is generated with the aid of an oxygen scavenging agent). Despite this technical failure it is evident that whatever the true oxygen sensitivity of electron transport and cytochrome oxidase activity in the SCG neuron, P50 values must be well below those seen in type-1 cells. In conclusion, we cannot account for our data in any way other than to propose that the apparent oxygen affinity for mitochondrial respiration in type-1 cells is lower than that seen in other cell types by up to two orders of magnitude.
levels low enough to inhibit either electron transport or cytochrome oxidase activity (save for complete anoxia, which is generated with the aid of an oxygen scavenging agent). Despite this technical failure it is evident that whatever the true oxygen sensitivity of electron transport and cytochrome oxidase activity in the SCG neuron, P50 values must be well below those seen in type-1 cells. In conclusion, we cannot account for our data in any way other than to propose that the apparent oxygen affinity for mitochondrial respiration in type-1 cells is lower than that seen in other cell types by up to two orders of magnitude.
Mechanisms for reduced oxygen affinity in cytochrome oxidase
To evaluate the oxygen sensitivity of cytochrome oxidase we employed artificial electron donors (TMPD and ascorbate) to maintain a supply of reduced cytochrome c and measured the effects of oxidase-mediated proton efflux upon ψm under conditions in which complexes I, III and IV are blocked. Under these conditions we again saw that hypoxia depolarised ψm, indicating inhibition of cytochrome oxidase activity (Fig. 6A and C).
Why cytochrome oxidase in type-1 cells should appear much more sensitive to hypoxia than in other cells is unknown. Since there is no precedent for this we can only offer conjecture. The suggestion that there is an alternative low-affinity form of cytochrome a3 (Mills & Jöbsis, 1972) seems unlikely as there is only one, mitochondrial, gene for subunit I of cytochrome oxidase (although there must remain the possibility of some form of post-translational modification). It is, however, the apparent Km that defines oxygen sensitivity of electron transport and this can be influenced by other factors. The high apparent affinity of cytochrome oxidase for oxygen is dependent upon kinetic trapping by electron transfer from the low spin haem to the iron–copper binuclear centre where oxygen is bound (the initial binding of oxygen being weak). As a consequence, the operational Km is dependent upon the rate of electron transfer and kinetic trapping (Verkhovsky et al. 1996). The oxygen sensitivity of cytochrome oxidase could therefore be influenced by other components of complex IV.
The apparent affinity of cytochrome oxidase could also be lowered through the presence of an endogenous competitive inhibitor. Nitric oxide can inhibit mitochondrial respiration with high affinity, i.e. in the nanomolar range (Brown & Cooper, 1994; Cooper & Giulivi, 2007). Knockout of neuronal nitric oxide synthase (nNOS), however, augments chemoreceptor responses to hypoxia (Kline et al. 2002). Knockout of enothelial NOS (eNOS) on the other hand blunts chemoreceptor oxygen sensitivity (Kline et al. 2000), yet oxygen sensitivity remains in isolated type-1 cells from control animals (so oxygen sensitivity is largely endothelium independent). CO can also inhibit cytochrome oxidase but its affinity is low (Cooper & Brown, 2008). Indeed, very high levels (>300 mmHg, approx. 280 μm) of exogenous CO are required to excite the carotid body via inhibition of cytochrome oxidase, an effect that is reversed by photo-dissociation (Lahiri et al. 1993a; Wilson et al. 1994). It seems unlikely that endogenous CO could be maintained at such levels moreover the response to hypoxia is not light sensitive (Lahiri et al. 1993a; Wilson et al. 1994).
Finally, we consider the possibility that hypoxia inhibits cytochrome oxidase via another oxygen sensing pathway either as an epiphenomenon or as an integral part of the O2 signal transduction pathway. It has recently been suggested that endogenous H2S production mediates acute oxygen sensing in blood vessels (Olson et al. 2006; Olson & Whitfield, 2010), fish gill chemoreceptors (Olson et al. 2008) and the mammalian carotid body (Peng et al. 2010; Olson, 2011) with H2S levels increasing during hypoxia (Olson et al. 2006). Knockout of one of the enzymes that can generate H2S appears to blunt chemoreceptor responses to hypoxia (Peng et al. 2010; Olson, 2011). H2S is a non-competitive inhibitor of cytochrome oxidase (Cooper & Brown, 2008). Micromolar levels of exogenous H2S inhibit mitochondrial metabolism and excite type-1 cells (Buckler, 2012). There is, however, uncertainty about whether endogenous H2S could reach such levels in isolated cells in vitro (Buckler, 2012).
There are therefore potential explanations for the unusual oxygen sensitivity of mitochondrial respiration in this tissue although there are uncertainties about the feasibility of some of them. Clearly more research is needed to identify the causes of this phenomenon.
Role of mitochondria in oxygen sensing
It is clear from the above discussion that mitochondrial function in type-1 cells is extraordinarily oxygen sensitive, particularly with respect to changes in NADH levels. This degree of oxygen sensitivity is not dissimilar to that of calcium signalling in type-1 cells (Fig. 7C). Indeed under some conditions the oxygen sensing range may even extend well into hyperoxia given that we were able to demonstrate changes in NADH between 150 and 75 mm Hg whereas normal carotid body tissue 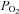 is unlikely to be greater than 60 mmHg (Lahiri et al. 1993b). While we cannot completely exclude the possibility that this is an epiphenomenon driven by some other unknown O2 sensor, it is difficult to reconcile our data with any single effect other than an inhibition of electron transport. Since all other interventions which inhibit electron transport also cause excitation in the carotid body, the most parsimonious explanation of all these data is that even if complex IV is not the primary site of oxygen sensing, modulation of electron transport is probably an integral part of the hypoxia transduction cascade. We have not examined the link between hypoxic inhibition of electron transport and chemoreceptor excitation in this study, but previous studies using a variety of chemical inhibitors of mitochondrial function indicate that inhibition of ATP synthesis appears to be the key event (Wyatt & Buckler, 2004). Changes in cytosolic nucleotide levels may then be detected by TWIK-related acid-sensitive potassium channels (TASK) directly and/or via an AMP kinase (Varas et al. 2007; Wyatt et al. 2007).
is unlikely to be greater than 60 mmHg (Lahiri et al. 1993b). While we cannot completely exclude the possibility that this is an epiphenomenon driven by some other unknown O2 sensor, it is difficult to reconcile our data with any single effect other than an inhibition of electron transport. Since all other interventions which inhibit electron transport also cause excitation in the carotid body, the most parsimonious explanation of all these data is that even if complex IV is not the primary site of oxygen sensing, modulation of electron transport is probably an integral part of the hypoxia transduction cascade. We have not examined the link between hypoxic inhibition of electron transport and chemoreceptor excitation in this study, but previous studies using a variety of chemical inhibitors of mitochondrial function indicate that inhibition of ATP synthesis appears to be the key event (Wyatt & Buckler, 2004). Changes in cytosolic nucleotide levels may then be detected by TWIK-related acid-sensitive potassium channels (TASK) directly and/or via an AMP kinase (Varas et al. 2007; Wyatt et al. 2007).
Whilst mitochondrial/metabolic signalling pathways could not account for all forms of acute oxygen sensing, e.g. that reported for ion channels in excised patches (Ganfornina & Lopez Barneo, 1991; Riesco Fagundo et al. 2001), it is of interest to note that mitochondria have been implicated in acute oxygen sensing in several other tissues, including adrenal chromaffin cells and pulmonary arterioles (Mojet et al. 1997; Buttigieg et al. 2008; Ward, 2008; Sylvester et al. 2012). The pathways involved in these tissues are various and do not necessarily depend upon metabolic signalling per se; changes in mitochondrial superoxide production have also been suggested (Archer et al. 1993; Waypa et al. 2001). It will nonetheless be interesting to see if the oxygen sensitivity of mitochondrial function in these other tissues is similarly enhanced.
Glossary
- ψm
mitochondrial membrane potential
- ETC
electron transport chain
- FCCP
carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone
- PMT
photomultiplier tube
- Rh123
rhodamine 123
- SCG
superior cervical ganglion
- TASK
TWIK-related acid-sensitive potassium channel
- TMPD
tetramethyl-p-phenylenediamine
Additional information
Competing interests
None.
Author contributions
All experiments were performed in the Department of Physiology Anatomy & Genetics, University of Oxford. K.J.B. conceived and designed the experiments, collected the data, was primarily responsible for data analysis and interpretation, and drafted the manuscript. P.T. assisted in data collection and the critical review and revision of the manuscript for important intellectual content.
Funding
This research was supported by the Medical Research Council Project UK (grant G101134).
Supplementary material
Supporting Information
References
- Anichkov S, Belen’kii M. Pharmacology of the Carotid Body Chemoreceptors. Oxford: Pergamon Press; 1963. [Google Scholar]
- Archer SL, Huang J, Henry T, Peterson D, Weir EK. A redox-based O2 sensor in rat pulmonary vasculature. Circ Res. 1993;73:1100–1112. doi: 10.1161/01.res.73.6.1100. [DOI] [PubMed] [Google Scholar]
- Brown GC, Cooper CE. Nanomolar concentrations of nitric oxide reversibly inhibit synaptosomal respiration by competing with oxygen at cytochrome oxidase. FEBS Lett. 1994;356:295–298. doi: 10.1016/0014-5793(94)01290-3. [DOI] [PubMed] [Google Scholar]
- Buckler KJ. A novel oxygen-sensitive potassium current in rat carotid body type I cells. J Physiol. 1997;498:649–662. doi: 10.1113/jphysiol.1997.sp021890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckler KJ. Effects of exogenous hydrogen sulphide on calcium signalling, background (TASK) K channel activity and mitochondrial function in chemoreceptor cells. Pflugers Arch. 2012;463:743–754. doi: 10.1007/s00424-012-1089-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckler KJ, Vaughan-Jones RD. Effects of mitochondrial uncouplers on intracellular calcium, pH and membrane potential in rat carotid body type I cells. J Physiol. 1998;513:819–833. doi: 10.1111/j.1469-7793.1998.819ba.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckler KJ, Vaughan Jones RD. Effects of acidic stimuli on intracellular calcium in isolated type I cells of the neonatal rat carotid body. Pflugers Arch. 1993;425:22–27. doi: 10.1007/BF00374499. [DOI] [PubMed] [Google Scholar]
- Buckler KJ, Vaughan Jones RD. Effects of hypoxia on membrane potential and intracellular calcium in rat neonatal carotid body type I cells. J Physiol. 1994;476:423–428. doi: 10.1113/jphysiol.1994.sp020143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckler KJ, Vaughan Jones RD. Effects of mitochondrial uncouplers on intracellular calcium, pH and membrane potential in rat carotid body type I cells. J Physiol. 1998;513:819–833. doi: 10.1111/j.1469-7793.1998.819ba.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttigieg J, Brown ST, Lowe M, Zhang M, Nurse CA. Functional mitochondria are required for O2 but not CO2 sensing in immortalized adrenomedullary chromaffin cells. Am J Physiol Cell Physiol. 2008;294:C945–956. doi: 10.1152/ajpcell.00495.2007. [DOI] [PubMed] [Google Scholar]
- Chen LB. Mitochondrial membrane potential in living cells. Annu Rev Cell Biol. 1988;4:155–181. doi: 10.1146/annurev.cb.04.110188.001103. [DOI] [PubMed] [Google Scholar]
- Cooper CE, Brown GC. The inhibition of mitochondrial cytochrome oxidase by the gases carbon monoxide, nitric oxide, hydrogen cyanide and hydrogen sulfide: chemical mechanism and physiological significance. J Bioenerg Biomembr. 2008;40:533–539. doi: 10.1007/s10863-008-9166-6. [DOI] [PubMed] [Google Scholar]
- Cooper CE, Giulivi C. Nitric oxide regulation of mitochondrial oxygen consumption II: molecular mechanism and tissue physiology. Am J Physiol Cell Physiol. 2007;292:C1993–2003. doi: 10.1152/ajpcell.00310.2006. [DOI] [PubMed] [Google Scholar]
- Duchen MR, Biscoe TJ. Mitochondrial function in type I cells isolated from rabbit arterial chemoreceptors. J Physiol. 1992a;450:13–31. doi: 10.1113/jphysiol.1992.sp019114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen MR, Biscoe TJ. Relative mitochondrial membrane potential and [Ca2+]i in type I cells isolated from the rabbit carotid body. J Physiol. 1992b;450:33–61. doi: 10.1113/jphysiol.1992.sp019115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emaus RK, Grunwald R, Lemasters JJ. Rhodamine 123 as a probe of transmembrane potential in isolated rat-liver mitochondria: spectral and metabolic properties. Biochim Biophys Acta. 1986;850:436–448. doi: 10.1016/0005-2728(86)90112-x. [DOI] [PubMed] [Google Scholar]
- Friel DD, Tsien RW. An FCCP-sensitive Ca2+ store in bullfrog sympathetic neurons and its participation in stimulus-evoked changes in [Ca2+]i. J Neurosci. 1994;14:4007–4024. doi: 10.1523/JNEUROSCI.14-07-04007.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganfornina MD, Lopez Barneo J. Single K+ channels in membrane patches of arterial chemoreceptor cells are modulated by O2 tension. Proc Natl Acad Sci U S A. 1991;88:2927–2930. doi: 10.1073/pnas.88.7.2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnaiger E, Lassnig B, Kuznetsov AV, Margreiter R. Mitochondrial respiration in the low oxygen environment of the cell. Effect of ADP on oxygen kinetics. Biochim Biophys Acta. 1998;1365:249–254. doi: 10.1016/s0005-2728(98)00076-0. [DOI] [PubMed] [Google Scholar]
- Herrington J, Park YB, Babcock DF, Hille B. Dominant role of mitochondria in clearance of large Ca2+ loads from rat adrenal chromaffin cells. Neuron. 1996;16:219–228. doi: 10.1016/s0896-6273(00)80038-0. [DOI] [PubMed] [Google Scholar]
- Heymans C, Bouckaert JJ, Dautrebande L. Sinus carotidien et reflexes respiratoires: sensibilite des sinus carotidiens aux substances chimiques. Action stimulante respiratoire reflexe du sulfure de sodium, du cyanure de potassium, de la nicotine et de la lobeline. Arch Int Pharmacodynam Ther. 1931;40:54–91. [Google Scholar]
- Kaelin WG, Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008;30:393–402. doi: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- Kline DD, Overholt JL, Prabhakar NR. Mutant mice deficient in NOS-1 exhibit attenuated long-term facilitation and short-term potentiation in breathing. J Physiol. 2002;539:309–315. doi: 10.1113/jphysiol.2001.014571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline DD, Yang T, Premkumar DR, Thomas AJ, Prabhakar NR. Blunted respiratory responses to hypoxia in mutant mice deficient in nitric oxide synthase-3. J Appl Physiol. 2000;88:1496–1508. doi: 10.1152/jappl.2000.88.4.1496. [DOI] [PubMed] [Google Scholar]
- Lahiri S, Iturriaga R, Mokashi A, Ray DK, Chugh D. CO reveals dual mechanisms of O2 chemoreception in the cat carotid body. Respir Physiol. 1993a;94:227–240. doi: 10.1016/0034-5687(93)90050-k. [DOI] [PubMed] [Google Scholar]
- Lahiri S, Rumsey WL, Wilson DF, Iturriaga R. Contribution of in vivo microvascular PO2 in the cat carotid body chemotransduction. J Appl Physiol. 1993b;75:1035–1043. doi: 10.1152/jappl.1993.75.3.1035. [DOI] [PubMed] [Google Scholar]
- McCormack JG, Denton RM. The role of Ca2+ ions in the regulation of intramitochondrial metabolism and energy production in rat heart. Mol Cell Biochem. 1989;89:121–125. [PubMed] [Google Scholar]
- Mills E, Jöbsis FF. Simultaneous measurement of cytochrome a3 reduction and chemoreceptor afferent activity in the carotid body. Nature. 1970;225:1147–1149. doi: 10.1038/2251147a0. [DOI] [PubMed] [Google Scholar]
- Mills E, Jöbsis FF. Mitochondrial respiratory chain of carotid body and chemoreceptor response to changes in oxygen tension. J Neurophysiol. 1972;35:405–428. doi: 10.1152/jn.1972.35.4.405. [DOI] [PubMed] [Google Scholar]
- Mojet MH, Mills E, Duchen MR. Hypoxia-induced catecholamine secretion in isolated newborn rat adrenal chromaffin cells is mimicked by inhibition of mitochondrial respiration. J Physiol. 1997;504:175–189. doi: 10.1111/j.1469-7793.1997.175bf.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan E, Lahiri S. Separation of carotid body chemoreceptor responses to O2 and CO2 by oligomycin and by antimycin A. Am J Physiol. 1982;242:C200–206. doi: 10.1152/ajpcell.1982.242.3.C200. [DOI] [PubMed] [Google Scholar]
- Mulligan E, Lahiri S, Storey BT. Carotid body O2 chemoreception and mitochondrial oxidative phosphorylation. J Appl Physiol. 1981;51:438–446. doi: 10.1152/jappl.1981.51.2.438. [DOI] [PubMed] [Google Scholar]
- Nair PK, Buerk DG, Whalen WJ. Cat carotid body oxygen metabolism and chemoreception described by a two-cytochrome model. Am J Physiol. 1986;250:H202–207. doi: 10.1152/ajpheart.1986.250.2.H202. [DOI] [PubMed] [Google Scholar]
- Obeso A, Almaraz L, Gonzalez C. Effects of cyanide and uncouplers on chemoreceptor activity and ATP content of the cat carotid body. Brain Res. 1989;481:250–257. doi: 10.1016/0006-8993(89)90801-9. [DOI] [PubMed] [Google Scholar]
- Olson KR. Hydrogen sulfide is an oxygen sensor in the carotid body. Respir Physiol Neurobiol. 2011;179:103–110. doi: 10.1016/j.resp.2011.09.010. [DOI] [PubMed] [Google Scholar]
- Olson KR, Dombkowski RA, Russell MJ, Doellman MM, Head SK, Whitfield NL, Madden JA. Hydrogen sulfide as an oxygen sensor/transducer in vertebrate hypoxic vasoconstriction and hypoxic vasodilation. J Exp Biol. 2006;209:4011–4023. doi: 10.1242/jeb.02480. [DOI] [PubMed] [Google Scholar]
- Olson KR, Healy MJ, Qin Z, Skovgaard N, Vulesevic B, Duff DW, Whitfield NL, Yang G, Wang R, Perry SF. Hydrogen sulfide as an oxygen sensor in trout gill chemoreceptors. Am J Physiol Regul Integr Comp Physiol. 2008;295:R669–680. doi: 10.1152/ajpregu.00807.2007. [DOI] [PubMed] [Google Scholar]
- Olson KR, Whitfield NL. Hydrogen sulfide and oxygen sensing in the cardiovascular system. Antioxid Redox Signal. 2010;12:1219–1234. doi: 10.1089/ars.2009.2921. [DOI] [PubMed] [Google Scholar]
- Ortega Saenz P, Pardal R, Garcia Fernandez M, Lopez Barneo J. Rotenone selectively occludes sensitivity to hypoxia in rat carotid body glomus cells. J Physiol. 2003;548:789–800. doi: 10.1113/jphysiol.2003.039693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng YJ, Nanduri J, Raghuraman G, Souvannakitti D, Gadalla MM, Kumar GK, Snyder SH, Prabhakar NR. H2S mediates O2 sensing in the carotid body. Proc Natl Acad Sci U S A. 2010;107:10719–10724. doi: 10.1073/pnas.1005866107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riesco Fagundo AM, Pérez García MT, Gónzalez C, López López JR. O2 modulates large-conductance Ca2+-dependent K+ channels of rat chemoreceptor cells by a membrane-restricted and CO-sensitive mechanism. Circ Res. 2001;89:430–436. doi: 10.1161/hh1701.095632. [DOI] [PubMed] [Google Scholar]
- Scandurra FM, Gnaiger E. Cell respiration under hypoxia: facts and artefacts in mitochondrial oxygen kinetics. Adv Exp Med Biol. 2010;662:7–25. doi: 10.1007/978-1-4419-1241-1_2. [DOI] [PubMed] [Google Scholar]
- Schofield CJ, Ratcliffe PJ. Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol. 2004;5:343–354. doi: 10.1038/nrm1366. [DOI] [PubMed] [Google Scholar]
- Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell. 2012;148:399–408. doi: 10.1016/j.cell.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen TCR, Hauss WH. Influence of dinitrophenol, dinitroortocresol and paranitrophenol upon the carotid sinus chemoreceptors of the dog. Arch Int Pharmacodyn Ther. 1939;63:251–258. [Google Scholar]
- Sylvester JT, Shimoda LA, Aaronson PI, Ward JP. Hypoxic pulmonary vasoconstriction. Physiol Rev. 2012;92:367–520. doi: 10.1152/physrev.00041.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varas R, Wyatt CN, Buckler KJ. Modulation of TASK-like background potassium channels in rat arterial chemoreceptor cells by intracellular ATP and other nucleotides. J Physiol. 2007;583:521–536. doi: 10.1113/jphysiol.2007.135657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkhovsky MI, Morgan JE, Puustein A, Wikstrom M. Kinetic trapping of oxygen in cell respiration. Nature. 1996;380:268–270. doi: 10.1038/380268a0. [DOI] [PubMed] [Google Scholar]
- Ward JP. Oxygen sensors in context. Biochim Biophys Acta. 2008;1777:1–14. doi: 10.1016/j.bbabio.2007.10.010. [DOI] [PubMed] [Google Scholar]
- Waypa GB, Chandel NS, Schumacker PT. Model for hypoxic pulmonary vasoconstriction involving mitochondrial oxygen sensing. Circ Res. 2001;88:1259–1266. doi: 10.1161/hh1201.091960. [DOI] [PubMed] [Google Scholar]
- Weir EK, Lopez-Barneo J, Buckler KJ, Archer SL. Acute oxygen-sensing mechanisms. N Engl J Med. 2005;353:2042–2055. doi: 10.1056/NEJMra050002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werth JL, Thayer SA. Mitochondria buffer physiological calcium loads in cultured rat dorsal root ganglion neurons. J Neurosci. 1994;14:348–356. doi: 10.1523/JNEUROSCI.14-01-00348.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams BA, Buckler KJ. Biophysical properties and metabolic regulation of a TASK-like potassium channel in rat carotid body type 1 cells. Am J Physiol Lung Cell Mol Physiol. 2004;286:L221–230. doi: 10.1152/ajplung.00010.2003. [DOI] [PubMed] [Google Scholar]
- Wilson DF, Erecinska M, Drown C, Silver IA. The oxygen dependence of cellular energy metabolism. Arch Biochem Biophys. 1979a;195:485–493. doi: 10.1016/0003-9861(79)90375-8. [DOI] [PubMed] [Google Scholar]
- Wilson DF, Mokashi A, Chugh D, Vinogradov S, Osanai S, Lahiri S. The primary oxygen sensor of the cat carotid body is cytochrome a3 of the mitochondrial respiratory chain. FEBS Lett. 1994;351:370–374. doi: 10.1016/0014-5793(94)00887-6. [DOI] [PubMed] [Google Scholar]
- Wilson DF, Owen CS, Erecinska M. Quantitative dependence of mitochondrial oxidative phosphorylation on oxygen concentration: a mathematical model. Arch Biochem Biophys. 1979b;195:494–504. doi: 10.1016/0003-9861(79)90376-x. [DOI] [PubMed] [Google Scholar]
- Wyatt CN, Buckler KJ. The effect of mitochondrial inhibitors on membrane currents in isolated neonatal rat carotid body type I cells. J Physiol. 2004;556:175–191. doi: 10.1113/jphysiol.2003.058131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt CN, Mustard KJ, Pearson SA, Dallas ML, Atkinson L, Kumar P, Peers C, Hardie DG, Evans AM. AMP-activated protein kinase mediates carotid body excitation by hypoxia. J Biol Chem. 2007;282:8092–8098. doi: 10.1074/jbc.M608742200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.