

Abstract
Sympathetic vascular transduction is commonly understood to act as a basic relay mechanism, but under basal conditions, competing dilatory signals may interact with and alter the ability of sympathetic activity to decrease vascular conductance. Thus, we determined the extent to which spontaneous bursts of muscle sympathetic nerve activity (MSNA) mediate decreases in forearm vascular conductance (FVC) and the contribution of local α-adrenergic receptor-mediated pathways to the observed FVC responses. In 19 young men, MSNA (microneurography), arterial blood pressure and brachial artery blood flow (duplex Doppler ultrasound) were continuously measured during supine rest. These measures were also recorded in seven men during intra-arterial infusions of normal saline, phentolamine (PHEN) and PHEN with angiotensin II (PHEN+ANG). The latter was used to control for increases in resting blood flow with α-adrenergic blockade. Spike-triggered averaging was used to characterize beat-by-beat changes in FVC for 15 cardiac cycles following each MSNA burst and a peak response was calculated. Following MSNA bursts, FVC initially increased by +3.3 ± 0.3% (P= 0.016) and then robustly decreased to a nadir of −5.8 ± 1.6% (P < 0.001). The magnitude of vasoconstriction appeared graded with the number of consecutive MSNA bursts; while individual burst size only had a mild influence. Neither PHEN nor PHEN+ANG infusions affected the initial rise in FVC, but both infusions significantly attenuated the subsequent decrease in FVC (–2.1 ± 0.7% and –0.7 ± 0.8%, respectively; P < 0.001 vs. normal saline). These findings indicate that spontaneous MSNA bursts evoke robust beat-by-beat decreases in FVC that are exclusively mediated via α-adrenergic receptors.
Key points
Sympathetic support of blood pressure demands the efficient control of vascular tone; however, little is known regarding how spontaneously occurring bursts of muscle sympathetic nerve activity (MSNA) dynamically influence forearm vascular conductance.
This study examined the extent to which spontaneous MSNA bursts evoke changes in forearm vascular conductance and blood pressure with and without local α-adrenergic blockade in young healthy men during supine rest.
We observed that under resting conditions, forearm vascular conductance increases briefly and then significantly decreases in association with the total amount of the preceding MSNA; however, during α-adrenergic blockade the decrease in vascular conductance is eliminated.
These results indicate that normal variations in spontaneous MSNA burst activity are systematically followed by transient and robust responses of forearm vasoconstriction and that this influence is mediated via α-adrenergic receptor mechanisms.
Introduction
Peripheral vascular responses to changes in muscle sympathetic nerve activity (MSNA) are critical for the effective regulation of arterial blood pressure (Fadel, 2008; Kiviniemi et al. 2011). Numerous studies have demonstrated that the human forearm vasculature significantly constricts in response to robust elevations in reflex-mediated MSNA (Lundvall & Edfeldt, 1994; Davy et al. 1998; Minson et al. 2000; Ray & Monahan, 2002). However, to our knowledge, no information is available describing the influence of spontaneously occurring MSNA on forearm vasoconstrictor responses. Unquestionably, sympathetic vasoconstriction at rest has an important role in forearm vascular tone because local infusion of phentolamine (a non-specific α-adrenoreceptor antagonist) significantly increases vascular conductance (Dinenno et al. 2002; Johansson et al. 2002). However, MSNA during normal resting conditions is not ‘all or none’; instead there is wide variability in both burst pattern and amplitude among young healthy individuals (Sundlof & Wallin, 1977; Fagius & Wallin, 1993; Joyner et al. 2010). Thus, it is still not known whether spontaneous oscillations in resting MSNA are able to induce consistent and meaningful changes in forearm vascular conductance (FVC).
A potential impediment to the ability of spontaneously occurring MSNA bursts to induce vasoconstriction is the complex milieu of vasoactive compounds acting to regulate resting vascular tone in the forearm circulation (Vanhoutte et al. 1981). In addition to adrenergic mechanisms (Dinenno et al. 2002), forearm basal tone depends upon the equilibrium of influences from vasoconstrictor substances such as angiotensin II (ANGII) (Saris et al. 2000), vasopressin and endothelin 1 (Cardillo et al. 1999) as well as vasodilator substances such as nitric oxide (Rosenmeier et al. 2003) and endothelium-derived hyperpolarizing factors (Ozkor et al. 2011). Importantly, vasodilator mechanisms have been shown to interact with varying degrees of sympathetic nerve activation to attenuate its normal vasoconstrictor effects (Kurjiaka & Segal, 1995). This interaction may be particularly influential for vascular beds such as the forearm, which has been shown to exhibit preferential responsiveness to vasodilator compounds (Newcomer et al. 2004). However, whether basal vasoactive mediators offset the potential vasoconstriction of spontaneously occurring MSNA bursts has not been tested under resting conditions.
With this background in mind, the purpose of the current study was to comprehensively examine the influence of spontaneous MSNA bursts on forearm vasculature. To do this, we first examined beat-by-beat sympathetic vascular transduction at rest in the forearm, as characterized by the spike-triggered changes in FVC following spontaneously occurring MSNA bursts. We then determined the extent to which local α-adrenergic receptor-mediated pathways contribute to observed FVC responses.
Methods
Subjects
A total of 22 young healthy men (age; 25 ± 1) volunteered to participate in two separate experimental protocols (19 subjects in protocol 1; seven subjects in protocol 2). Four subjects participated in both protocols. Subjects provided written informed consent and underwent a standard screening, including a health history questionnaire. All participants were healthy, non-obese, non-smokers and were not taking any medications. All subjects were recreationally active, engaging in low to moderate physical activity and none were competitively training. All study protocols were approved by the University of Missouri Health Sciences Institutional Review Board and were performed according to the Declaration of Helsinki. Subjects were instructed to report to the laboratory after an overnight fast and refrain for at least 12 h from exercise and caffeine.
General experimental measurements
Beat-by-beat changes of arterial blood pressure were measured from the left middle finger using a servo-controlled finger photoplethysmograph (Finometer; Finapres Medical Systems, Amsterdam, the Netherlands) with absolute values verified using an automated sphygmomanometer (Welch Allyn, Skaneatles Falls, NY, USA). Heart rate was measured via a lead II electrocardiogram (ECG; Quinton Q710, Bothell, WA, USA). Respiratory movements were detected with a strain-gauge pneumograph secured to the abdomen (Pneumotrace; UFI, Morro Bay, CA, USA).
Multiunit postganglionic MSNA was recorded using standard microneurographic techniques, as previously described (Vallbo et al. 1979; Padilla et al. 2010; Vianna et al. 2012; Fairfax et al. 2013). Briefly, a tungsten microelectrode was placed into the peroneal nerve near the left fibular head. Signals were amplified, filtered (bandwidth; 0.7–2.0 kHz), rectified and integrated (0.1 s time constant) to obtain mean voltage neurograms. MSNA was identified by the presence of spontaneous bursts with characteristic pulse synchronicity and by its responsiveness to end-expiratory breath holds, but not to arousal or skin stimulation. Previous studies have demonstrated synchronous outflow of MSNA burst activity throughout the skeletal muscle vasculature, being equivalent between contralateral limbs (Wallin et al. 1994) as well as between the arm and leg (Rea & Wallin, 1989; Lott et al. 2009). MSNA was quantified over the entire recording period as burst frequency (bursts min−1) and burst incidence (bursts·100 cardiac cycles−1). In addition, to account for variations in burst height, MSNA burst amplitudes were expressed as a percentage of the three largest bursts (assigned a mean value of 100 arbitrary units; AU) during the recording.
Brachial artery diameter and blood velocity were measured via duplex Doppler ultrasound, as previously described in detail by our laboratory (Padilla et al. 2011; Simmons et al. 2011). Briefly, the brachial artery was imaged longitudinally in the distal third of the upper arm with a high-resolution ultrasound system (Logiq P5; GE Medical Systems, Milwaukee, WI, USA). Diameter and velocity were continuously and simultaneously measured using a 12 MHz linear array transducer probe in pulsed-wave mode operating at a frequency of 5 MHz and insonation angle of 60°. Measurements were performed with the sample volume encompassing the entire vessel lumen, but not extending beyond it. To ensure acquisition of stationary images, the arm was secured at heart level over a vacuum pillow and the transducer was stabilized using a custom-designed clamp. Skin markings were used to verify that transducer placement was kept constant throughout the study.
Experimental protocols
Protocol 1: Characterization of forearm vascular conductance responses following muscle sympathetic nerve activity bursts
To determine the extent to which spontaneous bursts of MSNA evoke changes in FVC, all experimental measurements were continuously recorded during supine rest in 19 young healthy men. Before initiation of data collection, all measures were monitored for 10 min to ensure stability and suitability for analysis. Subsequently, all measurements were continuously recorded for 20–35 min, while the subject lay resting quiet and awake to determine the spike-triggered average changes following spontaneously occurring MSNA bursts.
Protocol 2: Role of α-adrenergic receptors in mediating forearm vascular conductance responses
To determine the α-adrenergic receptor contribution to changes in FVC following spontaneous MSNA bursts, experimental measurements were repeated in seven men during three sequential 20 min steady-state intra-arterial infusion conditions: Saline control (SAL); phentolamine (PHEN), to produce α-adrenergic blockade; and co-infusion of PHEN and ANGII (PHEN+ANGII), to produce α-adrenergic blockade and control for the elevation in resting blood flow from PHEN alone.
For this protocol, a 20 gauge, 5 cm (Model RA-04020, Arrow International, Reading, PA, USA) catheter was placed into the brachial artery of the non-dominant arm under aseptic conditions following local anaesthesia (2% lidocaine). The catheter was coupled to a three-port connector system: one port was linked to a pressure transducer (Deltran II, Utah Medical, Midvale, UT, USA; BP Amp, ADInstruments, Colorado Springs, CO, USA), and the remaining two ports were used for drug administration. An additional 30 min recovery period was provided following catheterization to permit full restoration of resting cardiovascular variables before initiating data collection. A continuous 2 ml h−1 SAL flush ensured patency of the arterial catheter line.
Before commencement of steady-state infusions, each subject's responsiveness to noradrenaline (Hospira, Lake Forest, IL, USA) at 20, 40 and 80 ng dl−1 min−1 was assessed to determine an adequate dosage to challenge α-adrenergic blockade. In addition, ANGII responsiveness to 6.25, 12.5 and 25 ng dl−1 min−1 was also assessed to approximate the dose needed to restore forearm blood flow (FBF) to resting values after PHEN infusion. Drugs were infused intra-arterially for 2 min per dose at increasing rates until the decrease in FBF reached a plateau. Drug administration was normalized per dl of forearm volume (12.4 ± 0.5 dl), as determined on a previous day via dual-energy X-ray absorptometry (Hologic QDR 4500A, Bedford, MA, USA). In most cases, the highest dose was not used due to robust vasoconstriction to middle doses of noradrenaline and ANGII. A minimum 10 min recovery period was used between each drug administration to allow for restoration of resting FBF.
Following dose–responses, MSNA was acquired and the extent to which spontaneous bursts of MSNA evoke changes in FVC was assessed during sequential 20 min steady-state infusions. Normal (0.9%) SAL was infused at a 2 ml min−1 infusion rate and was always performed first due to the >2 h elimination half-life of PHEN (Moore et al. 2008). Subsequently, PHEN (Oraverse, Septodont, Louisville, CO, USA) was infused for an initial 5 min period at 10 μg dl−1 min−1 to induce α-adrenergic blockade, after which the dose was decreased to a maintenance level of 5 μg dl−1 min−1 for the duration of the PHEN recording period (Casey et al. 2012; Barrett-O’Keefe et al. 2013). At the end of PHEN infusion, α-adrenoreceptor blockade was challenged with a 40 ng dl−1 min−1 noradrenaline infusion for 2 min. Finally, ANGII was co-infused at 12.5 ng dl−1 min−1 with PHEN (continued at 5 μg dl−1 min−1) and measurements began once FBF decreased to values similar to SAL.
Data analysis
Longitudinal Doppler ultrasound brachial artery images with corresponding blood velocity waveforms were acquired at 30 Hz into a custom Labview program. The video output (640 × 480 pixels) of the Doppler ultrasound machine was transmitted through a PCI-1411 video card (National Instruments, Austin, TX, USA). These video images from the ultrasound machine were embedded into an AVI file as data streams (33 points per image) together with ECG trigger pulses, arterial blood pressure waveforms and MSNA. Custom-designed edge detection and wall tracking software (LabVIEW; National Instruments), previously validated and described in detail (Padilla et al. 2011; Simmons et al. 2011; Fairfax et al. 2013), were used to determine beat-by-beat arterial diameters and Doppler velocities offline. Diameter and blood velocity measurements were used to calculate FBF as Vm·π·((D/2)2)·60 and FVC as FBF/MAP (ml min−1 mmHg−1); where Vm is mean blood velocity (cm s−1), D is arterial diameter (cm) and MAP is mean arterial pressure (mmHg).
Stroke volume values were estimated using Modelflow® software (Dyson et al. 2010; Kim et al. 2012), and aligned with LabVIEW program output via changes in cardiac interval. Cardiac output (CO) was estimated as the product of heart rate and stroke volume. Total vascular conductance (TVC) was calculated as CO divided by MAP. MAP was calculated as the integral of the arterial blood pressure waveform.
To characterize beat-by-beat sympathetic vascular transduction, spike-triggered averaging was used as previously described by our laboratory (Vianna et al. 2012; Fairfax et al. 2013). Briefly, for the 15 cardiac cycles following each MSNA burst, the percentage change in FVC (relative to the value of FVC during the burst) was calculated. Each MSNA burst was examined regardless of its proximity to other bursts. The average beat-by-beat percentage changes in FVC following all MSNA bursts were assessed for each subject and group mean changes were then determined. The peak or nadir of this mean response was used to provide an estimate of the overall vascular response, while the entire 15 heartbeat period was characterized beat-by-beat to provide a more detailed evaluation of the time course and magnitude of the responses. Conversely, to assess FVC responses in the absence of MSNA, the average beat-by-beat percentage changes in FVC following each heartbeat without an MSNA burst (i.e. non-bursts) was also assessed. These analyses were also performed on systemic cardiovascular variables: MAP, CO and TVC.
Because spontaneous MSNA bursts may occur in isolation or in consecutive sequence with other MSNA bursts, we examined the effect of burst patterning on the magnitude of FVC response. All MSNA bursts were segregated into four categories, each separated by ≥1 heartbeats lacking MSNA: singlet bursts (any isolated burst), couplet bursts (consecutive series of two bursts), triplet bursts (three consecutive bursts) and quadruplet bursts (any consecutive group of ≥4 bursts). The percentage beat-by-beat changes in FVC were calculated following every burst belonging to a given pattern and average responses were determined for each pattern. Additionally, the effect of normalized burst size on FVC response magnitude was determined. For this analysis, MSNA bursts were ranked by height and divided equally into four quartiles (Q1–Q4; smallest to largest), independent of burst pattern. The average percentage changes in FVC following every MSNA burst of a particular quartile were then calculated for each subject and a group mean was determined.
To assess the combined effect of varied burst pattern and size together, spontaneous MSNA was analysed as clusters of activity (Fairfax et al. 2013). Burst clusters were defined as any bursting activity separated on each side by ≥1 cardiac cycle(s) lacking MSNA. Clusters were first segregated into four groups, similar to burst patterns: singlet clusters (one isolated burst), couplet clusters (two consecutive bursts), triplet clusters (three consecutive bursts) and quadruplet clusters (≥4 consecutive bursts). Next, all clusters in a group were ranked by their sum of burst heights and then divided equally into four quartiles. Accordingly, a total of 16 MSNA cluster categories were assigned (four cluster groups, four quartiles each). Each cluster was considered a single event and thus, percentage changes in FVC were calculated from the first burst of each cluster (not the average change following each burst in the cluster).
To remove any systematic relationship with MSNA occurrence, a white noise sampling of randomly selected cardiac cycles was followed for 15 heartbeats to calculate percentage changes in FVC. The number of cardiac cycles chosen for white noise was matched to each subject's MSNA burst count, keeping the number of observations consistent within a subject.
Statistical analysis
Statistical analyses were performed using Sigmastat (version 3.0). Comparisons between burst pattern, size and cluster amplitude were considered using two-way repeated-measures ANOVA to test for differences between cardiac cycles, between conditions and for interaction between condition and cardiac cycles. A one-way ANOVA tested for differences between peak responses and between mean steady-state values among infusion conditions. Pearson correlations assessed the relationship between nadir changes in FVC and total amplitude of each MSNA burst cluster. Post hoc differences were determined using the Student–Neuman–Keuls test, comparing responses against the white noise control changes when appropriate. Significance was set a priori at P < 0.05 and data are expressed as means ±s.e.
Results
Protocol 1: Characterization of forearm vascular conductance responses following muscle sympathetic nerve activity bursts
Table 1 presents average values for cardiovascular, MSNA and brachial artery measurements during the entire resting period. Information regarding the number and distribution of individual MSNA bursts and MSNA clusters used for spike-triggered averaging over the same period is provided in Table 2.
Table 1.
Resting average neurocardiovascular measurements (protocol 1)
| Cardiovascular measurements | Vascular and neural measurements | ||
|---|---|---|---|
| SBP (mmHg) | 125 ± 3 | FBF (ml min−1) | 109.4 ± 10.3 |
| DBP (mmHg) | 71 ± 2 | FVC (ml min−1 mmHg−1) | 1.22 ± 0.10 |
| MAP (mmHg) | 90 ± 2 | Burst frequency, | 18 ± 2 |
| HR (bpm) | 60 ± 2 | Bursts min−1 | |
| CO (l min−1) | 6.7 ± 0.3 | Burst incidence, | 30 ± 3 |
| TVC (ml min−1 mmHg−1) | 74.7 ± 3.8 | Bursts 100 heart beats−1 | |
Values are means ±s.e. CO, cardiac output; DBP, diastolic blood pressure; FBF, forearm blood flow; FVC, forearm vascular conductance; HR, heart rate; MAP, mean arterial blood pressure; SBP, systolic blood pressure; TVC, total vascular conductance.
Table 2.
Muscle sympathetic nerve activity burst distribution (protocol 1)
| Count | Range | Percentage | |
|---|---|---|---|
| All bursts | 408 ± 49 | (75–881) | 100 ± 0% |
| Singlet bursts | 169 ± 21 | (58–406) | 45 ± 4% |
| Couplet bursts | 132 ± 16 | (16–322) | 32 ± 2% |
| Triplet bursts | 57 ± 12 | (0–237) | 13 ± 2% |
| Quadruplet bursts | 50 ± 17 | (0–325) | 10 ± 2% |
| Singlet clusters | 169 ± 21 | (58–406) | 59 ± 3% |
| Couplet clusters | 66 ± 8 | (8–161) | 29 ± 2% |
| Triplet clusters | 19 ± 4 | (0–79) | 9 ± 1% |
| Quadruplet clusters | 10 ± 3 | (0-65) | 4 + 1% |
Values are means ±s.e.
Effect of muscle sympathetic nerve activity burst occurrence on forearm vascular conductance
Figure 1 presents beat-by-beat percentage changes in FVC following: all MSNA bursts; cardiac cycles lacking MSNA; and randomly selected white noise. Interestingly, during the first two heartbeats following all MSNA bursts, FVC significantly increased (+1.32 ± 0.31%, P < 0.05). However, after this rise, FVC significantly decreased, changing from 1.51 ± 0.20 ml min−1 mmHg−1 to a subsequent nadir of 1.44 ± 0.19, representing a −4.3 ± 0.8% reduction following all MSNA bursts (P < 0.001). In contrast, cardiac cycles without MSNA (non-bursts) were followed by a delayed and significant increase in FVC, changing from 1.41 ± 0.17 ml min−1 mmHg−1 to a subsequent peak of 1.43 ± 0.17; a +1.8 ± 0.4% peak increase (P < 0.001). Importantly, following the randomly selected white noise, FVC demonstrated only inconsistent and insignificant (P > 0.05) changes.
Figure 1. Summary data of beat-by-beat percentage changes in forearm vascular conductance following all muscle sympathetic nerve activity bursts (filled circles), non-bursts (filled triangles) and white noise (open squares).
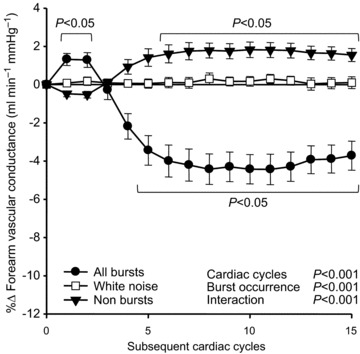
Brackets denote significant difference from percentage changes in white noise. Values are means ±s.e.
Effect of muscle sympathetic nerve activity burst pattern on forearm vascular conductance
Figure 2A displays beat-by-beat percentage changes in FVC following four MSNA burst patterns to account for variations in consecutive burst number. FVC following singlet bursts did not significantly decrease (P= 0.28). Couplet bursts were followed by a significant decrease in FVC, changing from 1.52 ± 0.21 ml min−1 mmHg−1 to 1.45 ± 0.21, and representing a −4.6 ± 1.3% nadir (P < 0.05). FVC also significantly decreased following triplet bursts, reducing from 1.65 ± 0.23 ml min−1 mmHg−1 to 1.52 ± 0.25; a nadir of −9.5 ± 1.6% (P < 0.05). Compared to triplet bursts, quadruplet bursts (≥4 consecutive bursts) were associated with a similar nadir decrease in FVC of −8.0 ± 1.4% (P= 0.76 vs. triplet). Overall, the influence of MSNA burst patterning was graded with FVC nadir magnitudes in that longer (triplet and quadruplet) patterns were associated with significantly greater FVC decreases relative to shorter (both singlet and couplet) patterns (Fig. 3).
Figure 2. Summary data of beat-by-beat percentage changes in forearm vascular conductance (FVC) following variations in spontaneous muscle sympathetic nerve activity (MSNA) burst pattern and size.
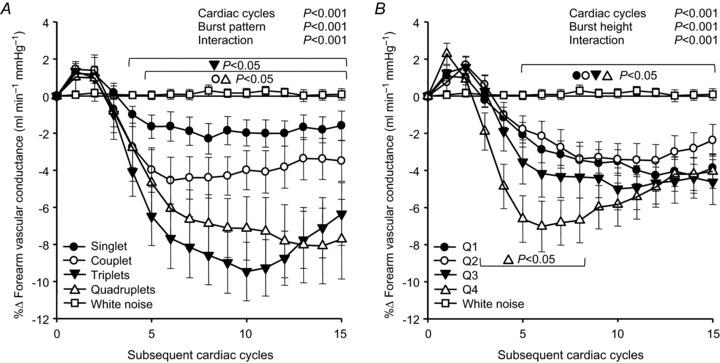
A, changes in FVC following consecutive MSNA bursts of increasing length (singlet, couplet, triplet and quadruplet). B, changes in FVC following MSNA bursts divided into quartiles of increasing size (Q1–Q4, respectively). White noise changes are provided for reference (open squares). Brackets denote significant difference from percentage changes in white noise. Values are means ±s.e.
Figure 3. Group data showing forearm vascular conductance nadir responses following variations in spontaneous MSNA burst pattern (left side; grey bars) and size (right side; white bars).
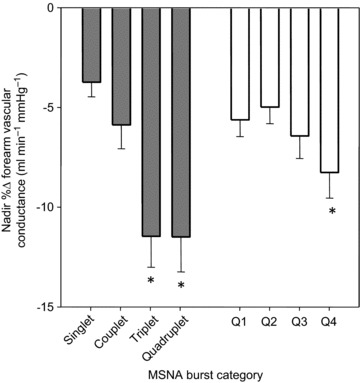
*P < 0.05 vs. singlet patterns or Q1 bursts, respectively. Values are means ±s.e. MSNA, muscle sympathetic nerve activity.
Effect of muscle sympathetic nerve activity burst size on forearm vascular conductance
Figure 2B demonstrates beat-by-beat changes in FVC following four quartiles of MSNA burst size to assess the influence of individual burst amplitudes on FVC responses, independent of the influence from patterns. All quartiles of MSNA burst height were associated with significant decreases in FVC (P < 0.05). FVC following the smallest 25% of all MSNA bursts (Q1) decreased to a −4.3 ± 0.8% nadir (P < 0.001). FVC changes following Q2 and Q3 MSNA bursts were similar to Q1, resulting in nadirs of −3.5 ± 0.9% and −5.0 ± 1.1%, respectively. The largest 25% of all MSNA bursts were followed by the greatest decreases in FVC, exhibiting a significantly greater reduction (−7.0 ± 1.3%; P < 0.05) than Q2 and Q1 MSNA bursts (Fig. 3).
Effect of muscle sympathetic nerve activity bursts on systemic cardiovascular variables
To evaluate the broad influences of MSNA, we additionally evaluated spike-triggered changes in systemic cardiovascular variables following MSNA bursts. Figure 4 demonstrates the average beat-by-beat changes in MAP, CO and TVC following all MSNA bursts. MAP gradually increased following all MSNA bursts, reaching a peak of +3.1 ± 0.4% (P < 0.05). This was associated with a brief rise in CO, attaining a +1.2 ± 0.3% peak (P < 0.05) during the second cardiac cycle and then quickly returning to baseline. Subsequently, TVC substantially decreased following MSNA bursts, reaching a nadir of –3.5 ± 0.4% (P < 0.05), which occurred in association with the peak increase of MAP. Similar to FVC, following non-bursting cardiac cycles TVC and MAP significantly increased exhibiting directionally opposite responses compared to those after MSNA bursts (data not shown). Additionally, no significant changes in any systemic cardiovascular variable were observed following the white noise control.
Figure 4. Summary data of beat-by-beat percentage changes in MAP (filled circles), cardiac output (open circles) and total vascular conductance (filled triangles) following spontaneous muscle sympathetic nerve activity bursts.
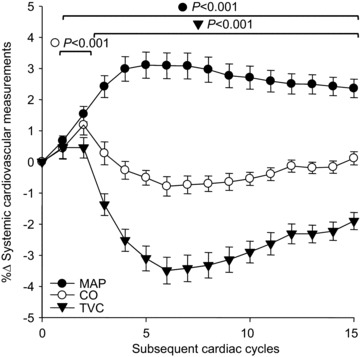
Brackets denote significant difference from percentage changes in each variable's respective white noise (not shown). Values are means ±s.e. CO, cardiac output; MAP, mean arterial pressure; TVC, total vascular conductance.
Protocol 2: Role of α-adrenergic receptors in mediating forearm vascular conductance responses
Table 3 displays the average cardiovascular measurements for the entire duration of each forearm infusion condition. MSNA burst frequency, burst incidence, and burst number and distribution were similar among infusion conditions and protocol 1. As expected, PHEN infusion significantly increased FBF and FVC (P < 0.05) compared to SAL infusion without affecting MAP or heart rate (P > 0.05). Noradrenaline challenge at the end of PHEN infusion did not significantly lower FBF (216 ± 37 ml min−1 vs. 203 ± 30; −4 ± 3%, P > 0.05), whereas the same dose of noradrenaline before PHEN infusion substantially lowered FBF (132 ± 26 ml min−1 vs. 44 ± 19; −70 ± 3%, P < 0.05), confirming the achievement of α-adrenergic blockade. Additionally, co-infusion of PHEN+ANGII successfully returned resting FBF close to that measured during SAL infusion (P > 0.05). Hence, our primary experimental condition (PHEN+ANGII) successfully matched the resting FBF of our control condition and abolished α-adrenergic receptor function. Similar conclusions were reached when comparing ANOVA results between SAL vs. PHEN and SAL vs. PHEN+ANGII. Thus, only PHEN+ANGII results are presented to avoid redundancy and provide the best physiological comparison to SAL.
Table 3.
Average neurocardiovascular measurements during forearm infusions (protocol 2)
| SAL | PHEN | PHEN + ANGII | |
|---|---|---|---|
| MAP (mmHg) | 93 ± 4 | 95 ± 4 | 100 ± 5 |
| HR (bpm) | 61 ± 3 | 64 ± 2 | 65 ± 2 |
| TVC (ml min−1 mmHg−1) | 69 ± 6 | 74 ± 5 | 71 ± 8 |
| FBF (ml min−1) | 110.9 ± 13.8 | 251.3 ± 33.9* | 149.7 ± 16.5 |
| FVC (ml min−1 mmHg−1) | 1.17 ± 0.11 | 2.59 ± 0.30* | 1.51 ± 0.16 |
| Burst frequency (bursts min−1) | 18 ± 3 | 20 ± 2 | 21 ± 2 |
| Burst incidence (Bursts 100 heart beats−1) | 30 ± 6 | 32 ± 4 | 33 ± 4 |
Values are means ±s.e. *P < 0.05 vs. SAL. FBF, forearm blood flow; FVC, forearm vascular conductance; HR, heart rate; MAP, mean arterial pressure; SAL, saline infusion; TVC, total vascular conductance.
Effect of α-adrenergic blockade on sympathetic vascular transduction
Figure 5 shows the beat-by-beat changes in FVC following: all MSNA bursts; cardiac cycles without MSNA; and white noise during SAL and PHEN+ANGII infusions. FVC significantly decreased following all MSNA bursts during SAL infusion (Fig. 5A), reaching a nadir of −5.8 ± 1.6% (P < 0.05). In comparison, during PHEN+ANGII infusion the decline of FVC following all MSNA bursts was eliminated (Fig. 5B), resulting in an insignificant –0.6 ± 0.8% nadir (P>0.05). A significant attenuation of these responses by PHEN+ANGII was also demonstrated when examined by variations in burst pattern and burst size (Fig. 6: P < 0.05). Interestingly, the early and significant increase in FVC (+3.3 ± 0.3%) observed during the first few cardiac cycles following all variations of MSNA during SAL infusion was unaffected by PHEN+ANGII (+3.2 ± 0.4%; P>0.05 vs. SAL). To characterize fully the influence of spontaneous MSNA bursts on FVC responses, the linear relationship between nadir changes in FVC following various MSNA burst clusters and the total amplitude contained within each corresponding cluster was compared during SAL and PHEN+ANGII infusions (Fig. 7). During SAL infusion, individual correlations between nadir FVC responses and total cluster size indicated a significant negative linear relationship (r=–0.38 ± 0.8; P < 0.05) that was abolished (r=+0.06 ± 0.2; P>0.05) by PHEN+ANGII infusion.
Figure 5. Summary data of beat-by-beat percentage changes in forearm vascular conductance following all spontaneous muscle sympathetic nerve activity bursts, non-bursts and white noise during saline infusion (A), and during phentolamine+angiotensin II co-infusion (α-adrenergic blockade; B).
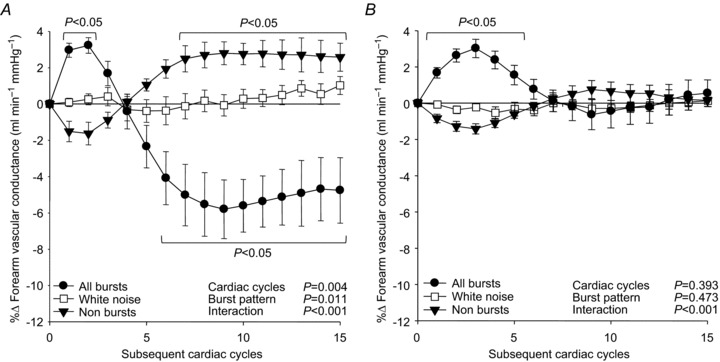
Brackets denote significant differences from percentage changes in white noise. Values are means ±s.e.
Figure 6. Group data during forearm infusions showing forearm vascular conductance peak and nadir responses following variations in burst pattern (left side, grey bars) and size (right side, white bars).
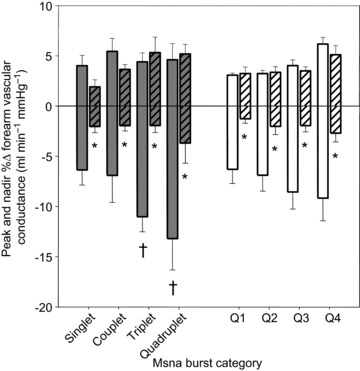
Saline infusion responses are shown as solid bars, and phentolamine+angiotensin II co-infusion responses are shown with hatched bars. *P < 0.05 vs. saline, †P < 0.05 vs. singlet patterns. Values are means ±s.e. MSNA, muscle sympathetic nerve activity.
Figure 7. Group data during forearm infusions showing the regression between MSNA burst clusters and nadir percentage changes in forearm vascular conductance.
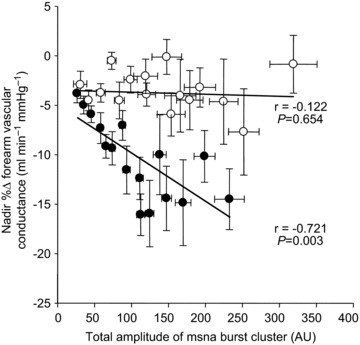
Filled circles show forearm vascular conductance responses during saline infusion, and open circles show responses during phentolamine+angiotensin II co-infusion (α-adrenergic blockade). Correlations displayed are derived from group means. Values are means ±s.e. MSNA, muscle sympathetic nerve activity.
Discussion
There are several novel findings from the current study. First, we demonstrated that spontaneously occurring MSNA bursts are systematically followed by biphasic changes in FVC as characterized by an immediate, brief and mild increase and then subsequently a robust decrease. The magnitude of the vasoconstrictor portion appeared graded with the preceding MSNA burst pattern (number of consecutive bursts) and cluster size (sum of consecutive MSNA activity), while burst size (height of individual bursts) appeared to only mildly influence the degree of vasoconstriction. Second, the decreases in FVC following spontaneous MSNA bursts were abolished by α-adrenergic blockade. Interestingly, the vasodilator portion of the biphasic response was unchanged with α-adrenergic blockade, indicating that α-adrenergic receptor function has no role in this particular element of the response. Lastly, FVC systematically rose following heartbeats without MSNA, further highlighting the temporal coupling of spontaneous MSNA and changes in vascular tone. Collectively, these results demonstrate that oscillations in spontaneous MSNA exert graded forearm vasoconstriction via α-adrenergic receptors in healthy young men.
We initially suspected that the presence of competing vasodilators and greater forearm vasodilator responsiveness (Newcomer et al. 2004) in combination with relatively modest responsiveness to α-adrenergic agonists (Pawelczyk & Levine, 2002) compared to the leg might obscure the ability of spontaneous MSNA bursts to evoke measurable vasoconstriction in the forearm. Although the degree of vasoconstriction following MSNA in the forearm was less than previously demonstrated in the leg (Fairfax et al. 2013), we observed significant forearm vasoconstrictor responses as well as efficient modulation of the magnitude of vasoconstriction by the natural variability in MSNA. Importantly, we also demonstrated that local α-adrenergic receptor function was necessary to mediate the decreases in FVC following spontaneous MSNA bursts. This strongly suggests that increased transmission of noradrenaline across the synaptic cleft causes the grading of FVC responses observed with changes in MSNA burst pattern, size and clustering. We speculate that each of these bursting variants are unique features by which the central nervous system can modulate the rate and amount of noradrenaline released (Esler et al. 1985; Kahan et al. 1988; Ninomiya et al. 1993) to permit in turn a variety of durations and strengths of ensuing vascular responses. Interestingly, burst size appeared to be the mildest effector of the degree of vasoconstriction with only the largest quartile (biggest MSNA bursts) resulting in greater vasoconstriction (Fig. 3). This result is in contrast to the more graded effect of burst size in a previous study examining beat-by-beat sympathetic vascular transduction in the leg (Fairfax et al. 2013). The reason for these differences is unclear but may have to do with the lower α-adrenergic receptor sensitivity in the arm compared to the leg (Pawelczyk & Levine, 2002). In this regard, it is plausible that it is not until α-receptors are exposed to a greater noradrenaline release associated with the largest bursts of MSNA that ensuing forearm vasoconstriction is augmented.
The white noise control condition strengthens our main findings by capitalizing on the specificity of spike-triggered averaging. Indeed, when the same data set was re-evaluated by scrambling the position of each white noise ‘burst’, we never identified any consistent changes in FVC, substantiating the significant responses observed following actual MSNA bursts. However, from the current data set, we cannot completely rule out the possibility that background vasodilator compounds may be restraining the magnitude of the observed forearm vasoconstrictor responses to spontaneous MSNA bursts. Although this question is of interest, it is beyond the scope of this study. Indeed, the purpose of our study was to examine the vasoconstrictor responses to unprovoked MSNA within the ‘normal’ milieu of vasoactive compounds in the forearm.
The early ‘dilator’ portion of the biphasic FVC response is intriguing because to our knowledge, there is no precedent for a systematic vasodilation to occur in synchrony with MSNA bursts in humans. Furthermore, this element of the vascular response is essentially absent from our previously published work in the leg (Fairfax et al. 2013). Nevertheless, its consistency and magnitude suggests that a genuine physiological event underlies this phenomenon. Although the exact mechanism for this unexpected early rise in FVC is unknown, there are several possible factors that warrant discussion.
(1) Arterial blood pressure has been shown to decrease ∼2 s before isolated MSNA bursts (Diedrich et al. 2013), providing a stimulus not only for the arterial baroreceptors to evoke MSNA bursts, but also for a possible myogenic dilation to occur in forearm vascular smooth muscle (Davis, 2012). Because myogenic responses occur much faster than adrenergic vasoconstriction, the fall in arterial blood pressure may set in motion myogenic vasodilation to produce the immediate rise in FVC. Subsequently, following a brief latency of two to three heartbeats to allow α-adrenergic receptor activation, ensuing vasoconstriction leads to a robust lowering of FVC.
(2) It is also plausible, despite the lesser affinity for β- versusα-adrenergic receptors, that the noradrenaline released with spontaneous MSNA bursts results in β-adrenergic vasodilation (Hart et al. 2011); however, the rapidity of the increase in FVC makes this unlikely.
(3) The early elevation in cardiac output may also contribute (Fig. 4), but as a prominent rise was not observed in the femoral artery following MSNA bursts this possibility is less likely (Fairfax et al. 2013).
(4) Likewise, although the inherent delay of transducing sympathetic action potentials into a vascular response may allow vascular conductance to increase before it decreases, this delay would also be expected to cause a rise in femoral artery conductance after MSNA bursts but was not present.
(5) Lastly, it is possible that a still unknown stimulus may activate vasodilator pathways in synchrony with MSNA bursts. Nevertheless, despite the number of possible mechanisms regarding the immediate dilator portion of the FVC response, we can conclude with certainty that α-adrenergic mechanisms are not involved because this element remained unchanged following α-adrenergic blockade.
The differences in our current findings of beat-by-beat sympathetic vasoconstriction in the forearm compared to our previous observation of leg vascular conductance following MSNA bursts (Fairfax et al. 2013) warrants consideration. First, in the forearm, a biphasic response pattern was consistently observed following all variations of MSNA, whereas this was not observed in the leg. This complex morphology of FVC changes required α-adrenergic blockade to determine if it was of sympathetic origin. We found that the decrease in FVC following MSNA bursts was completely due to α-adrenergic mechanisms; whereas the initial rise was unaffected. Thus, we provide novel evidence demonstrating, for the first time, heterogeneity in the beat-by-beat vascular conductance responses after MSNA bursts between limbs. Furthermore, the peak decrease in FVC following all MSNA bursts reported in the present study is significantly less than the decrease in leg vascular conductance after MSNA bursts (Fairfax et al. 2013) suggesting that beat-by-beat sympathetic vasoconstrictor responsiveness differs between limbs. These findings are in agreement with previous work using steady-state pharmacological interventions (Pawelczyk & Levine, 2002). Overall, our findings demonstrate that vascular responses to spontaneous MSNA bursts in the arm cannot be extrapolated to the leg and vice versa.
Another important finding of our study was that FVC consistently increased after non-bursting cardiac cycles: illustrating the significance of MSNA bursts for maintaining forearm vascular tone. Indeed, rather than demonstrating a blunted or absent vasoconstrictor response, a robust vasodilation was observed. Interestingly, the increase in FVC following non-bursts was abolished with local α-adrenergic blockade, suggesting that α-adrenergic function is an important contribution to this phenomenon. In association with FVC, TVC and MAP also increased following non-bursts (data not shown). Together, these results support a role for resting MSNA in the short-term maintenance of vascular tone and blood pressure. Indeed, even among young healthy men during supine rest, perhaps the model least demanding of sympathetic support, it appears that recurring MSNA is essential to maintain cardiovascular homeostasis. It is probable that any increased reliance on sympathetic mechanisms, such as augmented autonomic support with healthy ageing (Jones et al. 2001) or during adaptation to cardiovascular stress (Jordan et al. 2002), would manifest with an amplified increase in FVC following non-bursts. Previous evidence from our laboratory (Vianna et al. 2012) demonstrating augmented TVC increases following non-bursting heart beats in healthy older individuals lends support to this proposition. In this regard, the assessment of signal-averaged MSNA bursts and non-bursts provides a practical non-invasive index of sympathetic support of vascular tone and blood pressure for studies in healthy and diseased populations.
In summary, we have determined that FVC changes to spontaneously occurring MSNA bursts follow a biphasic response characterized by an immediate, brief and mild increase and then subsequently a robust decrease. The decreases following spontaneous MSNA bursts were mediated by α-adrenergic receptor mechanisms, providing physiological insight into beat-to-beat sympathetic vasoconstrictor action under basal conditions. These sympathetically mediated forearm vasoconstriction responses occur in synchrony with systemic cardiovascular changes, indicating that skeletal muscle vascular responses are integral to the regulation of resting cardiovascular haemodynamics. However, beat-by-beat vascular conductance responses after MSNA bursts are heterogeneous between limbs with biphasic changes and smaller decreases in the arm compared to the leg (Fairfax et al. 2013). Collectively, these results indicate that spontaneous MSNA bursts are capable of evoking robust beat-by-beat α-adrenergic receptor-mediated decreases in FVC that are graded to the natural variations in MSNA.
Acknowledgments
The time and effort expended by all the volunteer subjects is greatly appreciated. The authors thank Dr Frank Dinenno and his lab group for insight during the development of the arterial catheter-based study. This research was submitted in partial fulfilment of the requirements for the degree of Doctor of Philosophy for Seth T. Fairfax.
Glossary
- ANGII
angiotensin II
- CO
cardiac output
- D
Diameter
- ECG
electrocardiogram
- FBF
forearm blood flow
- FVC
forearm vascular conductance
- MAP
mean arterial pressure
- MSNA
muscle sympathetic nerve activity
- PHEN
phentolamine
- SAL
normal saline
- TVC
total vascular conductance
Additional information
Competing interests
None declared.
Author contributions
All experiments were performed at the University of Missouri, Columbia. S.T.F. completed all data analyses, figure preparation as well as contributing to all other aspects of this work. S.W.H. contributed to all data collection. D.P.C. and M.Y.Z. contributed to data collection of protocol 2. J.H.M. and P.C.D. II performed brachial arterial catheterizations for protocol 2. D.W.W. contributed to the experimental design and data collection for protocol 2. M.J.D. contributed to the conception of experiments, data interpretation and development of custom data collection software. P.J.F. contributed to the conception and design of experiments, all data collection and interpretation, and the drafting of the article. No author had any conflict of interest and all authors reviewed the article for intellectual content and approved the final version of the manuscript.
Funding
This work was supported by RO1 HL093167 (P.J.F.). M.J.D. was supported by P01HL-095486.
References
- Barrett-O’Keefe Z, Witman MA, McDaniel J, Fjeldstad AS, Trinity JD, Ives SJ, Conklin JD, Reese V, Runnels S, Morgan DE, Sander M, Richardson RS, Wray DW. Angiotensin II potentiates α-adrenergic vasoconstriction in the elderly. Clin Sci (Lond) 2013;124:413–422. doi: 10.1042/CS20120424. [DOI] [PubMed] [Google Scholar]
- Cardillo C, Kilcoyne CM, Waclawiw M, Cannon RO, 3rd, Panza JA. Role of endothelin in the increased vascular tone of patients with essential hypertension. Hypertension. 1999;33:753–758. doi: 10.1161/01.hyp.33.2.753. [DOI] [PubMed] [Google Scholar]
- Casey DP, Padilla J, Joyner MJ. a-adrenergic vasoconstriction contributes to the age-related increase in conduit artery retrograde and oscillatory shear. Hypertension. 2012;60:1016–1022. doi: 10.1161/HYPERTENSIONAHA.112.200618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MJ. Perspective: physiological role(s) of the vascular myogenic response. Microcirculation. 2012;19:99–114. doi: 10.1111/j.1549-8719.2011.00131.x. [DOI] [PubMed] [Google Scholar]
- Davy KP, Seals DR, Tanaka H. Augmented cardiopulmonary and integrative sympathetic baroreflexes but attenuated peripheral vasoconstriction with age. Hypertension. 1998;32:298–304. doi: 10.1161/01.hyp.32.2.298. [DOI] [PubMed] [Google Scholar]
- Diedrich A, Crossman AA, Beightol LA, Tahvanainen KU, Kuusela TA, Ertl AC, Eckberg DL. Baroreflex physiology studied in healthy subjects with very infrequent muscle sympathetic bursts. J Appl Physiol. 2013;114:203–210. doi: 10.1152/japplphysiol.00509.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinenno FA, Eisenach JH, Dietz NM, Joyner MJ. Post-junctional alpha-adrenoceptors and basal limb vascular tone in healthy men. J Physiol. 2002;540:1103–1110. doi: 10.1113/jphysiol.2001.015297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyson KS, Shoemaker JK, Arbeille P, Hughson RL. Modelflow estimates of cardiac output compared with Doppler ultrasound during acute changes in vascular resistance in women. Exp Physiol. 2010;95:561–568. doi: 10.1113/expphysiol.2009.050815. [DOI] [PubMed] [Google Scholar]
- Esler MD, Hasking GJ, Willett IR, Leonard PW, Jennings GL. Noradrenaline release and sympathetic nervous system activity. J Hypertens. 1985;3:117–129. doi: 10.1097/00004872-198504000-00003. [DOI] [PubMed] [Google Scholar]
- Fadel PJ. Arterial baroreflex control of the peripheral vasculature in humans: rest and exercise. Med Sci Sports Exerc. 2008;40:2055–2062. doi: 10.1249/MSS.0b013e318180bc80. [DOI] [PubMed] [Google Scholar]
- Fagius J, Wallin BG. Long-term variability and reproducibility of resting human muscle nerve sympathetic activity at rest, as reassessed after a decade. Clin Auton Res. 1993;3:201–205. doi: 10.1007/BF01826234. [DOI] [PubMed] [Google Scholar]
- Fairfax ST, Padilla J, Vianna LC, Davis MJ, Fadel PJ. Spontaneous bursts of muscle sympathetic nerve activity decrease leg vascular conductance in resting humans. Am J Physiol Heart Circ Physiol. 2013;304:H759–H766. doi: 10.1152/ajpheart.00842.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart EC, Charkoudian N, Wallin BG, Curry TB, Eisenach J, Joyner MJ. Sex and ageing differences in resting arterial pressure regulation: the role of the beta-adrenergic receptors. J Physiol. 2011;589:5285–5297. doi: 10.1113/jphysiol.2011.212753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson K, Eriksson M, Wahlqvist I, von zur Muhlen B, Lind L. Effects of blockade of alpha- and beta-adrenoceptors and neuropeptide Y(1) receptors, as well as brachial plexus blockade, on endothelium-dependent vasodilation in the human forearm. Clin Exp Pharmacol Physiol. 2002;29:603–607. doi: 10.1046/j.1440-1681.2002.03695.x. [DOI] [PubMed] [Google Scholar]
- Jones PP, Shapiro LF, Keisling GA, Jordan J, Shannon JR, Quaife RA, Seals DR. Altered autonomic support of arterial blood pressure with age in healthy men. Circulation. 2001;104:2424–2429. doi: 10.1161/hc4501.099308. [DOI] [PubMed] [Google Scholar]
- Jordan J, Shannon JR, Diedrich A, Black BK, Robertson D. Increased sympathetic activation in idiopathic orthostatic intolerance: role of systemic adrenoreceptor sensitivity. Hypertension. 2002;39:173–178. doi: 10.1161/hy1201.097202. [DOI] [PubMed] [Google Scholar]
- Joyner MJ, Charkoudian N, Wallin BG. Sympathetic nervous system and blood pressure in humans: individualized patterns of regulation and their implications. Hypertension. 2010;56:10–16. doi: 10.1161/HYPERTENSIONAHA.109.140186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahan T, Pernow J, Schwieler J, Wallin BG, Lundberg JM, Hjemdahl P. Noradrenaline release evoked by a physiological irregular sympathetic discharge pattern is modulated by prejunctional alpha- and beta-adrenoceptors in vivo. Br J Pharmacol. 1988;95:1101–1108. doi: 10.1111/j.1476-5381.1988.tb11744.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim A, Deo SH, Vianna LC, Balanos GM, Hartwich D, Fisher JP, Fadel PJ. Sex differences in carotid baroreflex control of arterial blood pressure in humans: relative contribution of cardiac output and total vascular conductance. Am J Physiol Heart Circ Physiol. 2012;301:H2454–H2465. doi: 10.1152/ajpheart.00772.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiviniemi AM, Frances MF, Tiinanen S, Craen R, Rachinsky M, Petrella RJ, Seppanen T, Huikuri HV, Tulppo MP, Shoemaker JK. alpha-Adrenergic effects on low-frequency oscillations in blood pressure and R-R intervals during sympathetic activation. Exp Physiol. 2011;96:718–735. doi: 10.1113/expphysiol.2011.058768. [DOI] [PubMed] [Google Scholar]
- Kurjiaka DT, Segal SS. Interaction between conducted vasodilation and sympathetic nerve activation in arterioles of hamster striated muscle. Circ Res. 1995;76:885–891. doi: 10.1161/01.res.76.5.885. [DOI] [PubMed] [Google Scholar]
- Lott ME, Hogeman C, Herr M, Bhagat M, Kunselman A, Sinoway LI. Vasoconstrictor responses in the upper and lower limbs to increases in transmural pressure. J Appl Physiol. 2009;106:302–310. doi: 10.1152/japplphysiol.90449.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundvall J, Edfeldt H. Very large range of baroreflex sympathetic control of vascular resistance in human skeletal muscle and skin. J Appl Physiol. 1994;76:204–211. doi: 10.1152/jappl.1994.76.1.204. [DOI] [PubMed] [Google Scholar]
- Minson CT, Halliwill JR, Young TM, Joyner MJ. Influence of the menstrual cycle on sympathetic activity, baroreflex sensitivity, and vascular transduction in young women. Circulation. 2000;101:862–868. doi: 10.1161/01.cir.101.8.862. [DOI] [PubMed] [Google Scholar]
- Moore PA, Hersh EV, Papas AS, Goodson JM, Yagiela JA, Rutherford B, Rogy S, Navalta L. Pharmacokinetics of lidocaine with epinephrine following local anaesthesia reversal with phentolamine mesylate. Anesth Prog. 2008;55:40–48. doi: 10.2344/0003-3006(2008)55[40:POLWEF]2.0.CO;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newcomer SC, Leuenberger UA, Hogeman CS, Handly BD, Proctor DN. Different vasodilator responses of human arms and legs. J Physiol. 2004;556:1001–1011. doi: 10.1113/jphysiol.2003.059717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninomiya I, Malpas SC, Matsukawa K, Shindo T, Akiyama T. The amplitude of synchronized cardiac sympathetic nerve activity reflects the number of activated pre- and postganglionic fibers in anaesthetized cats. J Auton Nerv Syst. 1993;45:139–147. doi: 10.1016/0165-1838(93)90125-e. [DOI] [PubMed] [Google Scholar]
- Ozkor MA, Murrow JR, Rahman AM, Kavtaradze N, Lin J, Manatunga A, Quyyumi AA. Endothelium-derived hyperpolarizing factor determines resting and stimulated forearm vasodilator tone in health and in disease. Circulation. 2011;123:2244–2253. doi: 10.1161/CIRCULATIONAHA.110.990317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padilla J, Young CN, Simmons GH, Deo SH, Newcomer SC, Sullivan JP, Laughlin MH, Fadel PJ. Increased muscle sympathetic nerve activity acutely alters conduit artery shear rate patterns. Am J Physiol Heart Circ Physiol. 2010;298:H1128–H1135. doi: 10.1152/ajpheart.01133.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padilla J, Simmons GH, Vianna LC, Davis MJ, Laughlin MH, Fadel PJ. Brachial artery vasodilatation during prolonged lower limb exercise: role of shear rate. Exp Physiol. 2011;96:1019–1027. doi: 10.1113/expphysiol.2011.059584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawelczyk JA, Levine BD. Heterogeneous responses of human limbs to infused adrenergic agonists: a gravitational effect. J Appl Physiol. 2002;92:2105–2113. doi: 10.1152/japplphysiol.00979.2001. [DOI] [PubMed] [Google Scholar]
- Ray CA, Monahan KD. Sympathetic vascular transduction is augmented in young normotensive blacks. J Appl Physiol. 2002;92:651–656. doi: 10.1152/japplphysiol.00788.2001. [DOI] [PubMed] [Google Scholar]
- Rea RF, Wallin BG. Sympathetic nerve activity in arm and leg muscles during lower body negative pressure in humans. J Appl Physiol. 1989;66:2778–2781. doi: 10.1152/jappl.1989.66.6.2778. [DOI] [PubMed] [Google Scholar]
- Rosenmeier JB, Fritzlar SJ, Dinenno FA, Joyner MJ. Exogenous NO administration and alpha-adrenergic vasoconstriction in human limbs. J Appl Physiol. 2003;95:2370–2374. doi: 10.1152/japplphysiol.00634.2003. [DOI] [PubMed] [Google Scholar]
- Saris JJ, van Dijk MA, Kroon I, Schalekamp MA, Danser AH. Functional importance of angiotensin-converting enzyme-dependent in situ angiotensin II generation in the human forearm. Hypertension. 2000;35:764–768. doi: 10.1161/01.hyp.35.3.764. [DOI] [PubMed] [Google Scholar]
- Simmons GH, Padilla J, Young CN, Wong BJ, Lang JA, Davis MJ, Laughlin MH, Fadel PJ. Increased brachial artery retrograde shear rate at exercise onset is abolished during prolonged cycling: role of thermoregulatory vasodilation. J Appl Physiol. 2011;110:389–397. doi: 10.1152/japplphysiol.00936.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundlof G, Wallin BG. The variability of muscle nerve sympathetic activity in resting recumbent man. J Physiol. 1977;272:383–397. doi: 10.1113/jphysiol.1977.sp012050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallbo AB, Hagbarth KE, Torebjork HE, Wallin BG. Somatosensory, proprioceptive, and sympathetic activity in human peripheral nerves. Physiol Rev. 1979;59:919–957. doi: 10.1152/physrev.1979.59.4.919. [DOI] [PubMed] [Google Scholar]
- Vanhoutte PM, Verbeuren TJ, Webb RC. Local modulation of adrenergic neuroeffector interaction in the blood vessel well. Physiol Rev. 1981;61:151–247. doi: 10.1152/physrev.1981.61.1.151. [DOI] [PubMed] [Google Scholar]
- Vianna LC, Hart EC, Fairfax ST, Charkoudian N, Joyner MJ, Fadel PJ. Influence of age and sex on the pressor response following a spontaneous burst of muscle sympathetic nerve activity. Am J Physiol Heart Circ Physiol. 2012;302:H2419–H2427. doi: 10.1152/ajpheart.01105.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallin BG, Burke D, Gandevia S. Coupling between variations in strength and baroreflex latency of sympathetic discharges in human muscle nerves. J Physiol. 1994;474:331–338. doi: 10.1113/jphysiol.1994.sp020025. [DOI] [PMC free article] [PubMed] [Google Scholar]