

Abstract
DNA hypermethylation of promoter CpG islands is associated with epigenetic silencing of tumor suppressor genes in oral squamous cell carcinomas (OSCCs). We used a methyl-CpG-binding domain protein capture method coupled with next-generation sequencing (MBDCap-seq) to survey global DNA methylation patterns in OSCCs with and without nodal metastasis and normal mucosa (total n = 58). Of 1462 differentially methylated CpG islands identified in OSCCs relative to normal controls, MBDCap-seq profiling uncovered 359 loci linked to lymph node metastasis. Interactive network analysis revealed a subset of these loci (n = 23), including the anaplastic lymphoma kinase (ALK) gene, are potential regulators and effectors of invasiveness and metastatic progression. Promoter methylation of ALK was preferentially observed in OSCCs without node metastasis, whereas relatively lower methylation levels were present in metastatic tumors, implicating an active state of ALK transcription in the latter group. The OSCC cell line, SCC4, displayed reduced ALK expression that corresponded to extensive promoter CpG island methylation. SCC4 treatment with demethylating agents induced ALK expression and increased invasion and migration characteristics. Inhibition of ALK activity in OSCC cells with high ALK expression (CAL27, HSC3 and SCC25), decreased cell growth and resulted in changes in invasive potential and mesenchymal marker expression that were cell-line dependent. Although ALK is susceptible to epigenetic silencing during oral tumorigenesis, overwriting this default state may be necessary for modulating invasive processes involved in nodal metastases. Given the complex response of OSCC cells to ALK inhibition, future studies are required to assess the feasibility of targeting ALK to treat invasive OSCCs.
Introduction
Oral squamous cell carcinoma (OSCC) is the major malignant disease of oral mucosa and is closely associated with betel quid chewing, chronic smoking and drinking (1–4). OSCC is also the sixth most common cancer worldwide with an average 5 year survival rate of ~60% (3–8). The incidence and severity of OSCC are increasing worldwide, especially in Taiwan (5). From 1981 to 2010, the standardized mortality rates increased from 2.5 to 8.1 per 100000 persons (9). OSCC patients are treated by surgery, chemotherapy, radiotherapy or combination. The treatment modality depends on the clinical staging, with invasion of regional lymph nodes being a major concern (10). Currently, there are no reliable biomarkers available for early detection of nodal disease beyond conventional oral examination nor are there predictive biomarkers for OSCC with invasive potential (10,11). Although OSCCs arise from the superficial mucosa in the oral cavity where they may be detected during routine clinical examination, definitive diagnosis tends to be deferred due to difficulty in differentiating them from other oral lesions with similar presentations. Although image analysis is available for presurgical diagnosis of neck lymph node metastasis, detection method for micrometastases and biomarkers of invasive tumors are lacking. Thus, the identification of novel biomarkers is urgently needed for early detection of aggressive disease and regional lymph node invasion (10,11).
Increased DNA methylation of multiple promoter CpG islands has frequently been observed in OSCCs and is an untapped resource of prognostic biomarkers for this disease (12,13). Working hand-in-hand with histone modifications, promoter methylation is mediated by DNA methyltransferases and polycomb repressor complex 2 that remodel compact chromatin structure for transcription silencing of tumor suppressor genes (14,15). There is increasing evidence, however, that hyper- and hypomethylated states of the same CpG islands can be seen in different tumors, highlighting the dynamics of epigenetic plasticity during cancer development (16). Because aberrant methylation patterns are stable and can inherently be transmitted from parental to daughter cells, hyper- and hypomethylated loci acquired during tumorigenesis are ideal biomarkers for diagnosis and prognosis of OSCCs.
In the present study, we used an affinity-based methylation capture assay coupled with next-generation sequencing to survey global changes of DNA methylation in OSCCs with and without neck lymph node metastasis (i.e. Met versus Non-met group). More than 2.4 billion sequence reads were processed through a bioinformatics pipeline, providing rich data sets for identifying aberrantly methylated loci in this cancer type. We cataloged a large set of hypermethylated CpG islands in OSCCs relative to normal mucosa controls. These aberrant events can lead to epigenetic silencing of multiple tumor suppressor genes and promote proliferation of transformed oral mucosal cells. Interestingly, we observed a subset of candidate CpG islands that were slightly methylated in the Met group relative to normal mucosal control, whereas the same loci were highly methylated in the Non-met group compared with normal controls. One such locus is the anaplastic lymphoma kinase (ALK), which we found to be frequently in a hypomethylated state in Met tumors relative to Non-met tumors. As a result of oncogenic mutations and fusions, overexpressed ALK greatly enhances Ras/Snail-mediated malignant transformation and invasion in several types of cancer (17,18). Here we report that hypomethylated ALK is a hallmark for active transcription of this gene within the context of widespread hypermethylation in OSCCs. Our data also suggest that ALK plays a pleiotropic role in modulating mesenchymal characteristics associated with aggressive growth and invasiveness of OSCC that may result in tumors with potential to invade regional critical structures and be refractive to traditional treatments. This epigenetically deregulated locus can be a potential prognostic biomarker for neck lymph node metastasis and for targeted treatment of OSCCs.
Materials and methods
Tissue specimens
Approved by the Institutional Review Boards, we collected tissue sections from archival formalin-fixed, paraffin-embedded blocks of 124 OSCC patients and 18 patients with benign oral mucosal lesion in the Dalin Tzu Chi General Hospital, Chiayi, Taiwan. Histological typing and staging were performed according to the American Joint Committee on Cancer guidelines (19). Clinicopathological data of studied tumors are presented in Supplementary Table S1, available at Carcinogenesis Online.
Cell culture conditions
Four OSCC cell lines were used in the present study: SCC4, CAL27 and SCC25 were obtained from ATCC and HSC3 was kindly provided by Dr Brian Schmidt. Cells were grown in Dulbecco’s modified Eagle’s medium culture medium with high glucose (Life Technologies, Grand Island, NY) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin at 37°C. For epigenetic inhibition studies, SCC4 cells were first treated with 5-aza-2′-deoxycytidine (DAC, 6 μM) and then with trichostatin A (TSA, 0.3 μM) (Sigma–Aldrich, St Louis, MO) following the protocol established previously by our laboratory (20). Control cells were treated with the 3 μM dimethyl sulfoxide. The potent and highly specific ALK inhibitor TAE684 (21) was purchased from Selleckchem (ThermoFisher, Pittsburg, PA). Treatment conditions with TAE684 are described in the corresponding section to the particular experiment.
DNA isolation and MBDCap-seq assay
Tumor and normal oral mucosa areas were mapped based on hematoxylin and eosin staining of paraffin blocks and macrodissected for DNA isolation (2 μg per sample) using the Pinpoint Slide DNA Isolation System™ (Zymo Research, Irvine, CA). Methylated DNA was eluted by the MethylMiner Methylated DNA Enrichment Kit (Life Technologies) following the manufacturer’s instructions. Briefly, genomic DNA sonicated to ~300 bp was captured by methyl-CpG-binding domain proteins and eluted in 1 M salt buffer for precipitation. Eluted DNA (>10 ng) was used to generate libraries for sequencing following the standard protocols from Illumina (San Diego, CA). MBDCap-seq libraries were sequenced using the Illumina HiSeq 2000 system as per manufacturer’s instructions. Image analysis and base calling were performed with the standard Illumina pipeline. Using the ELAND algorithm, unique reads (up to 50 bp reads) were mapped to the human reference genome (hg18), with up to two mismatches (see Supplementary Table S2, available at Carcinogenesis Online).
Bioinformatic analysis of next-generation sequencing data
Sequence reads mapped to unique genome locations were normalized to the sum of unique reads and other reads (e.g. repeat sequences). After normalization, unique reads were collected and directionally extended to 234 bp. The methylation level of each bin size was calculated by accumulating the reads number if the read was located in the bin. Then, methylation enrichment was normalized using the linear normalization method based on the uniquely mapped read number:
 |
(1) |
where  is the normalized read number of the ith bin,
is the normalized read number of the ith bin,  is the uniquely mapped read number of the ith bin and
is the uniquely mapped read number of the ith bin and  is the total uniquely mapped reads number. ‘INT’ function rounds the element (in the parenthesis) to the nearest integers toward minus infinity and ‘^’ means the power operator. For two groups A and B, the sample number is
is the total uniquely mapped reads number. ‘INT’ function rounds the element (in the parenthesis) to the nearest integers toward minus infinity and ‘^’ means the power operator. For two groups A and B, the sample number is  for group A and
for group A and  for group B. For a given region R (which includes m bin size and start at the sth bin), the average methylation level is
for group B. For a given region R (which includes m bin size and start at the sth bin), the average methylation level is
 |
(2) |
for group A or B (if G is A or B). In Equation (2),
is A or B). In Equation (2),  means the average methylation level of group
means the average methylation level of group  at region
at region 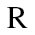 and
and  is the methylation levels of each sample of group
is the methylation levels of each sample of group 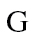 at region
at region 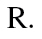 We then used paired t-test to compare if the methylation level of the region was significantly different between those two groups. Differentially methylated loci between OSCCs and normal controls and between the Met group and the Non-met group were analyzed by the above algorithm. Candidate loci of a gene network were further analyzed by Ingenuity Pathway Analysis software (Ingenuity Systems, Redwood City, CA).
We then used paired t-test to compare if the methylation level of the region was significantly different between those two groups. Differentially methylated loci between OSCCs and normal controls and between the Met group and the Non-met group were analyzed by the above algorithm. Candidate loci of a gene network were further analyzed by Ingenuity Pathway Analysis software (Ingenuity Systems, Redwood City, CA).
Pyrosequencing analysis
DNA (300 ng per sample) was subjected to bisulfite conversion using the EZ DNA Methylation™ Kit (Zymo Research). Primers used to amplify specific CpG island regions are shown in Supplementary Table S3, available at Carcinogenesis Online. CpG islands were defined by the following criteria: (i) they are at least 200 bp in length, (ii) G/C content was >50% and (iii) observed/expected CpG ratio (number of CpGs × length/number of C × G) was >0.6 (22). Methylated CpG sites were detected by the PyroMark Q96 MD System. Incomplete bisulfite conversion checkpoint was set as 5%. The methylation percentage of each interrogated CpG site was calculated and visualized using MultiExperiment Viewer v4.8 (Dana-Farber Cancer Institute, Boston, MA) and GraphPad Prism® (GraphPad Software).
Reverse transcription–polymerase chain reaction analysis
RNA was isolated from treated and control cells for reverse transcription–polymerase chain reactions (RT–PCRs). Real-time PCR was performed with SuperScript® III RT (Invitrogen™, Life Technologies) using the StepOnePlus™ Real-Time PCR Systems (Applied Biosystems®, Life Technologies). The ΔΔCt was calculated using the housekeeping gene GAPDH as a reference. Primer sequences for RT–PCR assays are shown in Supplementary Table S3, available at Carcinogenesis Online.
Protein analysis
Western blot analysis was performed as described previously (23). Briefly, 40–60 μg of protein was loaded on 10% bis/tris polyacrylamide gels under denaturing/reducing conditions and then transferred to polyvinylidene difluoride membranes in an iBlot electroblotter per manufacturers’ recommended conditions (Life Technologies). After blocking with tris-buffered saline containing 0.1% Tween-20 (TBST) and 5% milk for 1 h and then washing with TBST, membranes were incubated with primary antibody diluted in TBST (5% bovine serum albumin) for 1 h, 2 h or overnight (depending on antibody). Protein bands were visualized after 1 h incubation with secondary antibody (conjugated to horseradish peroxidase) in TBST (5% milk) followed by exposure to the enhanced chemiluminescence-based detection system and X-ray autoradiography (GE Healthcare; Amersham, Piscataway, NJ). Housekeeping factor GAPDH was used as loading control. Antibodies for E-cadherin (120 kDa) and fibronectin (230 kDa) were purchased from BD Biosciences (San Jose, CA). Antibodies for SNAIL1 (29 kDa) and GAPDH (40 kDa) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA).
Wound healing, invasion and proliferation assays
Wound healing assays of OSCC cell lines were performed by scratching the culture dish with 200 μl pipet tip and then observed cell migration 8 h later. Invasion assays were performed with seeding 2.5 × 104 cells on BD BioCoat™ Matrigel™ Matrix (BD Biosciences) and observed cell invasion through matrigel 20 h later. Cellular proliferation was assayed using the CellTiter-Glo® Luminescence Kit (Promega, Madison, WI) according to the manufacturer’s directions and as described earlier (23).
Statistical analysis
Student’s t-test was used to compare pyrosequencing, RT–PCR, wound healing and invasion results in different treatment and control groups. Statistical significance was assigned as *P < 0.05, **P < 0.01 and ***P < 0.001.
Results
Use of MBDCap-seq to survey global methylation changes in OSCCs
Two age-matched groups of OSCCs with and without neck lymph node metastasis (i.e. Met versus Non-met) were used for global methylation screening. As shown in Figure 1A and B, two representative OSCCs had lower gingival squamous cell carcinomas with moderately differentiated pT4a tumors. However, pathological examinations of lymph nodes revealed that the first patient was positive for metastasis, whereas the second patient had negative nodes (Figure 1C). Detailed clinicopathological information of the studied cohort (Met: 23, Non-met: 142 and control: 7) is presented in Supplementary Table S1, available at Carcinogenesis Online. Tumor areas were mapped based on hematoxylin and eosin staining of formalin-fixed, paraffin-embedded samples followed by macrodissection for DNA isolation. Control mucosal samples were obtained from healthy individuals.
Fig. 1.

Clinical photographs and pathological sections of oral cancer patients with or without neck lymph node metastasis. (A) and (A’) Images show OSCC with and without metastasis, respectively. Arrow indicates cancer lesion. (B) and (B’) Histological sections of primary tumors with and without metastasis, respectively (images from two representative tumors are shown, ×400). (C) and (C’) Histological sections of OSCCs with and without neck lymph node metastasis, respectively (images from two representative tumors are shown, ×400).
We first used MBDCap-seq to survey DNA methylation patterns in 23 Met tumors, 28 Non-met tumors and 7 normal control samples. Although the bisulfite-based next-generation sequencing has been used to generate whole-genome DNA methylation landscapes at single-base resolution (24,25), this approach is presently not feasible for comprehensive analysis of a large group of clinical samples due to the vast number of sequence reads required (>1 × 109 reads per sample). In contrast, the use of the methyl-DNA-binding protein column offers the advantage of direct elution of methylated DNA fragments without further bisulfite treatment or methylation-sensitive restrictions (26). In our hand, MBDCap-seq was recently used to successfully identify multiple hypermethylated CpG islands in cancer genomes (27). Methylated fragments, bound to the methyl-CpG-binding domain protein MBD2, were eluted for sequencing with the Illumina HiSeq 2000 sequencing system (Figure 2A). We processed 2 430 819 013 100 bp sequence reads from the 58 clinical samples, 62% of which were mapped to unique genome locations (Supplementary Table S2, available at Carcinogenesis Online). Because DNA methylation is expected to occur in GC-rich regions, the minimum coverage of 10 million unique reads was reported to provide sufficient sequence depth for methylation mapping of the genome (Figure 2B) (27). After data normalization, pairwise comparison within an 8 kb window was applied to identify differentially methylated CpG island loci in OSCCs by Student’s t-test. Technical repeats were conducted, and the results suggest that MBDCap-seq can reliably identify DNA methylation sites in OSCC genomes (Supplementary Figure S1, available at Carcinogenesis Online). Of 18 535 known promoter CpG islands analyzed, 1462 loci (8%) were found to be commonly hyper- or hypomethylated in OSCCs relative to mucosa controls (Figure 2C) (Supplementary Tables S4 and S5, available at Carcinogenesis Online). These loci displayed either CpG core methylation or CpG shore methylation (right, left or both flanks). The unique MBDCap-seq data set provides new opportunities to identify methylation biomarkers for OSCC prognostication.
Fig. 2.

MBDCap-seq analysis showing global differential methylation in OSCCs with and without metastasis. (A) Scheme outlining experimental procedures of MBDCap-seq using dissected tumor and control samples. DNA from sectioned paraffin-embedded tissue samples is subjected to MBD2 capture. After library construction and next-generation sequencing, data are analyzed to identify differentially methylated loci. (B) Summarized sequencing reads in normal samples and tumors with (Met) and without metastasis (Non-met). Each bar represents an individual sample. Blue and red bars indicate unique reads and repeat sequences, respectively. Dashed line indicates 10 million reads. (C) Methylation levels of loci, upstream 4 kb and downstream 4 kb from transcription start site of OSCC and normal samples. Left panel: hypermethylated loci in OSCC; right panel: hypomethylated loci in OSCC. (D) Methylation levels of loci of Met and Non-met samples (left panel: hypermethylated loci in Met; right panel: hypomethylated loci in Met).
Identification of differentially methylated CpG islands that are highly predictive of lymph node metastasis
Pairwise comparison using t-test between Met and Non-met tumors further identified 359 promoter CpG islands linked to lymph node metastasis, including 259 hypermethylated and 100 hypomethylated loci (Figure 2D and Supplementary Tables S6 and S7, available at Carcinogenesis Online). Cellular functions of these candidate loci are linked to cell death and apoptosis (e.g. DEDD2, GPR65, ARHGEF2, BMF, PDGFRA, CLTC and ALK), cell cycle regulation and cell proliferation (e.g. FGF3, FGF4, FGF19, TLN1 and CCND1) and cell migration and mobility (e.g. MYLK and TGFBR2) (28–32). Many of these loci are known to be involved in oncogenic signal transduction, including PI3K/AKT, NFκB and WNT/β-catenin pathways (33–36). Therefore, epigenetic deregulation of these candidate genes may contribute to oncogenic progression in OSCCs. Ingenuity Pathway Analysis further identified an interactive network of 23 loci, including DOK5, GAD2, HMGA2, F7, PTK6 and ALK, that were differentially methylated in OSCCs associated with nodal metastasis compared with non-metastatic tumors (Figure 3A–C). For example, the promoter of ALK was found to be methylated in 75% of non-metastatic OSCC, 58% of metastatic OSCC and 14% of normal tissue (Figure 3B).
Fig. 3.

Identification of differentially methylated loci in metastatic OSCCs. (A) Ingenuity Pathway Analysis identified a protein interaction network of 23 loci that includes ALK. Fourteen loci are hypermethylated and 11 loci hypomethylated in metastatic OSCCs. (B) Methylation heat map indicating differential methylation levels of each of the loci in this network. Note that ALK is hypomethylated in metastatic compared with non-metastatic tumors. (C) Differential methylation of DOK5, GAD2, HMGA2, ALK, F7 and PTK6 is shown in box-and-whisker graphs. Error bars indicate standard deviation. *P < 0.05 and **P < 0.01.
In silico analysis of The Cancer Genome Atlas data sets (http://cancergenome.nih.gov/) was conducted to determine whether promoter hypermethylation of ALK is associated with transcriptional repression. Because a specific oral cancer cohort is not available, we used the head and neck cancer cohort in The Cancer Genome Atlas for the analysis. In contrast to the common notion that promoter hypermethylation leads to gene silencing resulting in cancer progression, we found that hypomethylated ALK in head and neck cancer displayed increased expression levels of ALK relative to those of normal controls (P = 0.0068; Supplementary Figure S2A, available at Carcinogenesis Online). When using a 75 percentile cut-off for the high expression group, we found that elevated methylation levels are associated with decreased expression of ALK in tumors (P < 0.001; Supplementary Figure S2B, available at Carcinogenesis Online). The other 25% of the cohort had elevated expression of ALK with low methylation levels in its promoter CpG island.
To confirm the MBDCap-seq data, we conducted pyrosequencing analysis of the ALK locus, which we found to be frequently less methylated in Met tumors compared with its methylation status in Non-met tumors. Pyrosequencing primer sequences and locations are listed in Supplementary Figure S3, available at Carcinogenesis Online. Pyrosequencing results of 32 Met tumors, 92 Non-met tumors and 18 normal controls are presented in Figure 4A and B. Among 12 CpG sites interrogated within the ALK CpG island, hypomethylation was significantly observed in sites CpG #1 (P < 0.029), #5 (P < 0.004), #6 (P < 0.005), #7 (P < 0.002), #9 (P < 0.02) and #12 (P < 0.001) in the Met group relative to the Non-met group (Figure 4C). Lower levels of methylation suggest permissive transcriptional activity of ALK in Met tumors, whereas elevated methylation status in the Non-met group is indicative of epigenetic repression.
Fig. 4.

Pyrosequencing of ALK CpG sites confirms its hypomethylation in OSCC tumors with metastasis. (A) Heat map showing methylation percentage of each of the 12 ALK CpG sites for normal, non-metastatic and metastatic tumor samples. The Met group is hypomethylated relative to the Non-met group. Pyrosequencing primers containing interrogated CpG sites are indicated above. (B) Methylation of the CpG sites is represented in box-and-whisker graphs.
ALK expression is enhanced and invasion and motility are induced by epigenetic derepression
DNA methylation analysis of the ALK promoter’s 12 CpG sites was conducted in four OSCC cell lines (Figure 5A). Pyrosequencing data indicated that all the ALK CpGs were extensively methylated in SCC4 cells (Figure 5A), which appeared to be associated with reduced ALK expression in this cell line (Figure 5B, first lane). In contrast, partial DNA methylation (methylated at CpG #1–4) was observed in the other three oral cancer cell lines (CAL27, SCC25 and HSC3) exhibiting higher ALK expression (Figure 5A and B). Because metastatic development depends in part on the acquisition of mesenchymal phenotype by epithelial tumor cells, we examined the DNA methylation states of epithelial-to-mesenchymal transition (EMT) transcription factors, SNAI1, SNAI2, TCF3 and TWIST1, in the OSCC cell lines (Figure 5A). Our pyrosequencing data showed little (11–20%) to no methylation in these genes in the OSCC lines with the exception of TWIST1, which exhibited 41–65% methylation in its promoter region (Figure 5D). This suggests that TWIST1 expression may be regulated by epigenetic mechanisms.
Fig. 5.

Reversal of ALK methylation in OSCC cells leads to induction of ALK, cell motility and invasion. (A) Heat maps showing ALK, SNAI1, SNAI2, TCF3 and TWIST methylation status in OSCC cell lines. (B) Expression analysis of ALK mRNA in OSCC cell lines, as determined by real-time RT–PCR. (C) DAC (6 μM) and TSA (0.3 μM) treatments resulted in induction of ALK mRNA expression in SCC4 cells, determined by RT–PCR analysis (left panel). An induction in expression of EMT-related transcription factors SNAI1, SNAI2, TWIST1 and TCF3 is also observed. Western blot analysis was performed to examine the protein expression of SNAI1, fibronectin and E-cadherin (right panel). (D) DAC (6 μM) and TSA (0.3 μM) treatments led to a morphology change characteristic of an EMT-like phenotype in SCC4 cells. Note the change in cobblestone morphology, characteristic of epithelial cells, due to the treatment. (E) Wound healing assay, repeated five times, was performed to examine the effects of DAC and TSA treatments on cell motility (top panels). Invasion assay through matrigel invasion chambers was performed three times on control and DAC-treated SCC4 cells (bottom panels). Error bars represent standard deviation. *P < 0.05, **P < 0.01 and ***P < 0.001.
To confirm the association between ALK methylation and low expression in SCC4 cells, we sequentially treated SCC4 cells with a DNA demethylating agent, DAC (6 μM), and a histone deacetylase inhibitor, TSA (0.3 μM), for 4 days, which did not cause cytotoxicity in SCC4 cells (data not shown). We observed a strong induction of ALK expression in treated cells (Figure 5C). This was accompanied by induction of the RNA expression of SNAI1, SNAI2, TWIST1 and TCF3 analyzed in this cell line (Figure 5C). However, this treatment did not have an effect on the expression of SNAI1 protein or the mesenchymal factor fibronectin (Figure 5C). Treatment with DAC and TSA also resulted in disruption of SCC4 cell–cell contact characteristic of epithelial cells (Figure 5D) and increased cell mobility and invasiveness (Figure 5E). Combined, the data suggest that ALK expression may depend on the methylation status of its promoter and that removal of this epigenetic repression is associated with increased cellular invasiveness and motility. To further investigate the role of ALK in these processes implicated in metastatic growth, we examined the effects of inhibiting ALK activity on OSCC cell lines exhibiting elevated ALK expression as described below.
Inhibition of ALK activity diminishes cell growth and invasion in OSCC cells
We treated HSC3, CAL27 and SCC25 cells with the potent ALK inhibitor TAE684 (21) to determine the role of ALK on the oncogenic properties of OSCC cells. We first examined the effects of TAE684 on the growth of these cell lines. All three cells exhibited dose–response-dependent inhibition of cell growth, with HSC3 showing the most drastic decrease in cell growth (Figure 6A). On the other hand, these OSCC cell lines exhibited different responses to TAE684 in regards to invasiveness (Figure 6A). Invasion by HSC3 cells was most diminished by TAE684 at 20 nM compared with the other cell lines. This could be attributed to the more mesenchymal nature of HSC3, as shown by absence of strong cell–cell contact that is a characteristic of epithelial cells (Figure 6B). In addition, lower E-cadherin and higher fibronectin expression was observed in HSC3 cells compared with the other two cell lines (Figure 6C). Surprisingly, TAE684 increased the invasion of CAL27 cells in a dose-dependent manner (Figure 6A), a change that was associated with a strong induction of the mesenchymal transcription factor SNAI1 (Figure 6C). Although the data do not directly implicate ALK in inducing EMT in OSCCs, they do suggest that ALK may modulate the mesenchymal phenotype in oral cancer cells either by contributing to invasion or, in some oral cancer cell types, by initiating a mesenchymal-to-epithelial transition (MET) (37,38).
Fig. 6.

Inhibition of ALK activity leads to decreased cellular growth and a differential response in invasiveness in OSCC cell lines. OSCC cells HSC3, SCC25 and CAL27 were treated with the potent ALK inhibitor TAE864 at the indicated concentrations. (A) Cell growth was determined by the CellTiter-Glo® luminescence-based assay as described in Materials and methods for up to 96 h (upper panels). Invasion by HSC3, SCC25 and CAL27 was performed using transwell invasion chambers layered with matrigel (lower panels). Invasion was carried out for 20 h. (B) Microscopic images of HSC3, SCC25 and CAL27 cells. Note a mesenchymal morphologic appearance and absence of strong cell–cell contact in HSC3 cells. However, strong cell–cell contact characteristic of epithelial cells is observed in SCC25 and CAL27 cells. (C) Protein lysates were isolated from HSC3, SCC25 and CAL27 cells treated with TAE684 (20 and 100 nM) for subsequent western blot analysis of E-cadherin, SNAI1 and fibronectin. Treatment with TAE684 was carried out for 48 h. Error bars indicate standard deviation. *P < 0.05, **P < 0.01 and ***P < 0.001.
Discussion
Based on the current paradigm of oncogenesis, tumor suppressor genes are susceptible targets of epigenetic silencing that frequently leads to aberrant cell proliferation and tumor progression (15). DNA methyltransferases and polycomb complex proteins are important factors to modify chromatin structure of these susceptible loci into a permanent repressive state (14,15). However, it is presently unclear how other genes required for malignant growth and invasiveness are expressed in the global context of epigenetic repression. Our MBDCap-seq analysis detected a large set of hypermethylated loci in primary OSCCs, consistent with the described role of promoter hypermethylation in tumorigenesis (15). However, a subset of CpG islands (n = 100) were relatively less methylated in tumors exhibiting lymph node metastasis than in tumors without this metastasis (see Figure 2D). Even though both tumor groups showed higher methylation levels of the candidate loci than normal tissue, relative hypomethylation in tumors with metastasis compared with non-metastatic tumors suggests that some of the CpG islands may be partially protected against methylation in the former tumor group due to active transcription of these genes, which may prevent the access of DNA methyltransferases and polycomb complex proteins. Gene network analysis further uncovered a unique set of 11 hypomethylated loci implicated in tumor metastasis. Two of these genes, NCAM1 and CHL1, are known to encode oncogenic functions for cell adhesion, whereas five other genes, HIF3A, TRAF7, PTK6, TNFRSF10C and ALK, are potential regulators and effectors of signaling pathways involved in cellular stress, inflammation, cell migration and survival (39–43). Metastatic functions of the remaining genes, F7, CCDC92, SLC9A3 and IRS4, are presently unknown and can be explored in the future.
Our functional validation studies focused on one of these genes, the receptor tyrosine kinase ALK, which has not been described in OSCCs. Aberrantly expressed ALK can activate Ras signaling pathways through the direct binding of insulin receptor substrate 1 that triggers signaling interaction cascades in P13K or Raf/MAPK transduction pathways (44–46). Activation of these signaling pathways leads to altered activity and expression of downstream genes, including SNAI1, TCF/LEF, TWIST and E-Cadherin, which are known to regulate EMT-mediated cell migration and invasion (47,48).
Our evidence, however, indicated that ALK may play a role in modulating the mesenchymal characteristics of OSCCs rather than initiation of EMT. For example, ALK inhibitor TAE684 did not affect the expression of E-cadherin, whose repression is a hallmark of EMT establishment in oral cancer and other malignancies (37,38,49,50). Furthermore, ALK inhibition of all cell lines tested resulted in significant reduction of cell proliferation.
The effects of ALK inhibition on cell invasion and EMT gene expression resulted in different phenotypic responses by the various OSCC cell lines and seemed to depend on their inherent epithelial/mesenchymal characteristics. For example, HSC3 cells exhibit the most mesenchymal features among all OSCC cell lines tested. They express high levels of E-cadherin, Snail 1 and fibronectin. HSC3 cells are fast growing, highly invasive in vitro and readily invade and metastasize in nude mouse xenograft models (unpublished data). HSC3 treatment with the ALK inhibitor TAE684 resulted in dramatic inhibition of growth and invasion in these cells. In contrast, CAL27 cells grow more slowly and are moderately invasive in vitro and locally invasive in xenograft models, given lengthy periods of growth, and fail to express SNAIL1 and fibronectin. CAL27 treatment with TAE684 resulted in reduced proliferation but at a lesser degree than that observed in HSC3 cells. An unexpected finding was that ALK inhibition lead to increased invasion and expression of SNAIL1 in CAL27 cells, suggesting a potential role for ALK in MET in these cells. MET contribution to metastatic growth has been documented (37,38). Although EMT is involved in invasiveness and dissemination of cancer cells to distant sites, MET is required for the colonization of these sites by suppressing invasion and increasing proliferation (37). Lastly, SCC25 cells are very slow growing cells that are not invasive in xenograft models and take extensive periods of time to form tumors in vivo (~8 weeks) and do not express Snail 1 or fibronectin. TAE684 treatment of SCC25 cells yielded no observable effects on invasion and no increases in EMT gene expression. Combined, the data suggest that ALK has pleiotropic effects in OSCCs, contributing to growth and invasion at different stages of tumor progression leading to metastasis.
Upregulation of ALK can frequently be attributed to chromosomal rearrangements that bring hyperactive EML4 or FN1 promoter to fuse at the 5′-end of this gene (51,52). The oncogenic fusions may promote constitutive expression of this receptor tyrosine kinase for malignant transformation and invasion reported in anaplastic large-cell lymphoma (53), neuroblastoma (54), inflammatory myofibroblastic tumor (42), non-small cell lung cancer (51) and ovarian cancer (52). However, our initial study indicates that these oncogenic fusions may not occur in OSCCs, suggesting other transcription regulatory mechanisms may be responsible for ALK activation in oral cancer progression (Supplementary Figure S4, available at Carcinogenesis Online). One such transcription factor is the Paired-like Homeobox 2B protein known to bind the ALK promoter, providing a future direction for the mechanistic study in metastatic OSCCs (55).
In conclusion, low-level methylation of ALK and other candidate genes can be preferentially present in metastatic OSCCs. These loci are therefore candidate biomarkers for definitive diagnosis of aggressive OSCC with invasive potential and/or lymph node metastasis in oral cancer patients. Our data suggest that further studies are required in this group of patients to examine the suitability of epigenetic therapies designed to effectively reactivate tumor suppressor genes in advanced solid tumors, given our observation that ALK expression is induced by this class of agents. One alternative approach is the use of small molecule inhibitors targeting ALK to treat metastatic OSCCs. In this regard, it is important to further investigate the role of ALK in metastatic growth in OSCCs and the usability of such drugs in treating advanced stages of OSCC.
Supplementary material
Supplementary Table S1–S7 and Figures S1–S4 can be found at http://carcin.oxfordjournals.org/
Funding
Integrative Cancer Biology Program (R01CA069065, R01ES017594 and U54CA113001) and Cancer Center Support Grant (P30CA054174) of the National Institutes of Health; Cancer Therapy and Research Center Foundation.
Supplementary Material
Acknowledgements
The authors thank the kind gift of HSC3 cell line from Dr Brian Schmidt’s laboratory. We would also like to thank the Genomic Sequencing Facility of the Greehey Children’s Cancer Research Institute, University of Texas Health Science Center at San Antonio for next-generation sequencing.This work was supported by R01CA069065, R01ES017594, U54CA113001 (Integrative Cancer Biology Program), and P30CA054174 (Cancer Center Support Grant) of the National Institutes of Health and by generous gifts from the Cancer Therapy and Research Center Foundation.
Conflict of Interest Statement: None declared.
Glossary
Abbreviations:
- ALK
anaplastic lymphoma kinase
- DAC
5-aza-2′-deoxycytidine
- EMT
epithelial-to-mesenchymal transition
- MET
mesenchymal-to-epithelial transition
- OSCC
oral squamous cell carcinoma
- RT–PCR
reverse transcription–polymerase chain reaction
- TBST
tris-buffered saline containing 0.1% Tween-20
- TSA
trichostatin A.
References
- 1. Zygogianni A.G., et al. (2011). Oral squamous cell cancer: early detection and the role of alcohol and smoking. Head Neck Oncol., 3, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ekramuddaula F.M., et al. (2011). Evaluation of risk factors of oral cancer. Mymensingh Med. J., 20, 412–418 [PubMed] [Google Scholar]
- 3. Priebe S.L., et al. (2010). Oral squamous cell carcinoma and cultural oral risk habits in Vietnam. Int. J. Dent. Hyg., 8, 159–168 [DOI] [PubMed] [Google Scholar]
- 4. Zheng S., et al. (2010). Northwestern China: a place to learn more on oesophageal cancer. Part one: behavioural and environmental risk factors. Eur. J. Gastroenterol. Hepatol., 22, 917–925 [DOI] [PubMed] [Google Scholar]
- 5. Warnakulasuriya S. (2009). Global epidemiology of oral and oropharyngeal cancer. Oral Oncol., 45, 309–316 [DOI] [PubMed] [Google Scholar]
- 6. Parkin D.M., et al. (1988). Estimates of the worldwide frequency of sixteen major cancers in 1980. Int. J. Cancer, 41, 184–197 [DOI] [PubMed] [Google Scholar]
- 7. Nagpal J.K., et al. (2003). Oral cancer: reviewing the present understanding of its molecular mechanism and exploring the future directions for its effective management. Oral Oncol., 39, 213–221 [DOI] [PubMed] [Google Scholar]
- 8. Walker D.M., et al. (2003). The pathology of oral cancer. Pathology, 35, 376–383 [DOI] [PubMed] [Google Scholar]
- 9. Department of Health (2010). Executive Yuan (R.O.C.), Taiwan
- 10. Oliveira L.R., et al. (2011). Prognostic significance of immunohistochemical biomarkers in oral squamous cell carcinoma. Int. J. Oral Maxillofac. Surg, 40, 298–307 [DOI] [PubMed] [Google Scholar]
- 11. Viet C.T., et al. (2010). Understanding oral cancer in the genome era. Head Neck., 32, 1246–1268 [DOI] [PubMed] [Google Scholar]
- 12. Garcia M.P., et al. (2012). Epigenome and DNA methylation in oral squamous cell carcinoma. Methods Mol. Biol., 863, 207–219 [DOI] [PubMed] [Google Scholar]
- 13. Shaw R. (2006). The epigenetics of oral cancer. Int. J. Oral Maxillofac. Surg., 35, 101–108 [DOI] [PubMed] [Google Scholar]
- 14. Baylin S.B., et al. (2011). A decade of exploring the cancer epigenome—biological and translational implications. Nat. Rev. Cancer, 11, 726–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jones P.A., et al. (2007). The epigenomics of cancer. Cell, 128, 683–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lin H.J., et al. (2009). Seed in soil, with an epigenetic view. Biochim. Biophys. Acta, 1790, 920–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chiarle R., et al. (2008). The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat. Rev. Cancer, 8, 11–23 [DOI] [PubMed] [Google Scholar]
- 18. Kutok J.L., et al. (2002). Molecular biology of anaplastic lymphoma kinase-positive anaplastic large-cell lymphoma. J. Clin. Oncol., 20, 3691–3702 [DOI] [PubMed] [Google Scholar]
- 19. Edge S.B., et al. (2010). The American Joint Committee on Cancer: the 7th edition of the AJCC cancer staging manual and the future of TNM. Ann. Surg. Oncol., 17, 1471–1474 [DOI] [PubMed] [Google Scholar]
- 20. Hsu P.Y., et al. (2010). Estrogen-mediated epigenetic repression of large chromosomal regions through DNA looping. Genome Res., 20, 733–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lovly C.M., et al. (2011). Insights into ALK-driven cancers revealed through development of novel ALK tyrosine kinase inhibitors. Cancer Res., 71, 4920–4931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gardiner-Garden M., et al. (1987). CpG islands in vertebrate genomes. J. Mol. Biol., 196, 261–282 [DOI] [PubMed] [Google Scholar]
- 23. De La Garza E.M., et al. (2012). Raf-1, a potential therapeutic target, mediates early steps in endometriosis lesion development by endometrial epithelial and stromal cells. Endocrinology, 153, 3911–3921 [DOI] [PubMed] [Google Scholar]
- 24. Cokus S.J., et al. (2008). Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature, 452, 215–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Berman B.P., et al. (2012). Regions of focal DNA hypermethylation and long-range hypomethylation in colorectal cancer coincide with nuclear lamina-associated domains. Nat. Genet., 44, 40–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Robinson M.D., et al. (2010). Evaluation of affinity-based genome-wide DNA methylation data: effects of CpG density, amplification bias, and copy number variation. Genome Res., 20, 1719–1729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lan X., et al. (2011). High resolution detection and analysis of CpG dinucleotides methylation using MBD-Seq technology. PLoS One, 6, e22226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Valmiki M.G., et al. (2009). Death effector domain-containing proteins. Cell. Mol. Life Sci., 66, 814–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kim Y., et al. (2012). Platelet-derived growth factor receptors differentially inform intertumoral and intratumoral heterogeneity. Genes Dev., 26, 1247–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Azab A.K., et al. (2011). FGFR3 is overexpressed waldenstrom macroglobulinemia and its inhibition by Dovitinib induces apoptosis and overcomes stroma-induced proliferation. Clin. Cancer Res., 17, 4389–4399 [DOI] [PubMed] [Google Scholar]
- 31. Xu J., et al. (2008). Nonmuscle myosin light-chain kinase mediates neutrophil transmigration in sepsis-induced lung inflammation by activating beta2 integrins. Nat. Immunol., 9, 880–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang Y., et al. (2012). PDGF mediates TGFβ-induced migration during development of the spinous process. Dev. Biol., 365, 110–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Martelli A.M., et al. (2012). Two hits are better than one: targeting both phosphatidylinositol 3-kinase and mammalian target of rapamycin as a therapeutic strategy for acute leukemia treatment. Oncotarget, 3, 371–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Darnell J.E., Jr (2002). Transcription factors as targets for cancer therapy. Nat. Rev. Cancer, 2, 740–749 [DOI] [PubMed] [Google Scholar]
- 35. Ladelfa M.F., et al. (2011). Interaction of p53 with tumor suppressive and oncogenic signaling pathways to control cellular reactive oxygen species production. Antioxid. Redox Signal., 15, 1749–1761 [DOI] [PubMed] [Google Scholar]
- 36. Mimeault M., et al. (2010). Frequent deregulations in the hedgehog signaling network and cross-talks with the epidermal growth factor receptor pathway involved in cancer progression and targeted therapies. Pharmacol. Rev., 62, 497–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Krisanaprakornkit S., et al. (2012). Epithelial-mesenchymal transition in oral squamous cell carcinoma. ISRN Oncol., 2012, 681469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nieto M.A. (2011). The ins and outs of the epithelial to mesenchymal transition in health and disease. Annu. Rev. Cell Dev. Biol., 27, 347–376 [DOI] [PubMed] [Google Scholar]
- 39. Pili R., et al. (2003). Is HIF-1 alpha a valid therapeutic target? J. Natl., Cancer Inst., 95, 498–499 [DOI] [PubMed] [Google Scholar]
- 40. Ludyga N., et al. (2011). Impact of protein tyrosine kinase 6 (PTK6) on human epidermal growth factor receptor (HER) signalling in breast cancer. Mol. Biosyst., 7, 1603–1612 [DOI] [PubMed] [Google Scholar]
- 41. Tang L., et al. (2011). Expression of TRAF6 and pro-inflammatory cytokines through activation of TLR2, TLR4, NOD1, and NOD2 in human periodontal ligament fibroblasts. Arch. Oral Biol., 56, 1064–1072 [DOI] [PubMed] [Google Scholar]
- 42. Tothova Z., et al. (2012). Anaplastic lymphoma kinase-directed therapy in inflammatory myofibroblastic tumors. Curr. Opin. Oncol., 24, 409–413 [DOI] [PubMed] [Google Scholar]
- 43. Yamada T., et al. (2012). Paracrine receptor activation by microenvironment triggers bypass survival signals and ALK inhibitor resistance in EML4-ALK lung cancer cells. Clin. Cancer Res., 18, 3592–3602 [DOI] [PubMed] [Google Scholar]
- 44. Kuo A.H., et al. (2007). Recruitment of insulin receptor substrate-1 and activation of NF-kappaB essential for midkine growth signaling through anaplastic lymphoma kinase. Oncogene, 26, 859–869 [DOI] [PubMed] [Google Scholar]
- 45. Polgar D., et al. (2005). Truncated ALK derived from chromosomal translocation t(2;5)(p23;q35) binds to the SH3 domain of p85-PI3K. Mutat. Res., 570, 9–15 [DOI] [PubMed] [Google Scholar]
- 46. Valentinis B., et al. (2001). IGF-I receptor signalling in transformation and differentiation. Mol. Pathol., 54, 133–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cheng J.C., et al. (2012). EGF-induced EMT and invasiveness in serous borderline ovarian tumor cells: a possible step in the transition to low-grade serous carcinoma cells? PLoS One, 7, e34071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mulholland D.J., et al. (2012). Pten loss and RAS/MAPK activation cooperate to promote EMT and metastasis initiated from prostate cancer stem/progenitor cells. Cancer Res., 72, 1878–1889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kume K., et al. (2013). The transcription factor Snail enhanced the degradation of E-cadherin and desmoglein 2 in oral squamous cell carcinoma cells. Biochem. Biophys. Res. Commun., 430, 889–894 [DOI] [PubMed] [Google Scholar]
- 50. Hashimoto T., et al. (2012). Progression of oral squamous cell carcinoma accompanied with reduced E-cadherin expression but not cadherin s witch. PLoS One, 7, e47899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shaozhang Z., et al. (2012). Detection of EML4-ALK fusion genes in non-small cell lung cancer patients with clinical features associated with EGFR mutations. Genes Chromosomes Cancer, 51, 925–932 [DOI] [PubMed] [Google Scholar]
- 52. Ren H., et al. (2012). Identification of anaplastic lymphoma kinase as a potential therapeutic target in ovarian cancer. Cancer Res., 72, 3312–3323 [DOI] [PubMed] [Google Scholar]
- 53. Tabbo F., et al. (2012). ALK signaling and target therapy in anaplastic large cell lymphoma. Front. Oncol., 2, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Carpenter E.L., et al. (2012). Targeting ALK in neuroblastoma—preclinical and clinical advancements. Nat. Rev. Clin. Oncol., 9, 391–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bachetti T., et al. (2010). PHOX2B-mediated regulation of ALK expression: in vitro identification of a functional relationship between two genes involved in neuroblastoma. PLoS One, 5, pii: e13108 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.