

Abstract
Objective
To examine the monocyte-derived dendritic cell (DC) response to infectious HCV in a cell culture system.
Methods
Adherence-derived DCs were incubated with various titers of JFH-1HCV (genotype 2a) generated from transfected Huh 7.5 cells or co-incubated with Newcastle Disease Virus (NDV). Infection and the type 1 Interferon response were assessed by Real Time RT-PCR, morphology by light microscopy, and immunophenotype by flow cytometry.
Results
Our data demonstrated no viral replication or particle release from DC after HCV infection. Morphologically, monocytes showed a tendency to shift into immature DCs when cultured with HCV, as compared with control monocytes. This shift was confirmed by flow cytometry and appeared to be related to viral titers. There was also an increase in immature DC numbers. HCV infection induced IFNβ expression in DCs and the amount seemed to be inversely correlated with viral titers indicating that HCV has the capacity to negatively regulate such cells. However, IFNα does not appear to be affected by direct contact with the virus. A strong IFNβ signal induced by NDV in DC was substantially diminished by HCV.
Conclusions
HCV negatively affects the maturation of dendritic cells, and suppresses type 1 IFN response of DC. Our results suggest a mechanism of viral evasion of host immunity.
Keywords: Dendritic Cells, Hepatitis C Virus, JFH-1, type 1 IFN
Dendritic cells (DCs) are are a sparsely distributed, migratory group of leukocytes specialized in the uptake, transport, processing and presentation of epitopes to T cells (1–3). This defensive function is mediated by activating T helper cells (TH) to divide and secrete cytokines that define the adaptive response. In HCV, individuals with specific MHC class II haplotypes are more likely to clear the virus suggesting that APCs may be an important factor (4). Furthermore, DCs are being studied as therapeutic adjuvants (by pulsing or transducing with specific targets) for the treatment of many diseases, including HCV (5–14). Of these, some were reported to induce inflammatory responses, while others did not in a pattern of incomplete activation. Similarly, in studies evaluating DC function in chronic carriers, some have not found any abnormalities while others have found lower absolute numbers of DCs, impaired allo-stimulatory activity, reduced expression of CD86 (but not of HLA molecules), and a shift towards TH2 responses (15–23). There was also a correlation between the levels of HCV RNA circulating in the blood and those observed defects on DCs, also seen on chimpanzee models (24, 25). This was even more noticeable when antiviral treatment lowered the circulating levels of HCV RNA (24). Interestingly, although the quantity of DCs in peripheral blood mononuclear cells (PBMC) was lower, the responses of DCs to other pathogens (including responses to Lipopolysaccharide (LPS), tetanus toxoid or influenza) were unaffected (26–29). Furthermore, DCs from long term responders and normal DCs did not show impairment and the exogenous addition of IL-12 or IL-2 restored T cell proliferation (30). Overall, the data supports the notion that HCV does not cause general immune-suppression but may have a targeted effect since DC derived from chronic patients are able to display a normal morphology, phenotype and capacity to take up antigen and presented allostimulatory defects only in cells derived from non-responders to therapy (31, 32).
In 2005, the development of a new strain of HCV (JFH-1) that was able to replicate fully in vitro has allowed studies of virus-host interactions (33, 34). A recent study with pDCs showed a direct effect of the virus that modulates their response while a study on apoptotic body matured, monocyte-derived DC (MoDC) shows that the virus can’t induce DC maturation unless viral dsRNA is brought in via apoptotic bodies (35, 36). Here we report our research on the initial interaction of HCV with MoDC, from healthy donor PBMC. We show that this interaction has effects in monocytes and immature DCs, preventing a normal response in those cells. We also show that HCV downregulated the mature DCs TH1 pro-inflammatory response. Our results complement the current research being done on the role of HCV on DC and give a better understanding of the interaction between these cells and the virus which might provide insights into the mechanism(s) by which HCV develops chronic infection.
Materials and Methods
Isolation of PBMC and culture of monocytes and DC
Buffy coats (leukopac, PBL) were diluted three times its volume in 1× PBS pH 7.4 (Gibco, California, USA). The dilution was layered onto Lymphoprep (Axis-Shield, Norway) in a 2:1 ratio for Ficoll-Hypaque density gradient centrifugation and centrifuged for 25 minutes at 22 °C and 1200 rpm. PBMC were collected at the interface, washed twice and centrifuged each time for 10 minutes at 4 °C and 1200 rpm. Cell viability was assessed by trypan blue exclusion. All cultures of human PBMC and derived cells were maintained in RPMI 1640 medium (Sigma, Missouri, USA) supplemented with 2 mM L-glutamine (Life Technologies, Paisley, Scotland), 5000 U/ml penicillin (Sigma, Missouri, USA), 5000 U/ml streptomycin sulfate (Sigma, Missouri, USA), and 10% v/v fetal bovine serum (Gibco, California, USA) named cRPMI.
Monocytes were obtained by adhering 5×106 PBMC/well to a 6 well culture dish for 2 hours at 37°C. After aspirating the non-adherent cells, monocytes were washed with 1× PBS pH 7.4. Complete media was added to the remaining cells. To induce differentiation into DCs, monocytes were cultured with 50ng/ml GM-CSF (BD Biosciences) and 20ng/ml IL-4 (BD Biosciences) for 6 days. Maturation was induced with 1ug/ml LPS (Sigma) for 24 hours.
HCV Constructs and Viral Particle Generation
The linearized genomic pJFH-1 plasmid DNA was purified and used as a template for in vitro transcription using MEGAscript kit (Ambio, Autin, TX), delivered into Huh-7.5 cells by electroporation and cultured in cDMEM. Cells were passaged every 3–5 days and, at 21 days, supernatants were filtered and frozen. Viral titer (1×105 ffu/ml) was determined by the average number of NS5A-positive foci detected at the highest dilutions. Infection was done by adding supernatant or supernatant diluted in cDMEM for 24 hours before exchange. Control is uninfected-Huh7.5 supernatant.
NDV strain La Sota was added to uninfected CHO cells cultured in cDMEM for 2 days. Cells and media were subsequently exposed to 3 cycles of freeze/thaw treatment (i.e., −80°C and 32°C). The supernatant was centrifuged at 12,000rpm for 10 minutes to remove cellular debris and then aliquoted and frozen at −80°C until use.
ImmunoFluorescence
Incubated DCs were cytospun onto a coverslip (Fisherbrand) and Huh 7.5 cells were grown on them then fixed with acetone on ice. After three washes with 1×PBS, cells were incubated with mouse anti-HCV NS5A monoclonal antibody (generated by Dr. Johnson Lau in the Hybridoma Core Laboratory at the University of Florida, clone number HL1126) for 1 hour followed by goat anti-mouse IgG-FITC (Southernbiotech, Birmingham, AL) for another hour. Cells were examined under a fluorescent microscope (Olympus).
Reverse Transcription and Real Time Polymerase Chain Reaction
Total cellular RNA was isolated using Trizol (Invitrogen, Carlsbad, CA). Reverse transcription to obtain cDNA was performed using the Superscript II (i.e.: 50 U reverse transcriptase per reaction) first-strand synthesis for RT-PCR kit (Invitrogen) primed with oligo (dT)(Invitrogen) according to the manufacturer's instructions. Quantitative real-time fluorophore-labeled LUX primers pairs were obtained from Invitrogen (Supplemental table 1). The PCR conditions were: 50°C, 2’; 95°C, 2’ (95°C, 15”; 60°C, 30” (IFNs, HCV) or 62°C, 30” (GAPDH); and 72°C, 1 min) for 45 cycles. Reactions were monitored at every cycle during the annealing step on a spectrofluorometric thermal cycler (MJ Research DNA Engine Opticon® 2 thermal cycler, BIORAD). Results were analyzed with MJ Opticon Monitor 3.1 software from BIORAD.
Flow Cytometry
DCs were assayed by four-color immunofluorescence staining with a panel of directly conjugated antibodies. One million cells were stained with lineage (Lin) fluorescein isothiocyanate (FITC)-conjugated antibody cocktail (CD3, CD14, CD16, CD19, CD20, CD56) and HLA-DR PerCP-conjugated antibody (BD Biosciences, Heidelberg, Germany). The presences of maturation markers (CD80, CD83, CD86 and CD40) as well as the HCV binding receptor, CD81, were also measured. Pelleted cells were incubated for 30 min at 4°C with antibodies, washed with staining buffer (PBS + 2% BSA + 0.1% Na azide), and then fixed with 2% paraformaldehyde and stored at 4 °C. Cells were analyzed on a FACSCalibur flow cytometer (BD Biosciences, Heidelberg, Germany). Quantitation was done by the CellQuest software (BD Biosciences; version 3.2.1). Isotype-specific immunoglobulin controls were run for each fluorochrome. Twenty five thousand cells were analyzed for each sample.
Enzyme-linked Immunosorbent Assay (ELISA)
As described by the manufacturer, 96-well plates (Nunc-Immuno Plate, Roskilde, Denmark) were coated with 1 mg/ml of purified monoclonal antibody against human IL-12 (p70), IL-10, TNFα or IFNγ (Pierce-Endogen, Rockford, Illinois) in coating buffer (1× PBS, pH 7.4) at room temperature overnight. The plates were then incubated in blocking buffer (1× PBS, pH 7.4 with 1% bovine serum albumin [BSA]; Sigma, Axel, The Netherlands) for 2 hours at room temperature and washed in 0.05% Tween-20. Standard serial dilutions were added to the plates as well as 50uL of experimental supernatant in triplicate followed one hour later by biotin labeled anti-cytokine antibody. Recombinant human cytokines (hTNFα, hIFNγ, hIL-10, and hIL-12 (p70)) were used as standards. Cytokines were captured by the specific primary monoclonal antibody and detected by biotin-labeled specific secondary antibody (Pierce-Endogen, Rockford, Illinois) followed by strepavidin-horseradish peroxidase for 20 minutes and developed by adding the substrate TMB (Pierce-Endogen). Reaction was stopped by the addition of an equal volume of 0.18M H2SO4. The absorbance was read at 450 nm.
Autologous Proliferation Assay
Autologous PBMC were resuspended at 5×106/ml in sterile PBS and labeled for 15 minutes with 200nM CFSE according to manufacturer’s protocol (Molecular Probes, Eugene OR). Washed cells were then seeded on a 96 well plate at 5×105cells/well. Monocytes, immature DC or mature DC were added at the ratios 1:1, 1:10, 1:50 and 1:100. Positive control wells received lectin (PHA; 5ug/ml; Sigma, Missouri, USA) for non-specific stimulation of lymphocytes. Cells were cultured for 6 days at which time cells were harvested and stained for flow cytometry as described above for the following markers: CD3 APC, CD4 PerCP and CD8 PE.
Results
HCV Induces Phenotypical Differences in Monocytes and Immature DC but not on Mature DC
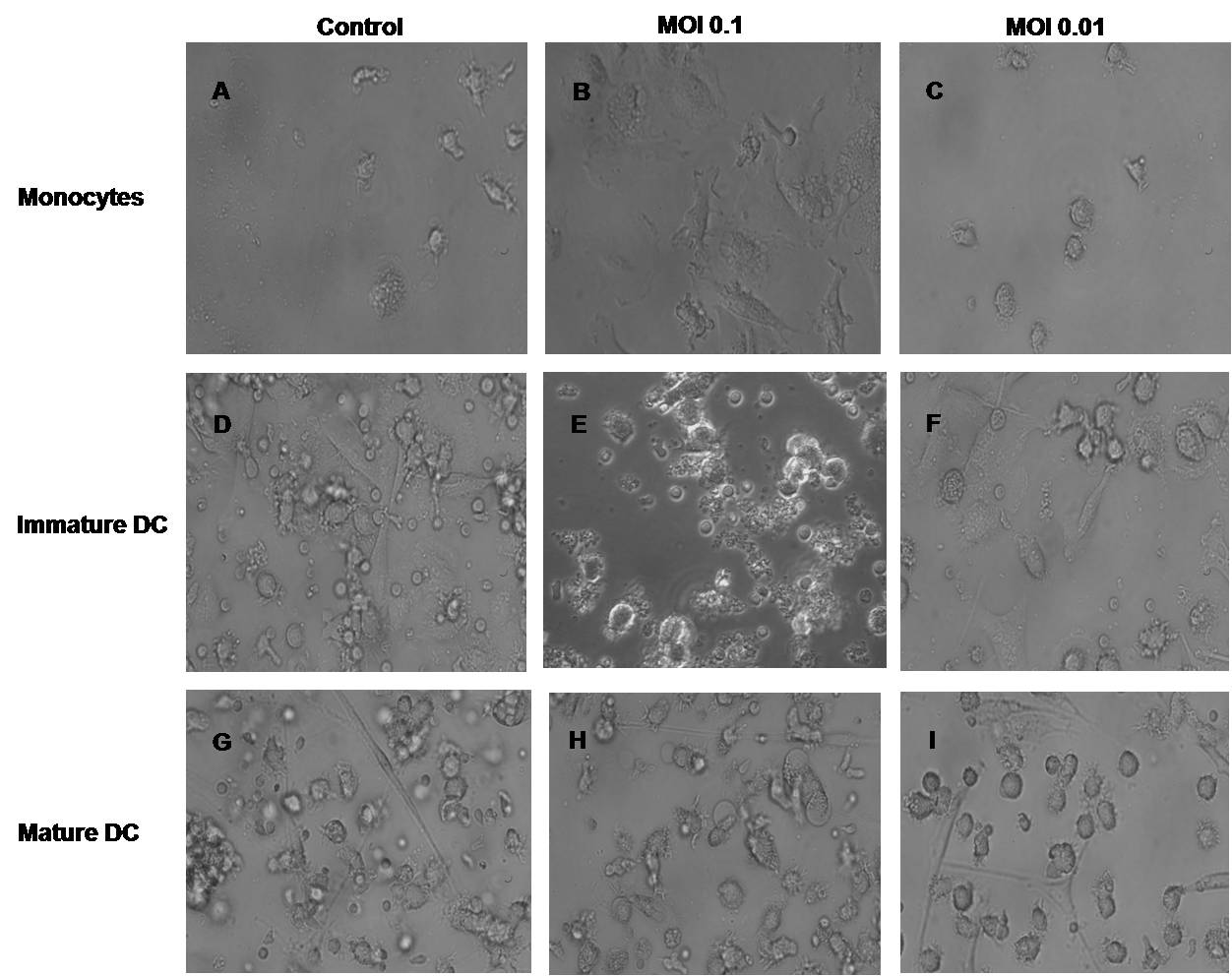
Since HCV has been reported to interfere with the function of DCs and these effects seem viral titer dependent, we decided to study the direct interaction of the virus on these cells. We cultured enriched monocytes, immature DCs or mature DC with either MOI of 0.1 or an MOI of 0.01(Supplemetal figure 1). LPS was chosen to induce maturation as the first results demonstrated an impaired DC allostimulatory function in chronic HCV infection were obtained with DCs treated with LPS (31). Furthermore, recent data reported a significant role of LPS in the pathogenesis of HCV infection, suggesting that the maturation stimulus may have physiological importance in vivo (37). We were able to observe a shift in the monocyte morphology, by light microscopy, to a more adherent phenotype with many processes after culture with a high MOI of HCV compared to control (Supplemental figure 2A and B). However, the morphology of monocytes after a low MOI of HCV did not change (Supplemental figure 2C). Immature DCs had a higher number of non-adherent clusters after addition of a high MOI of HCV but again not with low MOI (Supplemental figures 2D through F). Mature DCs did not show any morphological differences after addition of virus (Supplemental figure 2G through I).
To further characterize these observations we analyzed the cells by flow cytometry for the presence of DC (HLA-DR+Lin−). Monocytes are Lin+ because of the presence of CD14 which is also present in macrophages. They showed a phenotypical change towards DCs by losing the lineage marker during the first 24 hours of culture with a high MOI, reverting back to a Lin+ phenotype (might have returned to monocytes or become machrophages) after a week of culture, while a low MOI did not lose the marker but increased the amount of Lin+ cells at the beginning of the experiment (Figure 1A through D). Immature DC percentages when cultured with HCV (either MOI) increased but only the low MOI treated cells reverted back to the original percentages after 7 days of culture (Figures 1F through J). Mature cells showed no strong differences as compared to control (Figures 1K through P). These correlated with the morphological characteristics observed under the microscope. These cells remained immature since they did not up-regulate co-stimulatory molecules (CD40, CD80, CD83 or CD86) at either MOI (data not shown).
Figure 1. Phenotypical Characteristics of Monocytes and Dendritic Cells Incubated with HCV.
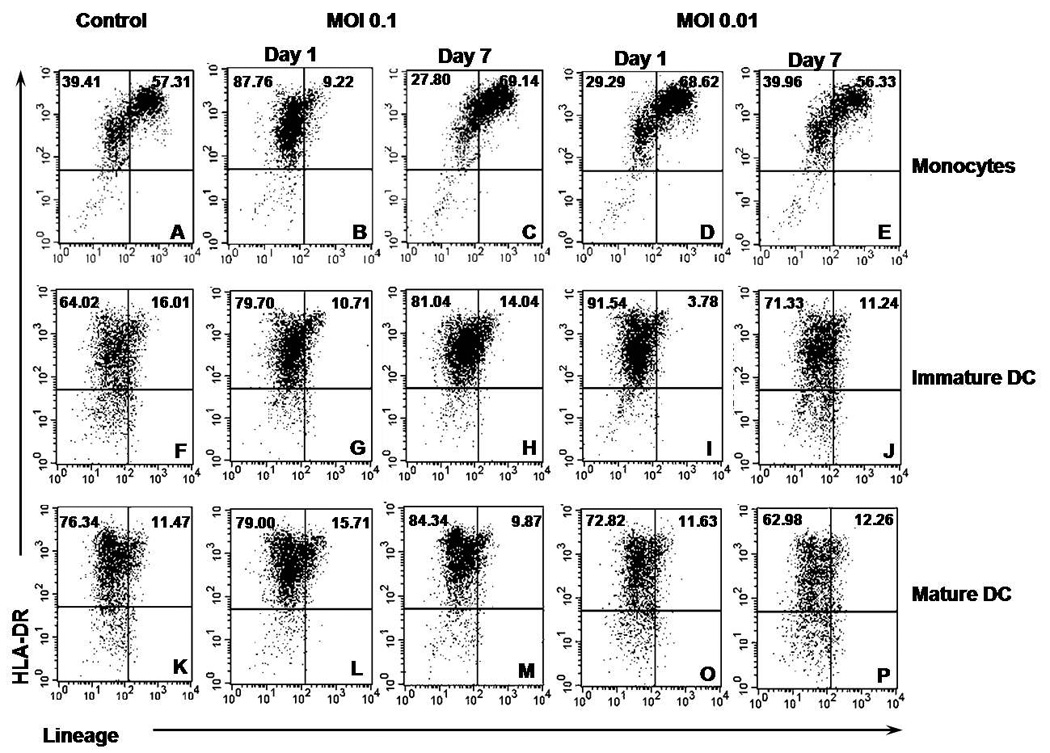
Monocytes or DC from healthy human PBMC incubated with HCV for one or seven days were stained for HLA-DR and lineage 1 cocktail and analyzed by flow cytometry. The percentage of cells per quadrant from the total analyzed is shown. Figures are representative of three separate experiments (three separate donors). A through E) Monocytes. F through J) Immature DCs. K through P) Mature DCs.
HCV affects the proliferation of T helper and CTLs at the basal level by affecting DC
In order to further understand our observations, we tested the ability of these cells to induce the proliferation of naïve T cell subsets in PBMC. We chose to study autologous cells since our observations indicated that HCV does not interfere with the maturation or activation of cells by other means, such as LPS, and because it is a way to really understand virus-specific responses (38). In the PBMC population we gated for T helper cells (CD3+CD4+, Figure 2 A through C) as well as the CTLs (CD3+CD8+, Figure 2D through F). As expected, the amount of proliferation of T helper cells on healthy PBMC did not change with increasing amounts of un-incubated monocytes while there was a slight increase in the basal PBMC proliferation in control immature and mature DCs that was dependent on the ratio of DCs added to the coculture. When monocytes or DCs were incubated there was some notable effects on T helper cells: 1) when co-cultured with monocytes there was an increase in proliferation but it was inversely correlated to the amount of virus; 2) there was a decrease in the amount of proliferation induced by co-culture with immature DCs; 3) contrary to immature DCs, there was an increase in the amount of proliferation induced by mature DC when they are incubated with HCV.
Figure 2. Immunostimulatory Capacity of Monocytes and Dendritic Cells Incubated with HCV.
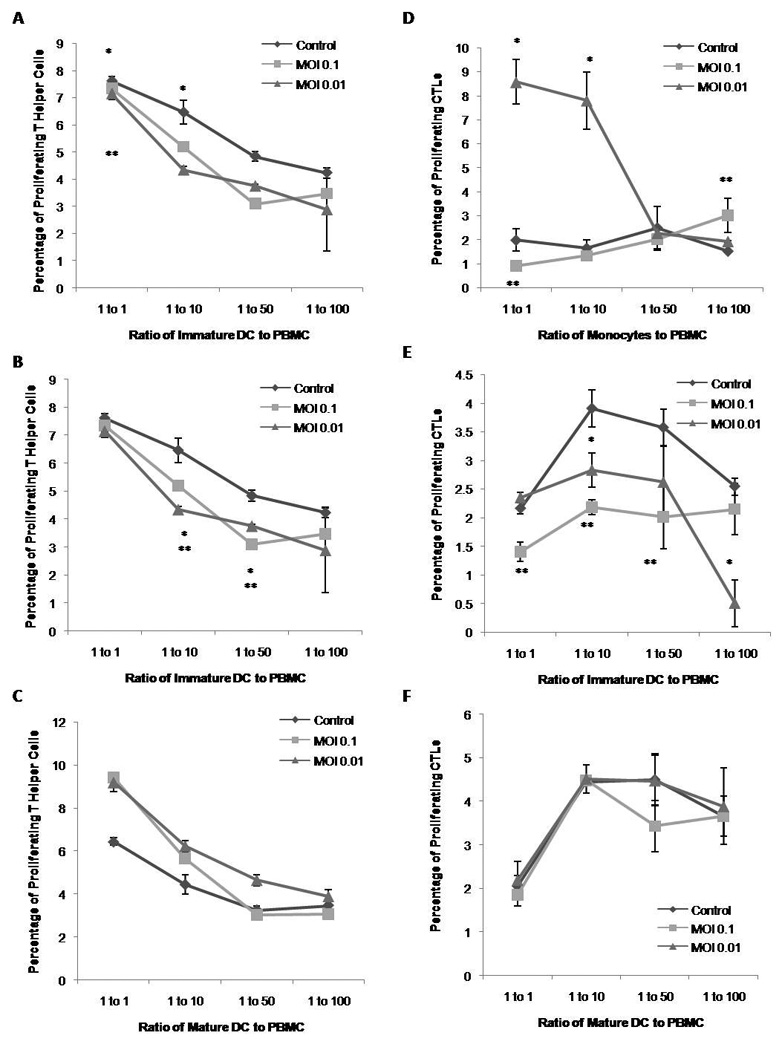
Monocytes or DC from healthy human PBMC incubated with HCV were co-cultured with autologous PBMC containing CFSE for seven days at 1:1, 1:10, 1:50 or 1:100 ratio of effector cells to proliferators. Cells were then also stained for CD3, CD4 and CD8, and analyzed by flow cytometry. Figures are representative of three separate experiments (three separate donors). A) Percentage of proliferating helper T cells (CD3+ CD4+) after stimulation with monocytes. B) Percentage of proliferating helper T cells after stimulation with immature DC. C) Percentage of proliferating helper T cells with mature DC. D) Percentage of proliferating CTL (CD3+ CD8+) after stimulation with monocytes. E) Percentage of proliferating CTL after stimulation with immature DCs. F) Percentage of proliferating CTL after stimulation with mature DCs.
As for CTLs in the PBMC, they are not directly affected by either cell since an increase in the ratio of monocytes or DCs did not change their proliferation but cocultured with these cells when incubated did have an effect in their proliferation. Monocytes up-regulated only at a low MOI of 0.01, similar to what was observed with T helper cells. Conversely, incubated, immature DC co-culture strongly downregulated the basal proliferation of CTLs in the culture. Since they are not dependent on the ratio of cells it might be an indirect effect likely from the T helper cells since they share a similar pattern.
HCV Affects Type 1 Interferon Responses in the Absence of Replication
In order to see if the cell recognizes HCV we studied the effects of the virus on the type I interferon responses (i.e.: IFNα2a and IFNβ) in monocytes, immature DCs and mature DCs. Monocytes did not produce IFNβ when exposed to HCV (Figure 3A). Furthermore, HCV downregulated the basal levels of expression in these cells with longer exposure to HCV resulting in the lowest expression of INFβ. DCs were able to express IFNβ in response to HCV, while mature DCs producing tenfold more than immature DCs (Figure 3B and C). In contrast, IFNα2a was produced consistently in the three groups. It was interesting to note that immature DCs had the highest expression of the three cell types (Figures 3D through F).
Figure 3. Type I IFN Production of Monocytes and Dendritic Cells Incubated with HCV-JFH.
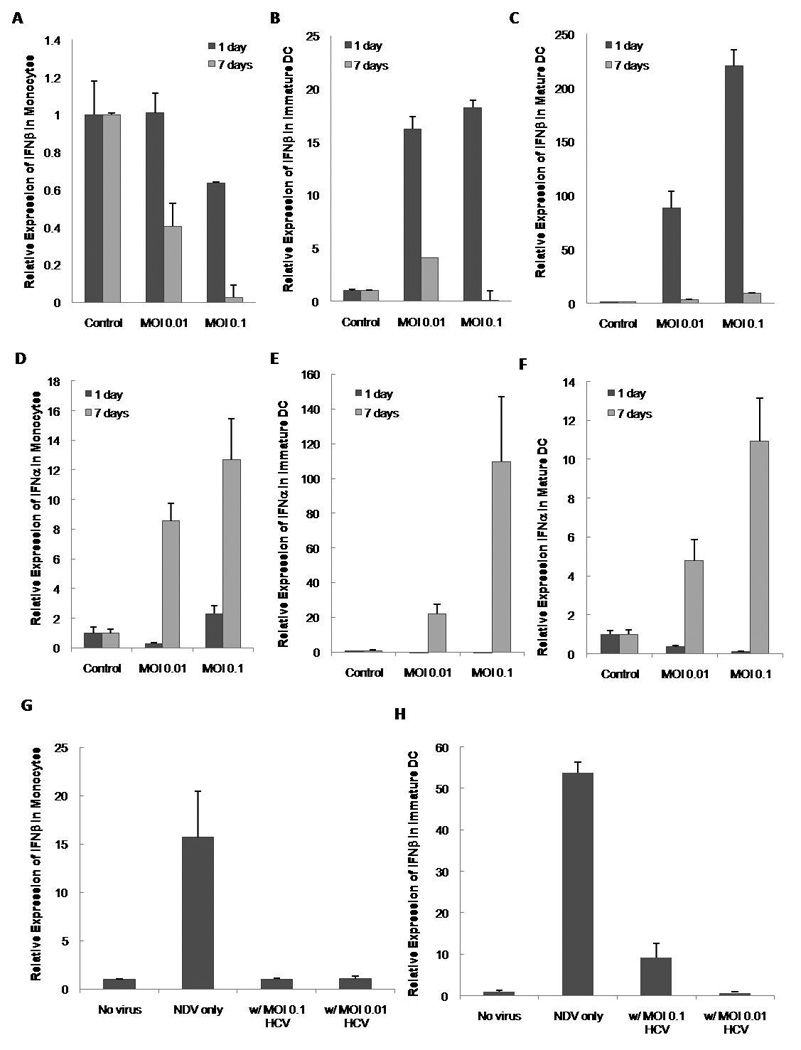
Monocytes (A and D), immature DC (B and E) and mature DC (C and F) were incubated with HCV and cells were collected at day 1 and day 7 post infection. Total RNA was isolated and IFN-β and IFNα2a mRNA expression was analyzed by real-time RT-PCR against unincubated controls and normalized against the housekeeping gene GAPDH. Monocytes (G) and immature DC (H) were either incubated by HCV alone or co-incubated with Newcastle Disease Virus (NDV). IFNβ was measured by Real Time RT-PCR. Bars represent the standard error (SEM) of three separate experiments (three separate donors).
These results indicate that HCV seems to downregulate IFNβ in monocytes and immature DCs. To understand this further, we tested the effects of this inhibition on the IFNβ produced by another virus. For this purpose we cultured monocytes and immature DC with Newcastle Disease Virus (NDV), HCV (either MOI) or both. Both monocytes and immature DCs were able to induce IFNβ in response to NDV with DC producing a much higher response than monocytes, as anticipated (Figure 3G and H). The presence of HCV significantly reduced the IFNβ produced in response to NDV.
We next measured the replication of HCV on all the cells by Real Time RT-PCR. As reported by others, HCV replicated in monocytes (39). Interestingly, DCs, (mature or immature) did not show signs of replication after seven days of culture (Figure 4A and B). To further corroborate that there was no replication we did immunofluorescence staining against NS5A on DCs. This assay also did not reveal the presence of HCV in these cells but did on the positive control (Huh7.5 cells with pJFH1, Figure 4C, D and E). The presence of CD81 was unchanged by the virus (data not shown).
Figure 4. Lack of HCV Replication on Dendritic Cells Incubated with HCV-JFH1.
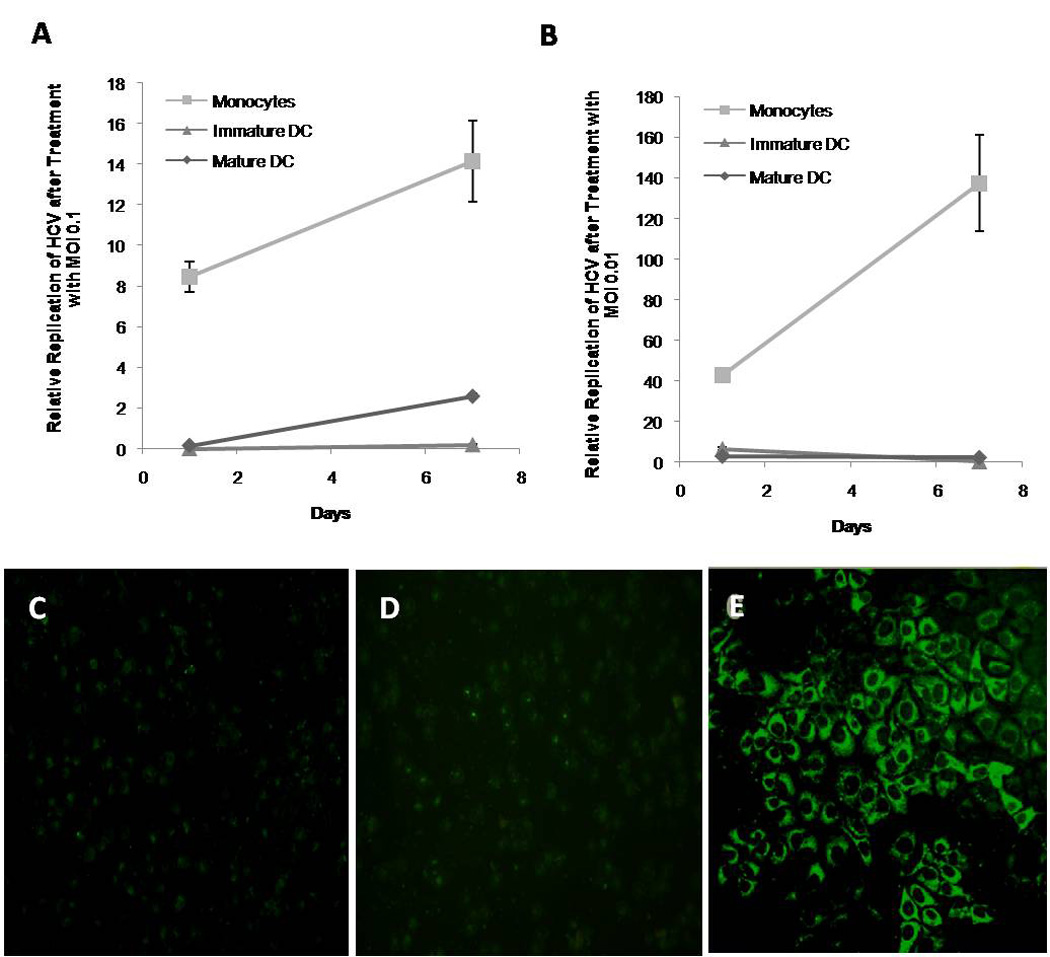
Incubated cells were analyzed for HCV replication by immunofluorescence and by Real Time RT-PCR. A) Relative HCV expression in monocytes, immature DC and LPS matured DC after infection with MOI=0.1 of HCV-JFH1 particles. B) Relative NS5A expression in monocytes, immature DC and LPS matured DC after infection with MOI=0.01 HCV-JFH1 particles. C) Unincubated immature DCs (negative control) were stained with NS5A with FITC conjugated secondary antibody. D) Immature DC incubated with MOI=0.1 HCV. E) Huh 7.5 cells previously transfected with HCV-JFH1 plasmid (virus source, positive control).
HCV Interferes with IL-12 and IFNγ production in monocytes and DCs
A strong type 1 response has been heralded as an important step towards clearance of HCV infection in acute and chronic treated HCV patients. Therefore we decided to study the role of HCV in the production of IL-12 as a marker for the initiation of a TH1 response. The supernatants of the experiments described in figure 4 were measured for IL-12. Monocytes did not have a reduced IL-12 production by day one with either concentration of virus. By day 7, MOI=0.1 did not show a significant difference while MOI=0.01 showed a decrease against the day 7 control monocytes as measured by paired student t-test (Figure 5A, P<0.05). Immature monocytes also did not show a significant change in the quantity of IL-12 by day 1 but, unlike monocytes, they produced less IL-12 with the higher titer of virus but significantly higher IL-12 with MOI=0.01 (Figure 5B).
Figure 5. Concentration of TH1 cytokines IL-12 and IFNγ produced by monocytes and DC after HCV infection.
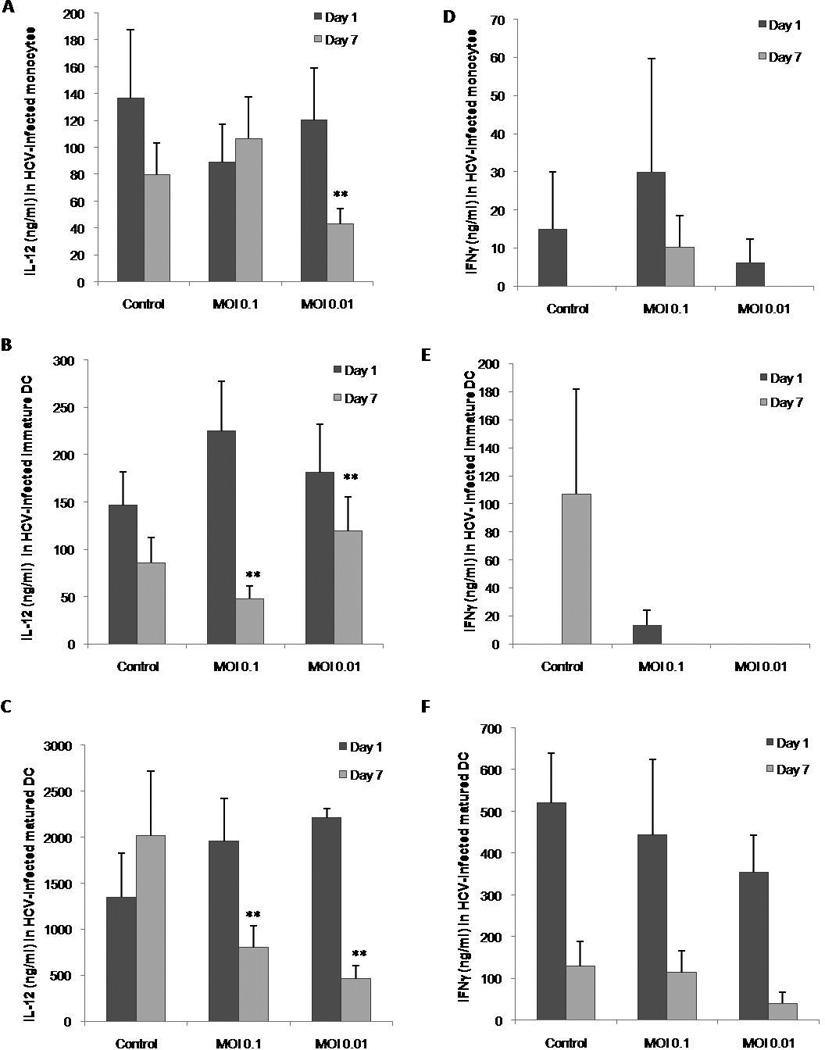
Supernatant from monocyte, immature DC or mature DC cultures with or without HCV were collected at 1 and 7 days post infection and analyzed by capture ELISA. A) Concentration of IL-12 produced by monocytes. B) Concentration of IL-12 produced by immature DCs. C) Concentration of IL-12 produced by mature DCs. D) Concentration of IFNγ produced by monocytes. E) Concentration of IFNγ produced by immature DCs. F) Concentration of IFNγ produced by mature DCs. Bars represent the standard error (SEM) of three separate experiments (three separate donors). * represents the significance of the comparison between the day 7 control and the test in a paired student t-test where p<0.05. ** represents the significance of the comparison between the day 7 control and the test in a paired student t-test where p<0.05.
Mature DCs were able to produce much higher amounts of cytokines in general as compared to the other two types of cells and they secreted significantly less IL-12 at any of the viral titers tested with MOI=0.01 being the lowest IL-12 between the two (Figure 5C). Since IL-12 is important for induction of the pro-inflammatory cytokine IFNγ we tested it to corroborate the effects of HCV on IL-12. None of the three cell types produced IFNγ in response to HCV as compared to control although mature DCs were able to produce more than any of the other cells Figures (5D through F).
IL-10 and TNFα in monocytes and DCs after HCV infection
IL-10 and TNFα are two cytokines that have been correlated in the pathogenesis of HCV in the liver of infected patients. Therefore we decided to measure the levels of these cytokines. Monocytes infected with HCV were able to produce a significant increase in the levels of IL-10 by day 7. There also seem to be a dose dependency since the higher HCV MOI produced a higher amount of IL-10 (Figure 6A). In contrast, immature DCs were able to significantly decrease the levels of IL-10 produced after infection with the higher MOI from day 1 and by both MOIs by day 7. By day 7 the levels of IL-10 expressed were similar and different titers did not reflect differences (Figure 6B). Mature DC did not show differences at day one but showed a significant decrease in expression at the lower MOI which was not significant in the higher titer (Figure 6C). Therefore HCV has a very contrasting role in the acute production of IL-10 in DCs as compared to monocytes.
Figure 6. Concentration of IL-10 andTNFα produced by monocytes and DC after HCV infection.
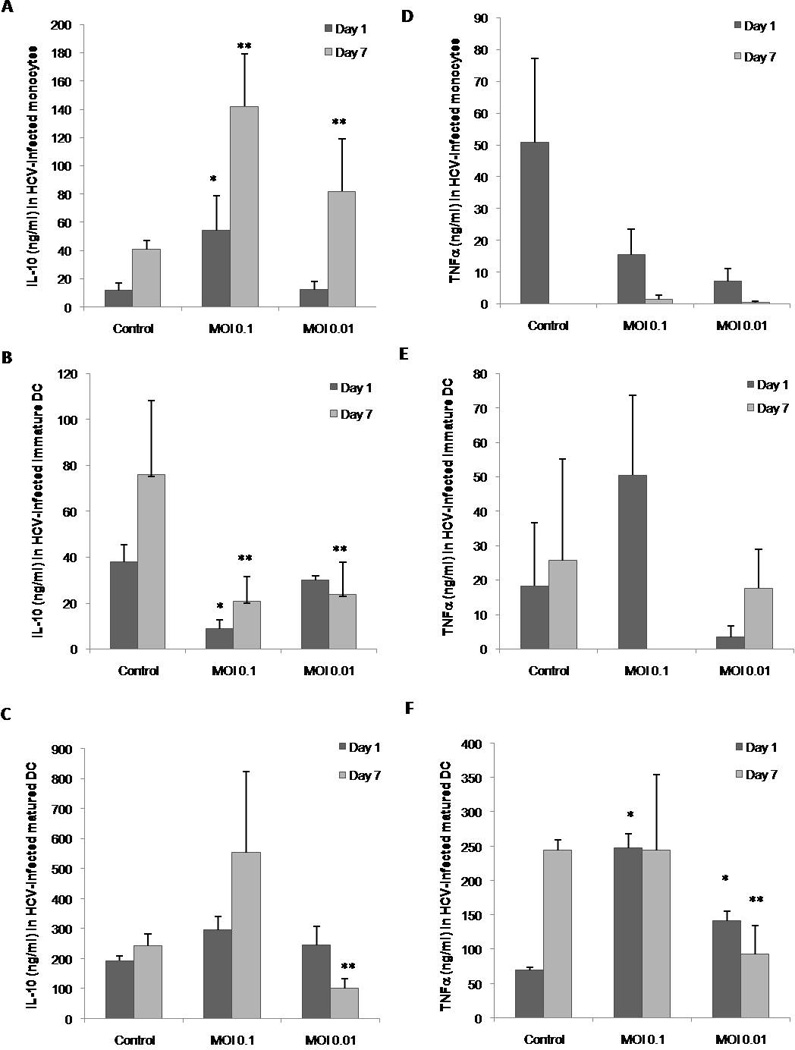
Supernatant from monocyte, immature DC or mature DC cultures with or without HCV were collected at 1 and 7 days post infection and analyzed by capture ELISA. A) Concentration of IL-10 produced by monocytes. B) Concentration of IL-10 produced by immature DCs. C) Concentration of IL-10 produced by mature DCs. D) Concentration of TNFα produced by monocytes. E) Concentration of TNFα produced by immature DCs. F) Concentration of TNFα produced by mature DCs. Bars represent the standard error (SEM) of three separate experiments (three separate donors). * represents the significance of the comparison between the day 7 control and the test in a paired student t-test where p<0.05. ** represents the significance of the comparison between the day 7 control and the test in a paired student t-test where p<0.05.
The pro-apoptotic cytokine TNFα in monocytes or immature DCs had a tendency to decrease while mature DCs showed a significant dose dependent increase on day 1 with a significant decrease, only in the lower MOI, by day 7 (Figures 6D though F). Therefore we found no specific correlation between monocytes and DCs after infection with HCV and chronic liver disease.
Discussion
One of the main questions regarding the defect in the function of DCs in HCV infection is whether it is a direct consequence of viral infection. While there is direct evidence about the replication of HCV in monocytes, the same cannot be said for DCs were detection of HCV genomes within isolated DCs has been inferred as indicative of replication (23, 31, 38, 40). Moreover, MoDCs responded to HCV with phenotypical and immunological changes, in the absence of replication, including the modulation of IFNβ. This phenomenon is corroborated by the results obtained when co-infecting with other virus. HCV impaired NDV-induced IFN production by DCs and monocytes indicating that HCV negatively regulates the induction of IFNβ by these cells in a way dependent on the amount of virus. Such an observation has also been made on chimpanzees, chronic and healthy patient cells were they saw an induction of type I IFNs and ISGs even in the incubation phase although lower on chronic patients (41). This correlates with data that shows the presence of HCV genomic RNA in MoDCs alters their immunostimulatory capacity and that the ability of DCs to stimulate T cells can be disrupted by downregulation of MHC I and co-stimulatory molecules, independent of replication in other viruses, which could suggest an initial mechanism that can lead to a chronic state (38, 42, 43). One explanation for this is that viral proteins can have immunomodulatory effects upon binding. Immature DCs for instance are capable of binding HCV-LPs in an envelope-specific, concentration dependent manner which induces DC activation (44, 45). One such binding mechanism suggested in the literature is the interaction of DC-SIGN with E2 which protects the virus from lysosomal degradation (38). Furthermore, several of the HCV proteins have been shown to directly affect the immune responses including the envelope and core proteins which have been shown to interfere with the type 1 IFN responses (46, 47). Wether this is the exact mechanisms of IFN downregulation even in the absence of replication is something that should be further elucidated in the future but it is likely a reason for the behavior we observed. I
The next logical question is which specific viral components could be responsible for the defects we observed. Different HCV gene products have been shown to interfere with the normal function of DCs (48, 49). The structural proteins (core, E1/E2), a likely culprit for our results based on the lack of replication or entry, have been shown to interfere with monocytes and DCs by upregulating IL-10 or TNFα (dependent on the cell type and in a dose dependent manner), hindering IL-12 production and allogeneic T cell stimulation (50, 51). This was linked to a defect in the IFNα production by stimulated pDCs, which can also help induce IL-10 (52). The effects on pDCs may have been due to apoptosis induction while on mDCs and MoDCs may have been due to the interference with the cells differentiation into an active status (38, 51, 53, 54). Furthermore, the defective responses induced by these structural proteins can result in poor cellular and humoral responses (50). Therefore, the immunomodulatory role of HCV may lie at the level of DC differentiation.
An interesting caveat was that our DCs were able to produce cytokines in response to the virus even if these responses were skewed. This correlates with other studies that show that same phenomenon (48–50, 55). Furthermore, a similar pattern was observed in DCs where the viral proteins were expressed by AAV vectors, perhaps due to maturation by the AAV itself (56, 57). Still, this depends on the type of maturation stimuli as well as the state of infection of the patient from where the cells were obtained (chronic versus healthy) (23, 38, 48, 49). More so, matured DCs should be taken with caution as a higher CD83 and CD86 with lower HLA-DR expression (as we observed) has been seen in the mDCs of HCV infected patients which correlates with liver inflammation (58).
Two things that make myeloid DCs the focus of our studies are that 1) pDC can be 10–50 times less efficient than mDCs and MoDCs at expanding both CD4+ and CD8+ naïve cells; 2) mDCs and MoDCs can secrete much higher levels of IL-12 which is important for shifting the balance towards a TH1 response since TH2 is associated with a chronic state of HCV infection; and 3) mDCs numbers are decreased in chronic patients that have not undergone anti-viral therapy also indicating the role of IFNα in maintaining DC populations (4, 38, 59–62). Sometimes, it seems is not the decrease in the number of DCs in patients but a shift in the expression of HLA-DR complemented with lower CD4+ T cell numbers (63). Monocytes are the main producers of IL-10 and TNFα, due to their numeric prevalence over DCs in blood, and these cytokines are known to inhibit the IFNα production by pDCs probably by inducing apoptosis (51, 64–66). In our system we found that HCV increased IL-10 production by monocytes and TNFα by mature DCs. Levels of IL-10 and TNFα are consistently higher in HCV infected patients and in DC after HCV infection and may be to blame for the apoptosis of pDC in infected patients (51). Recently, Shiina et al. and Ebihara et al. performed experiments similar to ours in which they found that direct interaction HCV does not affect matured mDCs or MoDCs (35, 36). While we show defects in MoDC this effects were more profound in immature DC or monocytes. Therefore, the maturation status of DC needs to be considered when studying the pathogenesis of HCV on these cells during infection. In conclusion, our results build upon recent reports show that HCV has a strategy to modulate the action of DCs by interfering with the type I IFN response and shifting to a TH2 response and show the importance of studying the behavior of immature DC in HCV infection.
Supplementary Material
Monocytes were isolated by adherence from healthy human PBMC. Immature DCs were obtained by culture of monocytes with GM-CSF and IL-4 for 6 days. Mature DCs were obtained by culturing Immature DC with LPS for 48 hours. Cells of each stage were incubated with HCV-JFH1, a genotype 2a strain of HCV that effectively replicates in culture at two different MOIs.
{kind=link}
Monocytes, immature DC and mature DC incubated with an MOI of 0.1 or an MOI=0.01 of HCV-JFH1for 7 days were observed at 100X magnification in a light microscope. Representative pictures of triplicate experiments (three separate donors) are shown. A) Control monocytes cultured without virus; B) monocytes incubated with MOI 0.1; C) monocytes incubated with MOI 0.01; D) immature DC without HCV; E) immature DC with MOI of 0.1 of HCV; F) immature DC with an MOI of 0.01 of HCV; G) LPS matured DC without virus; H) LPS matured DC incubated with MOI of 0.1 of HCV; I) LPS matured DC incubated with MOI of 0.01.
{kind=link}
Acknowledgements
We would like to express our gratitude to members of the Liu, Nelson, and Brantley laboratories for their continued support. We would also like to acknowledge the Center for Mammalian Genetics and Dr. Terada’s laboratory for allowing us the use of their equipment and Dr. Jingda Shi for his support with the Real Time PCR. Huh 7.5 cells were provided by Dr. Charles Rice (Rockefeller University, New York, NY) and the HCV-JFH1 plasmid was kindly provided by Dr. Takaji Wakita (Tokyo Metropolitan Institute for Neuroscience, Tokyo, Japan). Ms. Eksioglu is an NIH Ruth Kirshchtein predoctoral fellowship recipient: NIH-F31AI071353. This work was funded in part by grants NIH-DK002958 to Liu and NIH-A1061158 to Nelson.
Footnotes
Data was originally presented in part at the Keystone Symposium titled “Immunological Intervention in Human Disease” in Big Sky, MT, in January 2007.
References
- 1.Cella M, Sallusto F, Lanzavecchia A. Origin, maturation and antigen presenting function of dendritic cells. Curr Opin Immunol. 1997;9(1):10–16. doi: 10.1016/s0952-7915(97)80153-7. [DOI] [PubMed] [Google Scholar]
- 2.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392(6673):245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 3.Ardavin C, Martinez del Hoyo G, Martin P, et al. Origin and differentiation of dendritic cells. Trends Immunol. 2001;22(12):691–700. doi: 10.1016/s1471-4906(01)02059-2. [DOI] [PubMed] [Google Scholar]
- 4.Gowans EJ, Jones KL, Bharadwaj M, Jackson DC. Prospects for dendritic cell vaccination in persistent infection with hepatitis C virus. J Clin Virol. 2004;30(4):283–290. doi: 10.1016/j.jcv.2004.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Timmerman JM, Levy R. Dendritic cell vaccines for cancer immunotherapy. Annu Rev Med. 1999;50:507–529. doi: 10.1146/annurev.med.50.1.507. [DOI] [PubMed] [Google Scholar]
- 6.Everly JJ, Lonial S. Immunomodulatory effects of human recombinant granulocyte-macrophage colony-stimulating factor (rhuGM-CSF): evidence of antitumour activity. Expert Opin Biol Ther. 2005;5(3):293–311. doi: 10.1517/14712598.5.3.293. [DOI] [PubMed] [Google Scholar]
- 7.Banchereau J, Palucka AK. Dendritic cells as therapeutic vaccines against cancer. Nat Rev Immunol. 2005;5(4):296–306. doi: 10.1038/nri1592. [DOI] [PubMed] [Google Scholar]
- 8.Yu H, Huang H, Xiang J, Babiuk LA, van Drunen Littel-van den Hurk S. Dendritic cells pulsed with hepatitis C virus NS3 protein induce immune responses and protection from infection with recombinant vaccinia virus expressing NS3. J Gen Virol. 2006;87(Pt 1):1–10. doi: 10.1099/vir.0.81423-0. [DOI] [PubMed] [Google Scholar]
- 9.Encke J, Findeklee J, Geib J, Pfaff E, Stremmel W. Prophylactic and therapeutic vaccination with dendritic cells against hepatitis C virus infection. Clin Exp Immunol. 2005;142(2):362–369. doi: 10.1111/j.1365-2249.2005.02919.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang QC, Feng ZH, Zhou YX, Nie QH. Induction of hepatitis C virus-specific cytotoxic T and B cell responses by dendritic cells expressing a modified antigen targeting receptor. World J Gastroenterol. 2005;11(4):557–560. doi: 10.3748/wjg.v11.i4.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Racanelli V, Behrens SE, Aliberti J, Rehermann B. Dendritic cells transfected with cytopathic self-replicating RNA induce crosspriming of CD8+ T cells and antiviral immunity. Immunity. 2004;20(1):47–58. doi: 10.1016/s1074-7613(03)00353-4. [DOI] [PubMed] [Google Scholar]
- 12.Wang QC, Feng ZH, Zhou YX, Nie QH, Bai XF. [Acceleration of mixed lymphocyte reaction by HCV C-Fc gene-transferred murine dendritic cells] Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi. 2004;20(3):301–303. [PubMed] [Google Scholar]
- 13.Zhou Y, Lukes Y, Anderson J, Fileta B, Reinhardt B, Sjogren M. Hepatitis C virus E2 envelope protein induces dendritic cell maturation. J Viral Hepat. 2007;14(12):849–858. doi: 10.1111/j.1365-2893.2007.00879.x. [DOI] [PubMed] [Google Scholar]
- 14.Liang H, Russell RS, Yonkers NL, et al. Differential effects of hepatitis C virus JFH1 on human myeloid and plasmacytoid dendritic cells. J Virol. 2009;83(11):5693–5707. doi: 10.1128/JVI.02671-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kanto T, Inoue M, Miyazaki M, et al. Impaired function of dendritic cells circulating in patients infected with hepatitis C virus who have persistently normal alanine aminotransferase levels. Intervirology. 2006;49(1–2):58–63. doi: 10.1159/000087264. [DOI] [PubMed] [Google Scholar]
- 16.Longman RS, Talal AH, Jacobson IM, Rice CM, Albert ML. Normal functional capacity in circulating myeloid and plasmacytoid dendritic cells in patients with chronic hepatitis C. J Infect Dis. 2005;192(3):497–503. doi: 10.1086/431523. [DOI] [PubMed] [Google Scholar]
- 17.Kanto T, Inoue M, Miyatake H, et al. Reduced numbers and impaired ability of myeloid and plasmacytoid dendritic cells to polarize T helper cells in chronic hepatitis C virus infection. J Infect Dis. 2004;190(11):1919–1926. doi: 10.1086/425425. [DOI] [PubMed] [Google Scholar]
- 18.Wertheimer AM, Bakke A, Rosen HR. Direct enumeration and functional assessment of circulating dendritic cells in patients with liver disease. Hepatology. 2004;40(2):335–345. doi: 10.1002/hep.20306. [DOI] [PubMed] [Google Scholar]
- 19.Murakami H, Akbar SM, Matsui H, Horiike N, Onji M. Decreased interferon-alpha production and impaired T helper 1 polarization by dendritic cells from patients with chronic hepatitis C. Clin Exp Immunol. 2004;137(3):559–565. doi: 10.1111/j.1365-2249.2004.02550.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kakumu S, Ito S, Ishikawa T, et al. Decreased function of peripheral blood dendritic cells in patients with hepatocellular carcinoma with hepatitis B and C virus infection. J Gastroenterol Hepatol. 2000;15(4):431–436. doi: 10.1046/j.1440-1746.2000.02161.x. [DOI] [PubMed] [Google Scholar]
- 21.Li X, Lu S, Wang G, Yue B, Wang Z. Function of dendritic cell in chronic hepatitis C patients. Zhonghua Nei Ke Za Zhi. 2002;41(5):325–328. [PubMed] [Google Scholar]
- 22.Anthony DD, Yonkers NL, Post AB, et al. Selective impairments in dendritic cell-associated function distinguish hepatitis C virus and HIV infection. J Immunol. 2004;172(8):4907–4916. doi: 10.4049/jimmunol.172.8.4907. [DOI] [PubMed] [Google Scholar]
- 23.Saito K, Ait-Goughoulte M, Truscott SM, et al. Hepatitis C virus inhibits cell surface expression of HLA-DR, prevents dendritic cell maturation, and induces interleukin-10 production. J Virol. 2008;82(7):3320–3328. doi: 10.1128/JVI.02547-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsubouchi E, Akbar SM, Murakami H, Horiike N, Onji M. Isolation and functional analysis of circulating dendritic cells from hepatitis C virus (HCV) RNA-positive and HCV RNA-negative patients with chronic hepatitis C: role of antiviral therapy. Clin Exp Immunol. 2004;137(2):417–423. doi: 10.1111/j.1365-2249.2004.02544.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rollier C, Drexhage JA, Verstrepen BE, et al. Chronic hepatitis C virus infection established and maintained in chimpanzees independent of dendritic cell impairment. Hepatology. 2003;38(4):851–858. doi: 10.1053/jhep.2003.50426. [DOI] [PubMed] [Google Scholar]
- 26.Sarobe P, Lasarte JJ, Casares N, et al. Abnormal priming of CD4(+) T cells by dendritic cells expressing hepatitis C virus core and E1 proteins. J Virol. 2002;76(10):5062–5070. doi: 10.1128/JVI.76.10.5062-5070.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Piccioli D, Tavarini S, Nuti S, et al. Comparable functions of plasmacytoid and monocyte-derived dendritic cells in chronic hepatitis C patients and healthy donors. J Hepatol. 2005;42(1):61–67. doi: 10.1016/j.jhep.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 28.Longman RS, Talal AH, Jacobson IM, Albert ML, Rice CM. Presence of functional dendritic cells in patients chronically infected with hepatitis C virus. Blood. 2004;103(3):1026–1029. doi: 10.1182/blood-2003-04-1339. [DOI] [PubMed] [Google Scholar]
- 29.Tang TJ, Vukosavljevic D, Janssen HL, et al. Aberrant composition of the dendritic cell population in hepatic lymph nodes of patients with hepatocellular carcinoma. Hum Pathol. 2006;37(3):332–338. doi: 10.1016/j.humpath.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 30.Kanto T, Hayashi N, Takehara T, et al. Impaired allostimulatory capacity of peripheral blood dendritic cells recovered from hepatitis C virus-infected individuals. J Immunol. 1999;162(9):5584–5591. [PubMed] [Google Scholar]
- 31.Bain C, Fatmi A, Zoulim F, Zarski JP, Trepo C, Inchauspe G. Impaired allostimulatory function of dendritic cells in chronic hepatitis C infection. Gastroenterology. 2001;120(2):512–524. doi: 10.1053/gast.2001.21212. [DOI] [PubMed] [Google Scholar]
- 32.Auffermann-Gretzinger S, Keeffe EB, Levy S. Impaired dendritic cell maturation in patients with chronic, but not resolved, hepatitis C virus infection. Blood. 2001;97(10):3171–3176. doi: 10.1182/blood.v97.10.3171. [DOI] [PubMed] [Google Scholar]
- 33.Cai Z, Zhang C, Chang KS, et al. Robust production of infectious hepatitis C virus (HCV) from stably HCV cDNA-transfected human hepatoma cells. J Virol. 2005;79(22):13963–13973. doi: 10.1128/JVI.79.22.13963-13973.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wakita T, Pietschmann T, Kato T, et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med. 2005;11(7):791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shiina M, Rehermann B. Cell culture-produced hepatitis C virus impairs plasmacytoid dendritic cell function. Hepatology. 2008;47(2):385–395. doi: 10.1002/hep.21996. [DOI] [PubMed] [Google Scholar]
- 36.Ebihara T, Shingai M, Matsumoto M, Wakita T, Seya T. Hepatitis C virus-infected hepatocytes extrinsically modulate dendritic cell maturation to activate T cells and natural killer cells. Hepatology. 2008;48(1):48–58. doi: 10.1002/hep.22337. [DOI] [PubMed] [Google Scholar]
- 37.Caradonna L, Mastronardi ML, Magrone T, et al. Biological and clinical significance of endotoxemia in the course of hepatitis C virus infection. Curr Pharm Des. 2002;8(11):995–1005. doi: 10.2174/1381612024606983. [DOI] [PubMed] [Google Scholar]
- 38.Pachiadakis I, Pollara G, Chain BM, Naoumov NV. Is hepatitis C virus infection of dendritic cells a mechanism facilitating viral persistence? Lancet Infect Dis. 2005;5(5):296–304. doi: 10.1016/S1473-3099(05)70114-6. [DOI] [PubMed] [Google Scholar]
- 39.Caussin-Schwemling C, Schmitt C, Stoll-Keller F. Study of the infection of human blood derived monocyte/macrophages with hepatitis C virus in vitro. J Med Virol. 2001;65(1):14–22. doi: 10.1002/jmv.1095. [DOI] [PubMed] [Google Scholar]
- 40.Laporte J, Bain C, Maurel P, Inchauspe G, Agut H, Cahour A. Differential distribution and internal translation efficiency of hepatitis C virus quasispecies present in dendritic and liver cells. Blood. 2003;101(1):52–57. doi: 10.1182/blood-2002-03-0818. [DOI] [PubMed] [Google Scholar]
- 41.Miyazaki M, Kanto T, Inoue M, et al. Impaired cytokine response in myeloid dendritic cells in chronic hepatitis C virus infection regardless of enhanced expression of Toll-like receptors and retinoic acid inducible gene-I. J Med Virol. 2008;80(6):980–988. doi: 10.1002/jmv.21174. [DOI] [PubMed] [Google Scholar]
- 42.Bain C, Inchauspe G. Dendritic cells and hepatitis C virus. Pathol Biol (Paris) 2001;49(6):464–465. doi: 10.1016/s0369-8114(01)00166-3. [DOI] [PubMed] [Google Scholar]
- 43.Zhang T, Li Y, Ho WZ. Drug abuse, innate immunity and hepatitis C virus. Rev Med Virol. 2006;16(5):311–327. doi: 10.1002/rmv.508. [DOI] [PubMed] [Google Scholar]
- 44.Barth H, Ulsenheimer A, Pape GR, et al. Uptake and presentation of hepatitis C virus-like particles by human dendritic cells. Blood. 2005;105(9):3605–3614. doi: 10.1182/blood-2004-05-1952. [DOI] [PubMed] [Google Scholar]
- 45.Barth H, Schnober EK, Neumann-Haefelin C, et al. Scavenger receptor class B is required for hepatitis C virus uptake and cross-presentation by human dendritic cells. J Virol. 2008;82(7):3466–3479. doi: 10.1128/JVI.02478-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lin W, Kim SS, Yeung E, et al. Hepatitis C virus core protein blocks interferon signaling by interaction with the STAT1 SH2 domain. J Virol. 2006;80(18):9226–9235. doi: 10.1128/JVI.00459-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Taylor DR, Shi ST, Romano PR, Barber GN, Lai MM. Inhibition of the interferon-inducible protein kinase PKR by HCV E2 protein. Science. 1999;285(5424):107–110. doi: 10.1126/science.285.5424.107. [DOI] [PubMed] [Google Scholar]
- 48.Sarobe P, Lasarte JJ, Zabaleta A, et al. Hepatitis C virus structural proteins impair dendritic cell maturation and inhibit in vivo induction of cellular immune responses. J Virol. 2003;77(20):10862–10871. doi: 10.1128/JVI.77.20.10862-10871.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zabaleta A, Llopiz D, Arribillaga L, et al. Vaccination against hepatitis C virus with dendritic cells transduced with an adenovirus encoding NS3 protein. Mol Ther. 2008;16(1):210–217. doi: 10.1038/sj.mt.6300333. [DOI] [PubMed] [Google Scholar]
- 50.Torresi J, Bharadwaj M, Jackson DC, Gowans EJ. Neutralising antibody, CTL and dendritic cell responses to hepatitis C virus: a preventative vaccine strategy. Curr Drug Targets. 2004;5(1):41–56. doi: 10.2174/1389450043490677. [DOI] [PubMed] [Google Scholar]
- 51.Dolganiuc A, Chang S, Kodys K, et al. Hepatitis C virus (HCV) core protein-induced, monocyte-mediated mechanisms of reduced IFN-alpha and plasmacytoid dendritic cell loss in chronic HCV infection. J Immunol. 2006;177(10):6758–6768. doi: 10.4049/jimmunol.177.10.6758. [DOI] [PubMed] [Google Scholar]
- 52.Colonna M, Pulendran B, Iwasaki A. Dendritic cells at the host-pathogen interface. Nat Immunol. 2006;7(2):117–120. doi: 10.1038/ni0206-117. [DOI] [PubMed] [Google Scholar]
- 53.Takahashi H, Zeniya M. Do DCs influence the antiviral effect of interferon/ribavirin by changing their profile during the therapy? J Gastroenterol. 2006;41(8):816–817. doi: 10.1007/s00535-006-1863-5. [DOI] [PubMed] [Google Scholar]
- 54.Dolganiuc A, Kodys K, Kopasz A, et al. Hepatitis C virus core and nonstructural protein 3 proteins induce pro- and anti-inflammatory cytokines and inhibit dendritic cell differentiation. J Immunol. 2003;170(11):5615–5624. doi: 10.4049/jimmunol.170.11.5615. [DOI] [PubMed] [Google Scholar]
- 55.Thumann C, Schvoerer E, Abraham JD, et al. Hepatitis C virus structural proteins do not prevent human dendritic cell maturation. Gastroenterol Clin Biol. 2008;32(1 Pt 1):59–68. doi: 10.1016/j.gcb.2007.12.006. [DOI] [PubMed] [Google Scholar]
- 56.Liu Y, Zhou W, You C, et al. An autoimmune domain-reduced HCV core gene remains effective in stimulating anti-core cytotoxic T lymphocyte activity. Vaccine. 2006;24(10):1615–1624. doi: 10.1016/j.vaccine.2005.09.055. [DOI] [PubMed] [Google Scholar]
- 57.Li W, Li J, Tyrrell DL, Agrawal B. Expression of hepatitis C virus-derived core or NS3 antigens in human dendritic cells leads to induction of pro-inflammatory cytokines and normal T-cell stimulation capabilities. J Gen Virol. 2006;87(Pt 1):61–72. doi: 10.1099/vir.0.81364-0. [DOI] [PubMed] [Google Scholar]
- 58.Yonkers NL, Rodriguez B, Milkovich KA, et al. TLR ligand-dependent activation of naive CD4 T cells by plasmacytoid dendritic cells is impaired in hepatitis C virus infection. J Immunol. 2007;178(7):4436–4444. doi: 10.4049/jimmunol.178.7.4436. [DOI] [PubMed] [Google Scholar]
- 59.Shiina M, Kobayashi K, Kobayashi T, Kondo Y, Ueno Y, Shimosegawa T. Dynamics of immature subsets of dendritic cells during antiviral therapy in HLA-A24-positive chronic hepatitis C patients. J Gastroenterol. 2006;41(8):758–764. doi: 10.1007/s00535-006-1843-9. [DOI] [PubMed] [Google Scholar]
- 60.Miyatake H, Kanto T, Inoue M, et al. Impaired ability of interferon-alpha-primed dendritic cells to stimulate Th1-type CD4 T-cell response in chronic hepatitis C virus infection. J Viral Hepat. 2007;14(6):404–412. doi: 10.1111/j.1365-2893.2006.00814.x. [DOI] [PubMed] [Google Scholar]
- 61.Barnes E, Salio M, Cerundolo V, et al. Impact of alpha interferon and ribavirin on the function of maturing dendritic cells. Antimicrob Agents Chemother. 2004;48(9):3382–3389. doi: 10.1128/AAC.48.9.3382-3389.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Martini F, Sacchi A, Lalle E, et al. GB virus type C-driven protection in HIV/HCV coinfection: possible role of interferon gamma and dendritic cell activation. Gastroenterology. 2008;134(5):1631–1633. doi: 10.1053/j.gastro.2008.03.070. author reply 3. [DOI] [PubMed] [Google Scholar]
- 63.Mozer-Lisewska I, Dworacki G, Kaczmarek E, et al. Significance of alterations in PBMC immunophenotype of children with chronic viral hepatitis C-- the role of dendritic cells. Scand J Immunol. 2006;63(4):311–319. doi: 10.1111/j.1365-3083.2006.01741.x. [DOI] [PubMed] [Google Scholar]
- 64.Piazzolla G, Tortorella C, Schiraldi O, Antonaci S. Relationship between interferon-gamma, interleukin-10, and interleukin-12 production in chronic hepatitis C and in vitro effects of interferon-alpha. J Clin Immunol. 2000;20(1):54–61. doi: 10.1023/a:1006694627907. [DOI] [PubMed] [Google Scholar]
- 65.Jia HY, Du J, Zhu SH, et al. The roles of serum IL-18, IL-10, TNF-alpha and sIL-2R in patients with chronic hepatitis C. Hepatobiliary Pancreat Dis Int. 2002;1(3):378–382. [PubMed] [Google Scholar]
- 66.Gary-Gouy H, Lebon P, Dalloul AH. Type I interferon production by plasmacytoid dendritic cells and monocytes is triggered by viruses, but the level of production is controlled by distinct cytokines. J Interferon Cytokine Res. 2002;22(6):653–659. doi: 10.1089/10799900260100132. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Monocytes were isolated by adherence from healthy human PBMC. Immature DCs were obtained by culture of monocytes with GM-CSF and IL-4 for 6 days. Mature DCs were obtained by culturing Immature DC with LPS for 48 hours. Cells of each stage were incubated with HCV-JFH1, a genotype 2a strain of HCV that effectively replicates in culture at two different MOIs.
Monocytes, immature DC and mature DC incubated with an MOI of 0.1 or an MOI=0.01 of HCV-JFH1for 7 days were observed at 100X magnification in a light microscope. Representative pictures of triplicate experiments (three separate donors) are shown. A) Control monocytes cultured without virus; B) monocytes incubated with MOI 0.1; C) monocytes incubated with MOI 0.01; D) immature DC without HCV; E) immature DC with MOI of 0.1 of HCV; F) immature DC with an MOI of 0.01 of HCV; G) LPS matured DC without virus; H) LPS matured DC incubated with MOI of 0.1 of HCV; I) LPS matured DC incubated with MOI of 0.01.