

Abstract
The major tumour suppressor protein p53 plays an important role in maintaining mitochondrial content and function in skeletal muscle. p53 has been shown to reside in the mitochondria complexed with mitochondrial DNA (mtDNA); however, the physiological repercussions of mitochondrial p53 remain unknown. We endeavoured to elucidate whether an acute bout of endurance exercise could mediate an increase in mitochondrial p53 levels. C57Bl6 mice (n= 6 per group) were randomly assigned to sedentary, acute exercise (AE, 15 m min−1 for 90 min) or acute exercise + 3 h recovery (AER) groups. Exercise concomitantly increased the mRNA content of nuclear-encoded (PGC-1α, Tfam, NRF-1, COX-IV, citrate synthase) and mtDNA-encoded (COX-I) genes in the AE group, and further by ∼5-fold in the AER group. Nuclear p53 protein levels were reduced in the AE and AER groups, while in contrast, the abundance of p53 was drastically enhanced by ∼2.4-fold and ∼3.9-fold in subsarcolemmal and intermyofibrillar mitochondria, respectively, in the AER conditions. Within the mitochondria, the interaction of p53 with mtDNA at the D-loop and with Tfam was elevated by ∼4.6-fold and ∼3.6-fold, respectively, in the AER group. In the absence of p53, the enhanced COX-I mRNA content observed with AE and AER was abrogated. This study is the first to indicate that endurance exercise can signal to localize p53 to the mitochondria where it may serve to positively modulate the activity of the mitochondrial transcription factor Tfam. Our findings help us understand the mechanisms underlying the effects of exercise as a therapeutic intervention designed to trigger the pro-metabolic functions of p53.
Key points
p53 is one of several important proteins that regulate the synthesis and function of mitochondria in muscle, and mice lacking p53 have impaired aerobic capacity.
The role of p53 in response to physiological stress such as exercise has not been investigated.
We demonstrate that acute exercise induces the translocation of p53 to mitochondria, and promotes a subsequent interaction with mitochondrial transcription factor A (Tfam) and mitochondrial DNA (mtDNA) to positively affect mtDNA transcription.
This rapid effect on p53 translocation probably represents an early exercise response which can promote long-term beneficial mitochondrial and metabolic adaptations in muscle.
The results could have clinical implications in light of the growing recognition of exercise as an alternative therapy in cancer treatment, and the central role of p53 in halting tumorigenesis.
Introduction
The concept of utilizing physical exercise as a viable therapeutic modality for the treatment and management of age-associated chronic diseases often defined by dysregulated metabolism such as obesity, type 2 diabetes, cardiovascular diseases, dementia and cancer, is fast gaining acceptance (Warburton et al. 2010). While the overall health benefits of exercise are undeniable, the underlying molecular mechanisms mediating the protection conferred by exercise is only beginning to be amassed. Although a multitude of cellular conduits are involved, a fundamental adaptation is the increase in the mitochondrial oxidative capacity post exercise training. This increase in mitochondrial biogenesis brought about by endurance exercise is orchestrated by a host of transcriptional factors and co-activators that contrive together to coordinate improvements in oxidative capacity.
Affectionately known as the ‘Guardian of the Genome’ for its role in inducing cell-cycle arrest or cell death upon genotoxic stress signals, p53 is now reputed to play a vital role during cell metabolism, growth and development, and can be activated by physiological stressors to elicit an adaptive response (Saleem et al. 2011). p53 participates in regulating metabolism, mobilizing cellular anti-oxidant defence against physiological oxidative stress, and orchestrating a balance between the anabolic and catabolic pathways within the cell (Vousden & Lane, 2007). Interestingly, the cellular fate in response to p53 activation often hinges on its subcellular localization. For example, within the cytoplasm p53 inhibits autophagy, whereas in the nucleus it serves to activate autophagy through direct transcriptional activation of effector genes that promote autophagy (Maiuri et al. 2010). Similarly, in vitro studies have shown that within the mitochondrial matrix, p53 specifically binds to mtDNA polymerase γ ensuring mtDNA genomic integrity and maintenance (Achanta et al. 2005). p53 has also been purported to play a role in mtDNA transcription and translation by either binding directly (Heyne et al. 2004; Kulawiec et al. 2009) or indirectly via mitochondrial transcription factor A (Tfam) to mtDNA (Yoshida et al. 2003). p53 is not the only nuclear transcription factor to have been found interacting with the mitochondrial genome. Another important nuclear protein for mitochondrial biogenesis, peroxisome proliferator-activated receptor (PPAR)-γ co-activator-1α (PGC-1α), also reportedly resides within the mitochondria in a complex with mtDNA, where it may be involved in facilitating mtDNA transcription (Aquilano et al. 2010; Safdar et al. 2011). In addition, retinoic acid X recepor (RXR) and oestrogen receptors ERα/β also localize to the mitochondria, and have been implicated in mtDNA transcription (Casas et al. 2003; Chen et al. 2004). Clearly, the presence of bona fide nuclear proteins within mitochondria warrants further investigation into the physiological role of these proteins stationed in this organelle.
While much work has been done on p53 in cancer cell lines and in response to cell damaging signals, the physiological function of p53 within skeletal muscle and in response to physiologically relevant signals such as exercise remains unknown. We have previously demonstrated an increase in p53Ser15 phosphorylation content in response to acute contractile activity (Saleem et al. 2009). Here, we further investigated whether a physiological alteration in the cellular milieu, represented by an acute bout of exercise, could induce a change in the subcellular localization of p53 in murine skeletal muscle. Our findings indicate that p53 levels decreased substantially in the nucleus with exercise and recovery. We further illustrate that exercise preferentially shuttled p53 into the skeletal muscle mitochondria where it forms a complex at the D-loop region of mtDNA. These data suggest that the pro-metabolic/survival function of p53 in skeletal muscle can be differentially regulated in response to exercise.
Methods
Ethical approval
C57Bl/6J mice, bred in an institutional central animal facility (York University), were housed in micro-isolator cages in a temperature- and humidity-controlled room and maintained on a 12 h light–dark cycle with food and water ad libitum. All animal care protocols were submitted to the York University Animal Care Committee and were approved in accordance with the guidelines set forth by the Canadian Council on Animal Care. Animals were killed via cervical dislocation at the end of each experiment. p53 wild-type (WT) and knockout (KO) mice were acquired from Taconic Labs (Germantown, NY, USA).
Animal breeding and experimental design
At 3 months of age, C57Bl/6J mice (n= 6 per group) were matched for sex and body weight, and randomly assigned to sedentary (SED), acute bout of exercise (AE), or acute exercise followed by 3 h of recovery (AER) groups. All mice were acclimatized to the treadmill 2 weeks prior to the beginning of the experiment. The animals in both the AE and AER groups were then selected and subjected to an acute bout of treadmill running at 15 m min−1 for 90 min. All of the mice subjected to treadmill exercise were visibly exhausted at the end of the exercise as determined by their ability to withstand air and shock stimuli for greater than 5 s. The AE exercise group animals were killed immediately following exercise. The SED mice were killed by cervical dislocation at the same time as the AER group mice. Quadriceps femoris muscle was extracted from all mice and ∼70 mg was immediately snap frozen and stored at −80°C for subsequent mRNA expression. The remaining ∼200 mg of fresh quadriceps femoris was utilized for nuclear, cytosolic and mitochondrial fractionation. To assess the importance of p53, WT and p53 KO mice (n= 4 per group) were subjected to the same acute exercise challenge as documented above. Quadriceps femoris muscles were extracted and frozen for subsequent mRNA analysis.
RNA isolation
Total RNA was isolated from ∼70 mg of frozen muscle using TRI Reagent (Invitrogen) according to the manufacturer's instructions. RNA concentration and quality were measured using a Nanodrop 2000 (Thermo Scientific, Wilmington, DE, USA) and further verified with RNA gels.
mRNA expression analyses
The mRNA expression of PPAR-γ co-activator 1-α (PGC-1α), mitochondrial transcription factor A (Tfam), nuclear respiratory factor-1 (NRF-1), cytochrome c oxidase subunits I and IV (COX-I, COX-IV), citrate synthase (CS), glyceraldehye phosphate dehydrogenase (GAPDH) and p53 was quantified using a 7500 Real-Time PCR System (Applied Biosystems Inc., Foster City, CA, USA) and SYBR Green chemistry (PerfeCTa SYBR Green Supermix, ROX, Quanta BioSciences, Gaithersburg, MD, USA). First-strand cDNA synthesis from 2 μg of total RNA was performed with primers using SuperscriptIII transcriptase (Invitrogen) according to the manufacturer's directions. Forward and reverse primers (Table 1) for the previously mentioned genes were designed based on sequences available in GenBank using the MIT Primer 3 designer software (http://frodo.wi.mit.edu/), and were confirmed for specificity using the basic local alignment search tool (http://www.ncbi.nlm.nih.gov/BLAST/). β2 microglobulin was used as a control housekeeping gene, the expression of which did not alter between groups. All samples were run in duplicate simultaneously with negative controls that contained no cDNA. Melting point dissociation curves generated by the instrument were used to confirm the specificity of the amplified product. Primer efficiency curves were generated for each set to ensure 100 ± 2% efficiency.
Table 1.
Primer sequences based on gene transcripts available in GenBank
| Gene | Forward primer (5′→ 3′) | Reverse primer (5′→ 3′) |
|---|---|---|
| PGC-1α | ttccaccaagagcaagtat | cgctgtcccatgaggtatt |
| Tfam | gaagggaatgggaaaggtaga | aacaggacatggaaagcagat |
| NRF-1 | atccgaaagagacagcagaca | tggagggtgagatgcagagta |
| p53 | ccgacctatccttaccatcatc | ttcttctgtacggcggtctc |
| COX-I | ctagccgcaggcattactat | tgcccaaagaatcagaacag |
| COX-IV | ctccaacgaatggaagacag | tgacaaccttcttagggaac |
| CS | gcatgaagggacttgtgta | tctggcactcagggatact |
| GAPDH | aacactgagcatctccctca | gtgggtgcagcgaactttat |
| β2 microglobulin | ggtctttctggtgcttgtct | tatgttcggcttcccattct |
| mtDNA D-loop | agcccatgaccaacataactg | agactgtgtgctgtcctttca |
Mitochondrial fractionation
Briefly, ∼150 mg of skeletal muscle was minced, homogenized and subjected to differential centrifugation as previously documented to yield subsarcolemmal (SS) and intermyofibrillar (IMF) mitochondrial fractions (Saleem et al. 2009). The mitochondria were re-suspended in a small volume of resuspension buffer (100 mm KCl, 10 mm Mops, and 0.2% bovine serum albumin, pH 7.4, supplemented with protease inhibitor cocktail Complete, ETDA-free (Roche Applied Science, Mannheim, Germany)). All centrifugation steps were carried out at 4°C. Mitochondrial homogenates were analysed for protein content using the Bradford assay, and subsequently frozen at −80°C for further biochemical analysis including mitochondrial co-immunoprecipitation assay, mitochondrial chromatin immunoprecipitation (ChIP) assay and immunoblotting.
Mitochondrial co-immunoprecipitation assay
Mitochondrial co-immunoprecipitation assay was performed on isolated IMF mitochondrial fractions using Pierce Co-Immunoprecipitation Kit (Pierce, Rockford, IL, USA) according to the manufacturer's instructions. Briefly, mitochondrial fractions were homogenized in lysis buffer (0.025 m Tris, 0.15 m NaCl, 0.001 m EDTA, 1% NP-40, 5% glycerol, pH 7.4) supplemented with protease inhibitor cocktail Complete, ETDA-free (Roche Applied Science). The mitochondrial protein fraction (1 mg) was pre-cleared by incubation with control agarose resin to minimize non-specific binding. Anti-p53 (FL-393, Santa Cruz Biotechnology, Santa Cruz, CA, USA, 40 μg) was covalently coupled onto an amine-reactive resin. The pre-cleared lysates were subsequently incubated with antibody-coupled beads overnight at 4°C. Co-immunoprecipitates were washed, eluted and subsequently collected by centrifugation, boiled in 50 μl of lane marker sample buffer, and used for immunoblot analysis for Tfam. Anti-IgG antibody was used as a non-specific control.
mtDNA chromatin immunoprecipitation (ChIP) assay
ChIP assay was performed using an EZ-ChIP kit (Millipore, Billerica, MA, USA) according to the manufacturer's instructions. Mitochondrial fractions (1 mg) were cross-linked in 1% formaldehyde for 10 min at room temperature, and 10X glycine was added to stop fixation. Samples were then homogenized in 1 ml of SDS lysis buffer supplemented with protease inhibitor cocktail Complete, ETDA-free (Roche Applied Science). Chromatin was sheared by sonicating each sample on ice (output 20%, 4 times for 20 s, with a 20 s pause each time). Following centrifugation at 10,000 g at 4°C for 10 min, the supernate containing 1 mg of protein was diluted to 1 ml with ChIP dilution buffer. Anti-p53 (FL393, Santa Cruz Biotechnology, 10 μg) or anti-IgG antibody was added per sample and incubated overnight at 4°C. Anti-IgG antibodies were used as non-specific controls. Protein G-agarose (60 μl) was added and the sample was mixed for 1 h at 4°C with rotation. Precipitated samples were washed, and then eluted in 100 μl of elution buffer, and cross-linking was reversed by the addition of 8 μl of 5 m NaCl per sample followed by incubation at 65°C for 12 h. Co-immunoprecipitated DNA was purified according to the manufacturer's instructions. mtDNA D-loop region (Table 1) was quantified using a 7500 Real-Time PCR System and SYBR Green chemistry.
Nuclear and cytosolic fractionation
Nuclear and cytosolic fractions were prepared from freshly isolated skeletal muscle using a commercially available nuclear extraction kit (Pierce NE-PER). Briefly, 50–75 mg of skeletal muscle was minced and homogenized in Cytoplasmic Extraction Reagent I (CER I) buffer containing protease inhibitor cocktail Complete, EDTA-free. After a series of wash steps, nuclear proteins were extracted in high salt Nuclear Extraction Reagent (NER) buffer supplemented with protease inhibitors. The cytosolic fraction was spun at 100,000 g at 4°C for 60 min to obtain a pure cytosolic fraction.
Immunoblotting
Proteins were resolved on 8% or 12% SDS-PAGE gels depending on the molecular mass of the protein of interest. The gels were transferred onto enhanced chemiluminescent (ECL) nitrocellulose membranes, followed by blocking with 1–3% milk in Tris-buffered saline–Tween 20 (TBST) overnight at 4°C. Immunoblotting was carried out using mouse p53 (1:20, pAb421, a kind gift from Dr Sam Benchimol, York University), rabbit Tfam antibody (1:500, in-house), rabbit p53Ser15 (1:500, R&D systems, AF2887) and rabbit PGC-1α (1:500, Millipore, AB3242). With regard to PGC-1α, we have previously validated the use of this particular PGC-1α antibody in myotubes (Uguccioni & Hood, 2011), and it has also been used by other muscle physiology laboratories to detect PGC-1α (Hancock et al. 2008; Williams et al. 2009; Philp et al. 2011). We also further ascertained the antibody specificity by performing western blotting analysis using quadriceps femoris muscle from PGC-1α KO mice. Membranes were then incubated with the appropriate secondary antibody (Abcam) coupled to horseradish peroxidase at room temperature for 2 h. After incubation, membranes were washed three times in TBST, developed with an ECL kit, and quantified via densitometric analysis of the intensity of the signal with Sigma Scan Pro v.5 software (Jandel Scientific, San Rafael, CA, USA). Voltage-dependent anion channel (VDAC) and histone 2B (H2B) were used as loading controls for mitochondrial and nuclear fractions, respectively.
Statistical analyses
Data were analysed using one-way analysis of variance (ANOVA), except for Fig. 5D which was analysed using a two-way ANOVA, with the aid of Graph Pad 4.0 software. For all one-way ANOVA analyses, Tukey's post hoc test was used to identify individual differences when statistical significance was observed. Statistical significance was established at P≤ 0.05. Data are presented as mean ± standard error of the mean (SEM).
Figure 5. Acute exercise enhances the association of p53 with Tfam inside the mitochondria and promotes p53–mtDNA D-loop complex formation.
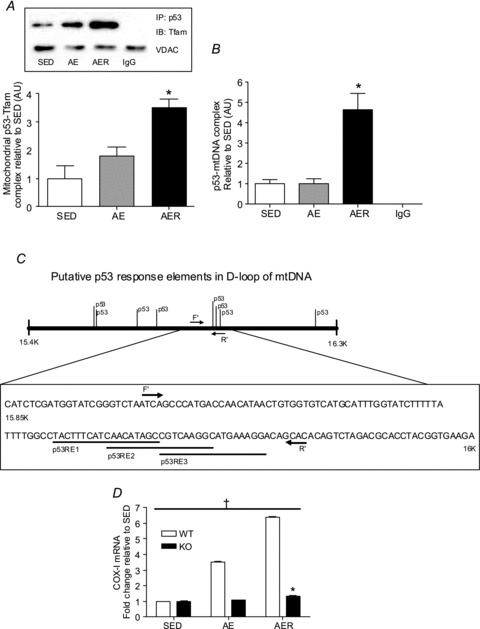
A, p53 was immunoprecipitated (IP) followed by immunoblotting (IB) for Tfam in isolated mitochondrial fractions from SED, AE and AER mice. p53–Tfam complex increased significantly in the AER group. *P < 0.05 AER vs. SED. IgG was used as a non-specific control for the co-immunoprecipitation assay. VDAC was used to ensure that equal amounts of mitochondrial fractions were used for co-IP experiments from each group of mice. Data are presented as a fold-increase over SED values. Error bars refer to SEM values. B, there is a significant increase in the p53 complexed at the D-loop region of the mtDNA in the AER group. *P < 0.05 AER vs. SED. Anti-IgG antibody was used as a non-specific control. Real-time amplification of signal from IgG group was below detectable limit. Data are presented as a fold-increase over SED values. Error bars refer to SEM values. C, schematic illustration of the D-loop sequence of mtDNA and putative p53 response elements (REs). The position of the forward (F′) and reverse (R′) primers used in mtDNA–ChIP analysis is identified, and the sequences of the three putative p53 REs within the amplicon amplified by the PCR reaction are shown. D, mRNA content of mtDNA-encoded COX-I subunit in wild-type (WT) and p53 knockout (KO) mice in SED, AE and AER conditions. β2 microglobulin was used as a housekeeping control gene. *P < 0.05 WT vs. KO, †P < 0.05 AE and AER vs. SED in WT mice. Data are presented as a fold-increase over SED mice. Error bars represent SEM values (n= 4 per group).
Results
Purity of cellular fractions, antibody specificity and cellular distribution of p53
The purity of nuclear and mitochondrial fractions and specificity of p53 antibody are of extreme importance for the subsequent subcellular localization analyses of p53 carried out in this study. We have previously established that the isolated mitochondrial subfractions are pure and functionally intact (Cogswell et al. 1993). To further assess the purity of the nuclear and mitochondrial fractions, we immunoblotted for the mitochondrial protein cytochrome c oxidase IV (COX-IV, Fig. 1A) and the nuclear protein histone 2B (H2B, Fig. 1B). COX-IV and H2B were only detected in mitochondrial and nuclear fractions, respectively. Furthermore, no cytosolic contamination was apparent in the nuclear and mitochondrial fractions as measured by GAPDH (Fig. 1C). We also confirmed the specificity of p53 antibody against the protein by measuring p53 expression in whole muscle homogenates from wild-type (WT) and p53 knockout (KO) mice (Fig. 1D). p53 was not detected in the muscle homogenates from KO mice (Fig. 1D) illustrating the specificity of the p53 antibody used in this study. The subcellular distribution of p53 in skeletal muscle was measured by probing for p53 in cytosolic, mitochondrial and nuclear fractions (Fig. 1E). p53 content was highest in the cytosolic fraction, followed by the nuclear and mitochondrial compartments in muscle (Fig. 1E).
Figure 1. Subcellular fraction purity, cellular distribution of p53, and antibody specificity of p53 and PGC-1α.
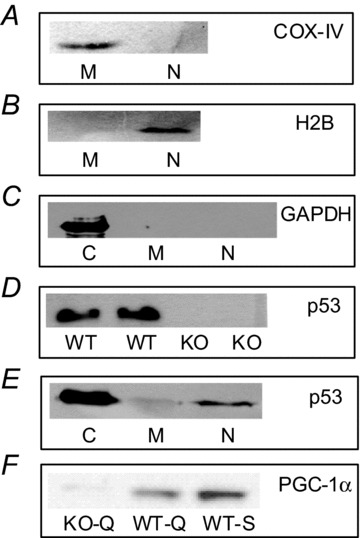
COX-IV (A), histone 2B (B) and GAPDH (C) were used as indicators of the purity of mitochondrial, nuclear and cytosolic fractions, respectively. D, specificity of p53 antibody as demonstrated by measuring p53 expression in whole muscle homogenates from p53 wild-type (WT) and knockout (KO) mice. E, subcellular pattern of p53 expression in cytosolic (C), mitochondrial (M) and nuclear (N) fractions. F, PGC-1α expression in KO quadriceps femoris (KO-Q, lane 1), WT quadriceps femoris (WT-Q, lane 2) and WT soleus (WT-S, lane 3) muscle homogenates.
Validation of PGC-1α antibody
To ascertain the specificity of the PGC-1α antibody, we performed immunoblotting using quadriceps femoris muscle from PGC-1α KO (Fig. 1F, lane 1), and WT mice (Fig. 1F, lane 2). We also included a soleus muscle homogenate as a positive control (Fig. 1F, lane 3), as it clearly depicts an increased abundance of PGC-1α in a slow, oxidative tissue such as soleus as opposed to a primarily fast glycolytic muscle such as quadriceps femoris. The ∼94 kDa band is absent in lane 1 confirming the antibody specificity. There is slight immunoreactivity in lane 1, but it is present at a higher molecular mass than the band in the WT lanes. We suspect it to be non-specific binding.
mRNA content of genes related to energy metabolism increases following an acute bout of endurance exercise
The transcript levels of nuclear DNA-encoded (PGC-1α, Tfam, NRF-1, COX-IV, CS) and mtDNA-encoded (COX-I) genes involved in mitochondrial biogenesis increased immediately following an acute bout of exercise (P < 0.05, Fig. 2). This enhancement in mRNA expression of these transcripts was maintained or further exacerbated after 3 h of recovery following the exercise (P < 0.05, Fig. 2). p53 expression decreased with acute exercise (AE), and the mRNA levels of the cytosolic glycolytic enzyme, GAPDH remained relatively unchanged (Fig. 2).
Figure 2. Changes in nuclear and mitochondrial DNA-encoded mRNA transcripts.
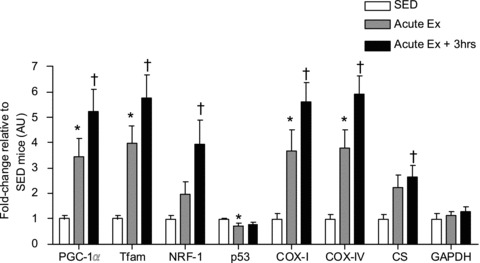
Transcriptional regulators of mitochondrial biogenesis (PGC-1α, NRF-1 and Tfam), components of the electron transport chain (COX-IV and COX-I), and mitochondrial enzyme citrate synthase (CS) mRNA expression increased in the AE and AER conditions. p53 mRNA levels decreased immediately post-exercise, and Glycolytic enzyme GAPDH mRNA content did not change with the experimental conditions. β2 microglobulin was used as a house keeping control gene. *P < 0.05 AE vs. SED, †P < 0.05 AER vs. SED. Data are presented as a fold-increase over SED mice. Error bars represent SEM values.
Acute exercise reduces nuclear, and enhances mitochondrial abundance of p53
p53 content decreased steadily after exercise by 56% in the nuclear fraction from the AER group (P < 0.05, Fig. 3A). In contrast, an elevation of PGC-1α expression was evident in the nucleus in the AE and AER conditions (P < 0.05, Fig. 3B), in line with previous literature (Wright et al. 2007). H2B served as a loading control and remained unchanged with the exercise treatments. A gradual increase in p53 levels within mitochondria was observed, by ∼2.4-fold (P < 0.05, Fig. 4A) in the subsarcolemmal (SS) mitochondrial fraction, and by ∼3.9-fold (P < 0.05, Fig. 4B) in the intermyofibrillar (IMF) mitochondrial fraction. The increase in p53 content occurred concomitantly with a gradual increase in p53Ser15 phosphorylation in mitochondrial fractions (Fig. 4C). Both the mtDNA transcription factor Tfam and VDAC expression remained unchanged with the exercise treatments (Fig. 4D).
Figure 3. Alterations in nuclear expression of p53 and PGC-1α with acute exercise.
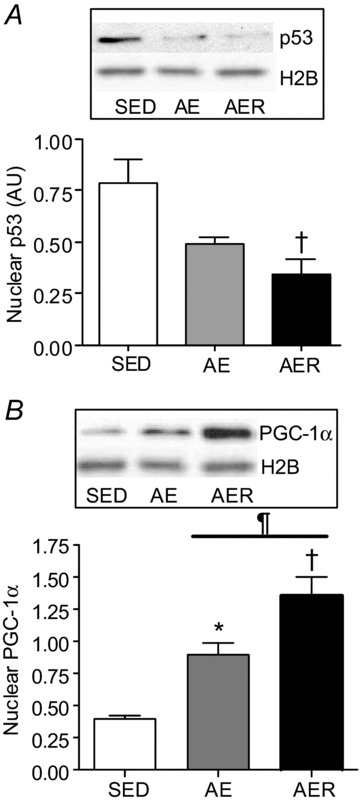
Nuclear content of p53 (A) steadily decreased, and that of PGC-1α (B) progressively increased in the AE and AER animal groups. Histone 2B was used as a loading control and did not change during the conditions. *P < 0.05 AE vs. SED, †P < 0.05 AER vs. SED. ¶P < 0.05 AER vs. AE. Data are presented as mean ± SEM.
Figure 4. Changes in mitochondrial Tfam and p53 content upon acute exercise.
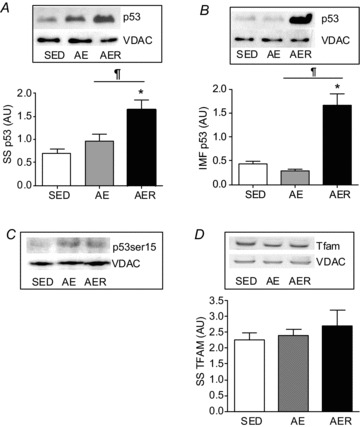
p53 content in SS mitochondria (A) and IMF mitochondria (B) increases significantly in the AER group. C, p53 phosphorylation at serine 15 displayed a gradual increase in AE and AER conditions in SS mitochondria compared to SED control (representative blot from n= 2). D, Tfam content did not alter immediately after AE, nor in the AER group. VDAC was used as loading control and did not alter between the experimental conditions. *P < 0.05 AER vs. SED. ¶P < 0.05 AER vs. AE. Data are presented as mean ± SEM values.
Mitochondrial p53 forms a complex with Tfam and binds to mtDNA at the D-loop region
Given the apparent translocation of p53 to the mitochondria with exercise, we posited that acute exercise would positively enhance this association of p53 with Tfam and mtDNA. To test our hypothesis we performed co-immunoprecipitation assays with the SS mitochondrial fraction from SED, AE and AER groups (Fig. 5A). Congruent with previous in vitro findings (Yoshida et al. 2003; Wong et al. 2009), we observed that p53 forms a complex with Tfam in skeletal muscle mitochondria (Fig. 5A). Since Tfam is primarily found in the mitochondrial matrix, this observation also confirmed the matrix-specific localization of p53 in mitochondrial fractions (Fig. 5A and B). Furthermore, we observed that the p53–Tfam complex increased progressively upon an acute bout of exercise by ∼3.6-fold in the AER group (P < 0.05, Fig. 5A). We also performed mtDNA co-immunoprecipitation assays to determine if p53 was bound to the D-loop of mtDNA, and whether this interaction was positively modified by acute exercise. We observed that in skeletal muscle mitochondria, p53 forms a complex with mtDNA within the D-loop region, and that this association is enhanced by ∼4.6-fold in the AER group in IMF mitochondria (P < 0.05, Fig. 5B).
Increase in mtDNA-derived COX-I mRNA post exercise is dependant upon p53 expression
The transcript levels of the mtDNA-encoded COX-I subunit increased immediately following an acute bout of exercise, and further with recovery in the WT mice (P < 0.05, Fig. 5D) as shown earlier. Remarkably, this exercise-induced adaptation in COX-I mRNA was completely abolished in the p53 KO mice (P < 0.05, Fig. 5D).
Discussion
The disruption of p53 expression carries grave physiological repercussions, as evident by aberrant mitochondrial bioenergetic efficiency, reduced mitochondrial biogenesis, greater fatigability and reduced exercise capacity observed in p53 KO animals (Matoba et al. 2006; Park et al. 2009; Saleem et al. 2009). If p53 plays a role in potentiating endurance exercise-induced mitochondrial adaptations, it is likely that p53 is post-translationally modified in response to exercise which may differentially regulate its subcellular localization post contractile activity. We and others have previously reported elevated p53Ser15 phosphorylation levels in response to acute contractile activity (Saleem et al. 2009; Bartlett et al. 2012), a modification that is classically linked to the increased stability and activity of the protein. Interestingly, we demonstrate here that p53Ser15 phosphorylation levels increase progressively with AE and AER conditions in the mitochondrial fractions, in line with the enhanced phosphorylation status of p53 as previously reported in whole muscle homogenates (Saleem et al. 2009; Bartlett et al. 2012). It is entirely plausible that the measured increase in Ser15 phosphorylation is due to the combined effect of the activation of p38 MAPK and AMPK, both of which are bona fide exercise-activated kinases (Akimoto et al. 2005; Ljubicic & Hood, 2009) and are known to post-translationally modulate p53 (She et al. 2000; Jones et al. 2005). In a recent paper, Bartlett et al. (2013) provided further suggestive evidence of a link between AMPK signalling and p53 phosphorylation in human skeletal muscle. Furthermore, since p53 is subject to a host of other post-translational modifications such as acetylation, ubiquitination, sumoylation and neddylation, it is possible that any of these may assist in trafficking p53 out of the nucleus and into the mitochondria. In fact, it has been previously demonstrated that mono-ubiquitinated p53 is shuttled out of the nucleus in a chromosome region maintenance 1 (CRM1)-dependent manner (Lohrum et al. 2001). Subsequent to this, mono-ubquitinated p53 is rapidly trafficked to the mitochondria where it undergoes deubiquitination by resident deubiquitinases such as HAUSP (Marchenko et al. 2007). Additionally, proteomic analyses have revealed that a large number of mitochondrially destined nuclear-encoded proteins lack canonical N-terminal mitochondrial targeting signals, but instead have cryptic internal signals. There is no discernable consistent pattern to the nature and location of these cryptic signals. Similar to many other proteins such as PGC-1α, RXR and ERα/β that have been identified within mitochondria, p53 also does not contain a canonical mitochondrial targeting signal, but it appears to possess a cryptic signal (Boopathi et al. 2008). Clearly, further research is required to carefully distil which post-translational modification is pivotal to the change in the subcellular address of p53 and its subsequent import into the mitochondria.
Apoptosis-inducing factor (AIF), synthesis of cytochrome c oxidase 2 (SCO2) and Tfam are important transcriptional targets of p53 through which it regulates cell metabolism. While AIF is known for its role in the induction of apoptosis, during basal conditions AIF contributes to efficient oxidative phosphorylation by promoting the proper assembly and function of mitochondrial respiratory complex I (Stambolsky et al. 2006). SCO2 has been shown to be vital for the proper assembly of subunit I of the mitochondrial cytochrome c oxidase complex, and re-expression of SCO2 quickly rescued the oxidative impairment in p53 KO cells (Matoba et al. 2006). A multitude of studies (Yoshida et al. 2003; Bourdon et al. 2007; Kulawiec et al. 2009; Lebedeva et al. 2009; Park et al. 2009) have demonstrated that the presence of p53 is a determinant of both Tfam expression and mtDNA content. Since p53 clearly targets the promoter regions of these genes and positively up-regulates their expression, we posited that p53 levels would increase in the nucleus upon an exercise stimulus in an effort to increase the transcription of the previously mentioned gene targets. However, p53 content reduced drastically with exercise, and even more so in the recovery period in the nuclear fraction. This is at odds with the expression pattern of the classic activator of mitochondrial biogenesis, PGC-1α, in which the content of PGC-1α steadily increased with exercise and recovery in the nuclear fraction as shown here, and by others (Wright et al. 2007). We hypothesize that the decrease in p53 content occurred so as to remove p53 from the vicinity of the plethora of gene targets that can induce cell death, or senescence, reactive outcomes often dictated by p53 in times of stress. Additionally, a recent study (Sahin et al. 2011) has indicated that p53 can transcriptionally suppress PGC-1α and PGC-1β expression, co-activators that are known to induce mitochondrial biogenesis. Therefore it is possible that the decrease in nuclear p53 content ensures that p53 does not suppress the activation of PGC-1α/β, so as not to impede the process of mitochondrial biogenesis.
It should also be noted that despite the apparent exodus of p53 from the nucleus, a basal level of p53 is still present in nuclei post exercise which may be sufficient to carry on its pro-metabolic role. Moreover, the idea that p53 is a transcriptional inhibitor of PGC-1α is not universally accepted. A recent study reported that p53 binds to the promoter of PGC-1α and up-regulates its expression during times of mild oxidative and nutritional stress (Aquilano et al. 2013). Thus, the interaction between p53 and PGC-1α appears to be specific to the cellular milieu, and it remains to be seen how exercise affects this relationship.
Another intriguing finding was the decrease in the mRNA level of p53, which was in vivid contrast to the significant elevations in the transcripts of other genes involved in mitochondrial biogenesis such as Tfam, NRF-1, PGC-1α, CS, COX I and COX IV. This could be due to the exquisite control exerted by p53 on its own mRNA expression. As illustrated previously (Mosner et al. 1995), with increasing p53 protein expression, there is a subsequent decrease in its mRNA expression as p53 binds to the 5′ untranslated region of its own mRNA and causes it to be degraded. Thus the decrease in mRNA expression that was observed in this study could simply be due to the ability of p53 to fine-tune its own expression.
The binding of p53-Tfam, and p53 with mtDNA post exercise is an exciting finding. It is possible that p53 acts as an accessory factor that may augment Tfam activity, an association highly dependent on altered energy demands such as during exercise. Tfam primarily exists as a mitochondrial transcription factor, but also functions to maintain mtDNA integrity and repair. We observed increasing amounts of Tfam bound to p53 in the mitochondria with exercise and 3 h recovery, despite a lack of change in the amount of total Tfam in the mitochondria. It is known that the majority of Tfam within the organelle is closely associated with mtDNA (Park & Larsson, 2011) at the D-loop region to allow Tfam to regulate mtDNA transcription (Larsson et al. 1998). We have previously shown that chronic contractile activity results in increased Tfam binding to the mtDNA D-loop region, indicative of the ability of chronic exercise to modify mtDNA transcription (Gordon et al. 2001). Interestingly, p53 has been shown to increase the binding of Tfam to damaged DNA (Yoshida et al. 2003; Wong et al. 2009) and it is entirely plausible that it potentiates the binding of Tfam to the D-loop as well. We also observed an elevated amount of p53 bound to the D-loop of mtDNA, either directly or via Tfam, in the mitochondrial fractions following exercise and recovery. Therefore, we hypothesize that p53 may function as a mitochondrial transcription factor, positively modulating mtDNA-encoded gene expression either directly, or via modulation of Tfam activity at the D-loop region. To assess the plausibility of this hypothesis, we subjected the D-loop region 15400 bp–16299 bp of the Mus musculus mitochondrial genome (NCBI reference number: NC_005089.1) to a patch analysis using the Consite software http://asp.ii.uib.no:8090/cgi-bin/CONSITE/consite. Bioinformatic analysis revealed putative p53 response elements (REs), which were identified and plotted against the mtDNA D-loop sequence. This is in line with previous studies documenting the interaction of p53 with mtDNA (Heyne et al. 2004; Kulawiec et al. 2009), and corresponds with several reports that have identified common nuclear transcription factors and co-activators such as SIRT1, ERα/β, retinoid X receptor and PGC-1α that also reside in the mitochondria, and are involved in regulating mtDNA transcription (Casas et al. 2003; Chen et al. 2004; Aquilano et al. 2010; Safdar et al. 2011). Further support for a significant role of p53 in regulating mtDNA transcription can be acquired from observing the large increase in COX-I mRNA expression in wild-type mice, which was completely abrogated in the p53 knockout animals. This indicates that the increase in intra-mitochondrial p53 with exercise is indeed functional in facilitating mtDNA transcription. In addition, p53 can mediate mtDNA genomic stability directly, via its inherent base excision repair activity, and indirectly by (1) promoting the binding of Tfam to damaged mtDNA, and (2) by enhancing the function of mtDNA polymerase γ, the sole mtDNA repair enzyme (Saleem et al. 2011). Therefore, it is possible that exercise induces p53-Tfam-mediated maintenance and repair of mtDNA, exclusively or concomitantly with enhanced mtDNA transcription.
Thus, the nuclear depletion and mitochondrial accumulation of p53 subsequent to an acute bout of exercise highlights the fascinating diverse transcriptional and non-transcriptional modes of regulating oxidative metabolism as commandeered by p53 in a physiological setting. Clearly, the action of p53 on the mitochondrial genome may represent an important conduit by which it facilitates the coordinated expression of the nuclear and mitochondrial genomes in promoting mitochondrial biogenesis and mtDNA stability in muscle. The tumour suppressor protein p53 plays an indispensible role in halting tumorigenesis, is important in mediating oxidative metabolism, and as indicated by our data, responds to an exercise stimulus. Together with the current awareness of exercise as a feasible, cost-effective and safe therapy to reduce the incidence and progression of cancer (Friedenreich & Orenstein, 2002; Warburton et al. 2010) and other metabolic diseases, our results may carry important clinical implications. Further work that delineates the signalling mechanisms involved, and that focuses on elucidating the effect of exercise training on p53 function and regulation is clearly warranted.
Acknowledgments
The authors would like to acknowledge Dr. Michael Connor for use of his Nanodrop instrument, and Dr. Sam Benchimol for the generous gift of the p53 antibody.
Glossary
- AE
acute exercise
- AER
acute exercise plus recovery
- ChIP
mitochondrial chromatin immunoprecipitation
- COX
cytochrome c oxidase
- CRM1
chromosome region maintenance 1
- CS
citrate synthase
- GAPDH
glyceraldehyde phosphate dehydrogenase
- H2B
histone 2B
- KO
knockout
- IMF
intermyofibrillar
- mtDNA
mitochondrial DNA
- NRF-1
nuclear respiratory factor-1
- PGC-1α
PPAR-γ coactivator-1α
- PPAR
peroxisome proliferator-activated receptor
- RXR
retinoic acid X receptor
- SED
sedentary
- SS
subsarcolemmal
- Tfam
mitochondrial transcription factor A
- VDAC
voltage-dependent anion channel
- WT
wild-type
Additional information
Competing interests
None.
Author contributions
A.S. and D.A.H. conceived and designed the experiments, and wrote the manuscript. A.S. collected, analysed and interpreted the data. Both authors approved the final version of the manuscript. All the experiments were carried out at York University.
Funding
A.S. was funded by a NSERC CGS scholarship. D.A.H. holds a Canada Research Chair in Cell Physiology. This research was funded by an NSERC grant to D.A.H.
References
- Achanta G, Sasaki R, Feng L, Carew JS, Lu W, Pelicano H, Keating MJ, Huang P. Novel role of p53 in maintaining mitochondrial genetic stability through interaction with DNA Pol γ. EMBO J. 2005;24:3482–3492. doi: 10.1038/sj.emboj.7600819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akimoto T, Pohnert SC, Li P, Zhang M, Gumbs C, Rosenberg PB, Williams RS, Yan Z. Exercise stimulates Pgc-1α transcription in skeletal muscle through activation of the p38 MAPK pathway. J Biol Chem. 2005;280:19587–19593. doi: 10.1074/jbc.M408862200. [DOI] [PubMed] [Google Scholar]
- Aquilano K, Baldelli S, Pagliei B, Cannata S, Rotilio G, Ciriolo MR. p53 orchestrates the PGC-1α-mediated antioxidant response upon mild redox and metabolic imbalance. Antioxid Redox Signal. 2013;18:386–399. doi: 10.1089/ars.2012.4615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aquilano K, Vigilanza P, Baldelli S, Pagliei B, Rotilio G, Ciriolo MR. Peroxisome proliferator-activated receptor γ co-activator 1α (PGC-1α) and sirtuin 1 (SIRT1) reside in mitochondria: possible direct function in mitochondrial biogenesis. J Biol Chem. 2010;285:21590–21599. doi: 10.1074/jbc.M109.070169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett JD, Hwa JC, Jeong TS, Louhelainen J, Cochran AJ, Gibala MJ, Gregson W, Close GL, Drust B, Morton JP. Matched work high-intensity interval and continuous running induce similar increases in PGC-1α mRNA, AMPK, p38, and p53 phosphorylation in human skeletal muscle. J Appl Physiol. 2012;112:1135–1143. doi: 10.1152/japplphysiol.01040.2011. [DOI] [PubMed] [Google Scholar]
- Bartlett JD, Louhelainen J, Iqbal Z, Cochran AJ, Gibala MJ, Gregson W, Close GL, Drust B, Morton JP. Reduced carbohydrate availability enhances exercise-induced p53 signalling in human skeletal muscle: implications for mitochondrial biogenesis. Am J Physiol Regul Integr Comp Physiol. 2013;304:R450–R458. doi: 10.1152/ajpregu.00498.2012. [DOI] [PubMed] [Google Scholar]
- Boopathi E, Srinivasan S, Fang JK, Avadhani NG. Bimodal protein targeting through activation of cryptic mitochondrial targeting signals by an inducible cytosolic endoprotease. Mol Cell. 2008;32:32–42. doi: 10.1016/j.molcel.2008.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourdon A, Minai L, Serre V, Jais JP, Sarzi E, Aubert S, Chrétien D, de Lonlay P, Paquis-Flucklinger V, Arakawa H, Nakamura Y, Munnich A, Rötig A. Mutation of RRM2B, encoding p53-controlled ribonucleotide reductase (p53R2), causes severe mitochondrial DNA depletion. Nat Genet. 2007;39:776–780. doi: 10.1038/ng2040. [DOI] [PubMed] [Google Scholar]
- Casas F, Daury L, Grandemange S, Busson M, Seyer P, Hatier R, Carazo A, Cabello G, Wrutniak-Cabello C. Endocrine regulation of mitochondrial activity: involvement of truncated RXRα and c-Erb Aα1 proteins. FASEB J. 2003;17:426–436. doi: 10.1096/fj.02-0732com. [DOI] [PubMed] [Google Scholar]
- Chen JQ, Eshete M, Alworth WL, Yager JD. Binding of MCF-7 cell mitochondrial proteins and recombinant human estrogen receptors α and β to human mitochondrial DNA estrogen response elements. J Cell Biochem. 2004;93:358–373. doi: 10.1002/jcb.20178. [DOI] [PubMed] [Google Scholar]
- Cogswell AM, Stevens RJ, Hood DA. Properties of skeletal muscle mitochondria isolated from subsarcolemmal and intermyofibrillar regions. Am J Physiol Cell Physiol. 1993;264:C383–C389. doi: 10.1152/ajpcell.1993.264.2.C383. [DOI] [PubMed] [Google Scholar]
- Friedenreich CM, Orenstein MR. Physical activity and cancer prevention: etiologic evidence and biological mechanisms. J Nutr. 2002;132:3456S–3464S. doi: 10.1093/jn/132.11.3456S. [DOI] [PubMed] [Google Scholar]
- Gordon JW, Rungi AA, Inagaki H, Hood DA. Effects of contractile activity on mitochondrial transcription factor A expression in skeletal muscle. J Appl Physiol. 2001;90:389–396. doi: 10.1152/jappl.2001.90.1.389. [DOI] [PubMed] [Google Scholar]
- Hancock CR, Han DH, Chen M, Terada S, Yasuda T, Wright DC, Holloszy JO. High-fat diets cause insulin resistance despite an increase in muscle mitochondria. Proc Natl Acad Sci U S A. 2008;105:7815–7820. doi: 10.1073/pnas.0802057105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyne K, Mannebach S, Wuertz E, Knaup KX, Mahyar-Roemer M, Roemer K. Identification of a putative p53 binding sequence within the human mitochondrial genome. FEBS Lett. 2004;578:198–202. doi: 10.1016/j.febslet.2004.10.099. [DOI] [PubMed] [Google Scholar]
- Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, Birnbaum MJ, Thompson CB. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell. 2005;18:283–293. doi: 10.1016/j.molcel.2005.03.027. [DOI] [PubMed] [Google Scholar]
- Kulawiec M, Ayyasamy V, Singh KK. p53 regulates mtDNA copy number and mitocheckpoint pathway. J Carcinog. 2009;8:8. doi: 10.4103/1477-3163.50893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson NG, Wang J, Wilhelmsson H, Oldfors A, Rustin P, Lewandoski M, Barsh GS, Clayton DA. Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat Genet. 1998;18:231–236. doi: 10.1038/ng0398-231. [DOI] [PubMed] [Google Scholar]
- Lebedeva MA, Eaton JS, Shadel GS. Loss of p53 causes mitochondrial DNA depletion and altered mitochondrial reactive oxygen species homeostasis. Biochim Biophys Acta. 2009;1787:328–334. doi: 10.1016/j.bbabio.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ljubicic V, Hood DA. Specific attenuation of protein kinase phosphorylation in muscle with a high mitochondrial content. Am J Physiol Endocrinol Metab. 2009;297:E749–E758. doi: 10.1152/ajpendo.00130.2009. [DOI] [PubMed] [Google Scholar]
- Lohrum MA, Woods DB, Ludwig RL, Balint E, Vousden KH. C-terminal ubiquitination of p53 contributes to nuclear export. Mol Cell Biol. 2001;21:8521–8532. doi: 10.1128/MCB.21.24.8521-8532.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiuri MC, Galluzzi L, Morselli E, Kepp O, Malik SA, Kroemer G. Autophagy regulation by p53. Curr Opin Cell Biol. 2010;22:181–185. doi: 10.1016/j.ceb.2009.12.001. [DOI] [PubMed] [Google Scholar]
- Marchenko ND, Wolff S, Erster S, Becker K, Moll UM. Monoubiquitylation promotes mitochondrial p53 translocation. EMBO J. 2007;26:923–934. doi: 10.1038/sj.emboj.7601560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matoba S, Kang JG, Patino WD, Wragg A, Boehm M, Gavrilova O, Hurley PJ, Bunz F, Hwang PM. p53 regulates mitochondrial respiration. Science. 2006;312:1650–1653. doi: 10.1126/science.1126863. [DOI] [PubMed] [Google Scholar]
- Mosner J, Mummenbrauer T, Bauer C, Sczakiel G, Grosse F, Deppert W. Negative feedback regulation of wild-type p53 biosynthesis. EMBO J. 1995;14:4442–4449. doi: 10.1002/j.1460-2075.1995.tb00123.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park CB, Larsson NG. Mitochondrial DNA mutations in disease and aging. J Cell Biol. 2011;193:809–818. doi: 10.1083/jcb.201010024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JY, Wang PY, Matsumoto T, Sung HJ, Ma W, Choi JW, Anderson SA, Leary SC, Balaban RS, Kang JG, Hwang PM. p53 improves aerobic exercise capacity and augments skeletal muscle mitochondrial DNA content. Circ Res. 2009;105:705–712. doi: 10.1161/CIRCRESAHA.109.205310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philp A, Chen A, Lan D, Meyer GA, Murphy AN, Knapp AE, Olfert IM, McCurdy CE, Marcotte GR, Hogan MC, Baar K, Schenk S. Sirtuin 1 (SIRT1) deacetylase activity is not required for mitochondrial biogenesis or peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) deacetylation following endurance exercise. J Biol Chem. 2011;286:30561–30570. doi: 10.1074/jbc.M111.261685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safdar A, Little JP, Stokl AJ, Hettinga BP, Akhtar M, Tarnopolsky MA. Exercise increases mitochondrial PGC-1 content and promotes nuclear-mitochondrial cross-talk to coordinate mitochondrial biogenesis. J Biol Chem. 2011;286:10605–10617. doi: 10.1074/jbc.M110.211466. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Sahin E, Colla S, Liesa M, Moslehi J, Muller FL, Guo M, Cooper M, Kotton D, Fabian AJ, Walkey C, Maser RS, Tonon G, Foerster F, Xiong R, Wang YA, Shukla SA, Jaskelioff M, Martin ES, Heffernan TP, Protopopov A, Ivanova E, Mahoney JE, Kost-Alimova M, Perry SR, Bronson R, Liao R, Mulligan R, Shirihai OS, Chin L, DePinho RA. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature. 2011;470:359–365. doi: 10.1038/nature09787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleem A, Adhihetty PJ, Hood DA. Role of p53 in mitochondrial biogenesis and apoptosis in skeletal muscle. Physiol Genomics. 2009;37:58–66. doi: 10.1152/physiolgenomics.90346.2008. [DOI] [PubMed] [Google Scholar]
- Saleem A, Carter HN, Iqbal S, Hood DA. Role of p53 within the regulatory network controlling muscle mitochondrial biogenesis. Exerc Sport Sci Rev. 2011;39:199–205. doi: 10.1097/JES.0b013e31822d71be. [DOI] [PubMed] [Google Scholar]
- She QB, Chen N, Dong Z. ERKs and p38 kinase phosphorylate p53 protein at serine 15 in response to UV radiation. J Biol Chem. 2000;275:20444–20449. doi: 10.1074/jbc.M001020200. [DOI] [PubMed] [Google Scholar]
- Stambolsky P, Weisz L, Shats I, Klein Y, Goldfinger N, Oren M, Rotter V. Regulation of AIF expression by p53. Cell Death Differ. 2006;13:2140–2149. doi: 10.1038/sj.cdd.4401965. [DOI] [PubMed] [Google Scholar]
- Uguccioni G, Hood DA. The importance of PGC-1α in contractile activity-induced mitochondrial adaptations. Am J Physiol Endocrinol Metab. 2011;300:E361–E371. doi: 10.1152/ajpendo.00292.2010. [DOI] [PubMed] [Google Scholar]
- Vousden KH, Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol. 2007;8:275–283. doi: 10.1038/nrm2147. [DOI] [PubMed] [Google Scholar]
- Warburton DE, Charlesworth S, Ivey A, Nettlefold L, Bredin SS. A systematic review of the evidence for Canada's Physical Activity Guidelines for Adults. Int J Behav Nutr Phys Act. 2010;7:39. doi: 10.1186/1479-5868-7-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams DB, Sutherland LN, Bomhof MR, Basaraba SA, Thrush AB, Dyck DJ, Field CJ, Wright DC. Muscle-specific differences in the response of mitochondrial proteins to β-GPA feeding: an evaluation of potential mechanisms. Am J Physiol Endocrinol Metab. 2009;296:E1400–E1408. doi: 10.1152/ajpendo.90913.2008. [DOI] [PubMed] [Google Scholar]
- Wong TS, Rajagopalan S, Freund SM, Rutherford TJ, Andreeva A, Townsley FM, Petrovich M, Fersht AR. Biophysical characterizations of human mitochondrial transcription factor A and its binding to tumor suppressor p53. Nucleic Acids Res. 2009;37:6765–6783. doi: 10.1093/nar/gkp750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright DC, Han DH, Garcia-Roves PM, Geiger PC, Jones TE, Holloszy JO. Exercise-induced mitochondrial biogenesis begins before the increase in muscle PGC-1α expression. J Biol Chem. 2007;282:194–199. doi: 10.1074/jbc.M606116200. [DOI] [PubMed] [Google Scholar]
- Yoshida Y, Izumi H, Torigoe T, Ishiguchi H, Itoh H, Kang D, Kohno K. p53 physically interacts with mitochondrial transcription factor A and differentially regulates binding to damaged DNA. Cancer Res. 2003;63:3729–3734. [PubMed] [Google Scholar]