

Abstract
Targeted T cells are emerging as effective non-toxic therapies for cancer. Multiple elements, however, contribute to the overall pathogenesis of cancer through both distinct and redundant mechanisms. Hence, targeting multiple cancer-specific markers simultaneously could result in better therapeutic efficacy. We created a functional chimeric antigen receptor—the TanCAR, a novel artificial molecule that mediates bispecific activation and targeting of T cells. We demonstrate the feasibility of cumulative integration of structure and docking simulation data using computational tools to interrogate the design and predict the functionality of such a complex bispecific molecule. Our prototype TanCAR induced distinct T cell reactivity against each of two tumor restricted antigens, and produced synergistic enhancement of effector functions when both antigens were simultaneously encountered. Furthermore, the TanCAR preserved the cytolytic ability of T cells upon loss of one of the target molecules and better controlled established experimental tumors by recognition of both targets in an animal disease model. This proof-of-concept approach can be used to increase the specificity of effector cells for malignant versus normal target cells, to offset antigen escape or to allow for targeting the tumor and its microenvironment.
Keywords: bispecific chimeric antigen receptor, CAR, cancer immunotherapy, molecular modeling, T-cell therapy
Introduction
Chimeric antigen receptors (CARs) are artificial molecules that redirect the specificity of T cells to predetermined antigens.1,2,3 The prototype CAR was first described by Eshhar and colleagues in 1993, in which specific activation and targeting of T cells was mediated through molecules consisting of a target-antigen–specific antibody domain and the γ- or ζ-signaling subunits of the Fc epsilon receptor or T cell receptor, respectively.2 Since then, many groups have devised CAR molecules with single tumor-directed specificities and enhanced signaling endodomains;4,5,6 CAR T cell-based clinical trials are currently underway, with some highly promising early results.6,7,8,9,10
Rendering an individual T cell bispecific could have substantial functional implications that would likely translate into therapeutic benefits. Downregulation or mutation of target antigens is commonly observed in cancer cells, creating antigen-loss escape variants; a bispecific T cell could thus offset tumor escape.11 Furthermore, this bispecificity could enable simultaneous targeting of tumor cells and elements in the tumor microenvironment thereby augmenting T cell activation and function by increasing avidity and by broadening their therapeutic reach.12 To accomplish such bispecificity, we constructed a CAR in which two distinct antigen recognition domains are present in tandem on a single transgenic receptor.
The folding of an amino acid chain into highly organized, biologically functional three-dimensional protein structures, such as a CAR, continues to be a challenge in the design of novel protein molecules.13 In particular, protein misfolding, mispairing, and malfunction/dysfunction have traditionally impeded the wide scale production of molecules with multiple specificities.14 Advances in computational modeling methods, through characterization of the underlying energy landscapes as well as the dynamics of the polypeptide chains, have made structure prediction, analysis and design of a novel protein molecule, such as a tandem CAR, more feasible.15,16 Furthermore, docking routines have recently made it possible to predict, with high accuracy, the interface between two candidate molecules in a manner that could help to elucidate their functionality.17
We used computational modeling tools to predict the functionality of a novel single CAR molecule that can mediate bispecific activation and targeting of T cells. This tandem CAR (TanCAR) recognizes each target molecule individually and facilitates synergistic activation and functionality when both are encountered simultaneously. Such findings could have important therapeutic implications.
Results
Requirements for a bispecific “tandem” CAR: The TanCAR
We designed a proof-of-concept molecule, the TanCAR, to simultaneously target the B-cell antigen CD19 and the human epidermal growth factor receptor 2 (HER2/neu; also known as ErbB-2, CD340, and p185).
The extracellular domain of the TanCAR was designed to include a CD19-specific single chain antibody variable fragment (scFv) followed by a Gly-Ser linker, a HER2-specific scFv (FRP5) and another Gly-Ser tandem repeat hinge.18 Gly-Ser tandem repeats are highly flexible and non-cleavable thus would allow for near-free motion of the TanCAR subunits. Because of the respective lengths of HER2 (632aa/125Å) and CD19 (280aa/65Å) extracellular domains and the knowledge that FRP5 binds to the distal-most 4 loops of HER2 (W. Wels; unpublished data), we placed HER2-scFv in the juxta-membrane position and the CD19-scFv in the distal position to allow for more relaxed and potentially simultaneous binding. The intracytoplasmic domain of the proposed TanCAR molecule consists of a CD28/CD3-ζ signaling moiety, as previously described (Figure 1).19
Figure 1.

Designing a bispecific tandem chimeric antigen receptor (TanCAR) molecule. representation of the proposed chimeric antigen receptor molecule extracellular domains engaging the two targets; HER2 and CD19.
Computational docking of the proposed TanCAR design to target molecules
Predictive molecular modeling was used to interrogate the hypothetical TanCAR structure and provide rational basis for constructing a potentially functional molecule. To determine if the aforementioned molecular arrangement was possible, we created a TanCAR structural model using the protein-structure modeling webserver, ModWeb.20 Four structural templates with >58% sequence identity were identified, from which a model for residues 39–285 of TanCAR was constructed (Figure 2a).
Figure 2.

Docking platforms predict favorable binding potential of TanCAR to target molecules. Compilation of structure and docking data of (a) hypothetical structure of both FRP5-derived scFv and the CD19-specific scFv joined with a 20 amino acid Gly-Ser linker; (b) most favorable docking models of FRP5-derived scFv and the distal 200 amino acid residues of the extracellular domain of HER2; (c) most favorable docking models of CD19-scFv and the extracellular domain of CD19, and combined docking of (d) HER2 and (e) CD19 and the whole TanCAR exodomain, individually.
The next step was to assess how this TanCAR might interact with CD19 and HER2, individually. Although the structure of HER2 is known,21 no structure of CD19 is currently available. As such, a homology model for CD19 was generated using the 3MJG crystal structure as a structural template for residues 1–272 within the Modeller software.22 Using a combination of Patchdock and FireDock,23,24 automated tools for docking and refining two structures based on shape complementarity, the docking of HER2- and CD19-scFv's to their respective target molecules was simulated (Figure 2b,c). Thereafter, the whole TanCAR extracellular-domain model was docked pairwise to both structures, individually (Figure 2d,e). Results from the pairwise dockings were screened based on the overall score and agreement with known interaction sites from peptide spotting experiments in the case of HER2 (Supplementary Figure S1).25
From the in silico docking of these molecules, it appeared that the potential interactions of TanCAR with the target molecules could accommodate the intended bispecificity, and as such, we used this arrangement as an initial model to explore the ability of TanCAR to interact with the target molecules individually.
Construction, delivery, and expression of the TanCAR-encoding transgene
The modeled design of the TanCAR extracellular domain, composed of the CD19- and HER2-scFv fragments in tandem and separated by a linker, was assembled on Clone Manager, modified to introduce desired and remove unwanted restriction enzyme sites and optimized for maximum protein production using GeneOptimizer software.26 This exodomain was custom synthesized as a DNA fragment and subcloned in frame into an SFG retroviral vector containing a short hinge, CD28 transmembrane and signaling domains and the signaling domain of the CD3ζ-chain (Figure 3a).27 The resulting TanCAR transgene was then expressed in 293T cells as previously described.28 By flow cytometry, we determined ~89% and ~59% of 293T cells to surface express the TanCAR using Fab-specific antibody (binds both CD19-scFv and HER2-scFv) and a HER2-Fc protein (binds HER2-scFv only), respectively (Supplementary Figure S2 and Figure 3b). Similarly, >70% of T cells transduced using the 293T cells retroviral supernatant surface expressed the TanCAR (Figure 3c). Specific detection of the juxta-membrane HER2-scFv confirmed that the surface expression of the TanCAR extracellular domain in its entirety.
Figure 3.

Construction and surface expression of the TanCAR molecule. (a) pSFG vector construct encoding the TanCAR; (b) detection of the surface expression of the TanCAR using a Fab-specific antibody and FRP5-specific HER2-Fc protein on 293T cells; and (c) on T cells. See Supplementary Figure S2 for description of the labeling strategy.
TanCAR-expressing T cells distinctly recognize each target antigen
To test the dual functionality of the TanCAR T cells against CD19 and HER2, we first confirmed surface expression of these target antigens on a panel of human cancer cell lines using flow cytometry. Raji Burkitt lymphoma cells uniformly expressed the B-lineage marker CD19 but lacked detectable HER2 (Figure 4a).6 Conversely, Daoy medulloblastoma cells uniformly expressed HER2 but did not express CD19.28 MDA-MB-468 breast cancer cells were negative for both.28
Figure 4.

The TanCAR T cells distinctly recognize individual target molecules. (a) Flow cytometric analysis of the surface expression of the target antigens, HER2 and CD19, on a panel of human cancer cell lines used for functional testing; (b) cytotoxicity assay showing recognition and killing of HER2-positive Daoy cells and efficient blocking of this lysis using a soluble HER2 fragment; (c) similarly, TanCAR T cells recognized CD19-positive Raji cells and this lysis was blocked using the CD19 Ab 4G7; (d) in cocultures, TanCAR T cells secreted IFN-γ as well as IL-2 upon encounter of HER2- and CD19-positive target cell above the non-transduced T-cell control (NT). No cytokines were secreted in coculture with the HER2- and CD19-null target cell MDA-MB-468. (b–d) Shown are representative plots of three or more experiments done in triplicates.
In 4-hour 51Cr-release assays, TanCAR T cells recognized and killed HER2-expressing Daoy cells but not MDA-MB-468 cells. Non-transduced T cells from the same donor had no lytic activity, excluding an allogeneic response. Up to 95% of lysis could be blocked by soluble HER2 protein (Figure 4b), indicating that specific recognition occurred due to HER2 binding. Similarly, CD19-expressing Raji cells were killed by TanCAR T cells but not CD19 negative MDA-MB-468 cells. CD19-specific MAb 4G7 blocked cytolytic activity against Raji cells by up to 70% (Figure 4c).
In cocultures, TanCAR T cells secreted Interferon-γ (IFN-γ) and Interleukin-2 (IL-2) upon encountering HER2- or CD19-positive target cells. No cytokines were secreted in coculture with MDA-MB-468 (Figure 4d). These results indicate that TanCAR is bispecific for CD19 and HER2, and mediates activation and targeting of T cells upon encounter of either antigen alone.
Enhanced functionality upon simultaneous encounter of both target antigens and preserved TanCAR T-cell–induced cytolysis in a model of antigen loss
To investigate the hypothetical potential for simultaneous binding of the TanCAR molecule to both target antigens we visualized and aligned the pairwise dockings in UCSF's Chimera and evaluated the ensemble docking for global energy, agreement with interaction sites and steric clashes.29 Indeed, this composite algorithm yielded a sterically possible favorable docking combination in which HER2 and CD19 structures were both bound to the TanCAR without any clashes (Figure 5a). Based on this docking model, both the HER2 and CD19 are arranged such that their N-termini are essentially orientated in the same direction; hypothetically, this is the tumor cell direction. As the CD19 structure is ~50% of the size of HER2, there is a separation of ~93Å between the N-termini of both molecules. Differences in orientation of CD19 and HER2 along the cell membrane, as well as variations in the cell membrane itself, could account for the difference between the size and orientation of the receptors but would still allow for the simultaneous docking of both HER2 and CD19 to the TanCAR.
Figure 5.
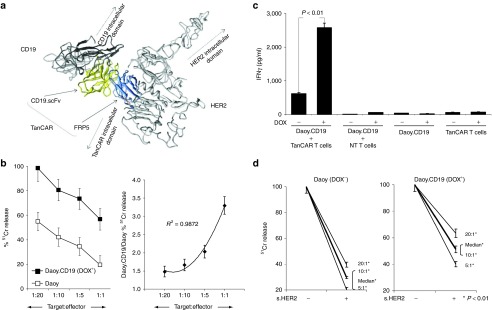
Enhanced cytolytic function upon simultaneous recognition of two antigens and preserved TanCAR T cell-induced cytolysis in a model of antigen loss. (a) Collective modeling of the simultaneous docking of the TanCAR to both HER2 and CD19. (b) In cytotoxicity assays, we saw consistently higher killing after the induction of CD19 at various tumor to T cell ratios. This was synergistic; with an exponential trend following a higher order equation; and was more prominent in higher tumor to T cell ratios (right panel). (c) Similarly, induction of CD19 (DOX+) in Daoy.TET.CD19 and T cell cocultures resulted in a more than fourfold increase in IFN-γ release as detected by ELISA (P < 0.01). (d) We modeled the scenario in which tumor cells downregulate the target antigen, by blocking HER2 in CD19-induced (DOX+) and CD19 null (DOX−) Daoy.TET.CD19 cells using a soluble HER2 fragment (s.HER2). Although soluble HER2 successfully induced substantial blocking of HER2-mediated killing in DOX− cells at various tumor to T cell ratios, it resulted only in a partial decrease in the cytolytic effect of TanCAR T cells in DOX+. P values were significant at T cell to tumor cell ratios of 1:20, 1:10, and 1:5. Shown are representative plots of three experiments done in triplicates.
To study the kinetics of activation of TanCAR T cells in vitro and in vivo in the presence of both target molecules, we used a tetracycline-inducible system (Tet-On 3G) to conditionally express a truncated non-signaling CD19 molecule on Daoy cells (Daoy.TET.CD19).30 In the presence of doxycycline (DOX+), 60–85% of the endogenously HER2-positive Daoy.TET.CD19 cells expressed CD19 and the reporter gene mCherry (Supplementary Figure S3a). In cytotoxicity assays, we saw consistently higher killing after the induction of CD19 for all tumor/T-cell ratios tested. This enhancement of cytolysis followed an exponential trend with a higher order equation, an effect that became more evident at higher tumor/T cell ratios (Figure 5b). This indicates a synergistic effect of simultaneous recognition of both target antigens on the ability of T cells to lyse target cells. Consistent with these results, we observed fourfold increase (P < 0.01) in IFN-γ secretion upon induction of CD19 (Figure 5c).
Downregulation or mutation of target antigens is a common process in cancer cells creating antigen-loss escape variants. Such variants are responsible for persistence of tumor cells at very low frequencies, and their outgrowth culminates in relapse. We simulated the downregulation of a targeted antigen by blocking HER2 recognition in CD19-induced (DOX+) Daoy.TET.CD19 cells using a soluble HER2 fragment. Although the competitor successfully impaired HER2-mediated killing in these cells (up to 90%), bispecific TanCAR T cells were less affected by soluble HER2 fragment blocking because of maintained recognition of CD19 (Figure 5d).
Simultaneous targeting of two antigens enhances the in vivo antitumor activity of TanCAR T cells
We used a xenograft model to assess the potential advantage of simultaneously targeting two antigens in established tumors. We inoculated Daoy.TET.CD19 xenografts in the flanks of SCID mice. The induction of CD19 by intraperitoneal doxycycline was confirmed in a subset of animals (Supplementary Figure S3b). The tumors were allowed to establish for 3 weeks, after which animals were randomly assigned into four groups. All four groups of animals had similar tumor volumes after randomization (mean tumor volume 94 µl; SD 28 µl). Two groups received intraperitoneal doxycycline (DOX+) whereas the other two received phosphate-buffered saline (PBS; DOX−). Within DOX+ and DOX− groups, five animals each received one intratumoral injection of 10,000 TanCAR T cells/µl tumor volume. Administration of PBS or doxycycline alone induced minimal or no alteration of the tumor growth pattern. By contrast, TanCAR T cells resulted in significant delay in tumor progression that was further increased by induction of CD19 (Figure 6a). Kaplan–Meier survival studies after 60 days from treatments showed that mice administered PBS with and without doxycycline had a median survival of 28 and 31 days, respectively. In contrast, mice treated with TanCAR T cells had a median survival of 44 days (P < 0.01). Furthermore, mice administered TanCAR T cells and doxycycline had a median survival of >60 days with 50% of the mice surviving >80 days (P < 0.001; Figure 6b). Hence, simultaneous recognition of two antigens enhanced the in vivo antitumor activity of adoptively transferred TanCAR T cells.
Figure 6.

Simultaneous targeting of two antigens enhances the in vivo antitumor activity of adoptively transferred TanCAR T cells. (a) Daoy.TET.CD19 xenografts were established for 3 weeks in the flanks of SCID mice, and then animals were randomized into four groups. Administration of PBS into the tumor and/or systemic doxycycline induced minimal or no alteration of the tumor growth pattern. By contrast, treatment with TanCAR T cells resulted in a significant delay in tumor progression that was further enhanced by induction of CD19 expression in the DOX+ group. (b) Kaplan–Meier survival curve: analysis performed 60 days after PBS or T-cells injection. Mice treated with TanCAR T cells had a significantly longer survival probability in comparison with control mice. Furthermore, induction of CD19 by the administration of doxycycline resulted in enhanced antitumor activity of adoptively transferred TanCAR T cells.
Discussion
We created a functional CAR, the TanCAR, a novel artificial molecule that mediates bispecific activation and targeting of T cells. We demonstrated the feasibility of cumulative integration of structure and docking simulation data using computational tools to interrogate the design and predict the functionality of such a complex bispecific molecule. Our prototype TanCAR induced distinct T cell reactivity against each of two target molecules, and produced synergistic enhancement of effector functions when both antigens were simultaneously encountered. Furthermore, the TanCAR preserved the cytolytic ability of T cells upon loss of one of the target molecules and better controlled established experimental tumors by recognition of both targets in an animal disease model.
An approach using effector cells with multiple specificities could have a number of benefits as cancer therapy. First, simultaneous targeting of multiple tumor antigens could overcome antigen escape by providing an alternative killing pathway if there is antigen downregulation or deletion. Second, such effector cells would exhibit a broader spectrum of specificities allowing for targeting of heterogeneous antigens, such as those present on the tumor cells and within the tumor microenvironment, thereby enhancing tumor control by damaging the tumor complex. Lastly, the simultaneous encounter of several antigens should enhance T cell activation through increased avidity, a particular benefit when tumors express only modest levels of individual antigens.12,31
The potential benefits of bispecific antibodies and related molecules have led to an intensive investigation and definite successes of various designs and specificities. Indeed, Blinatumomab, a CD3/CD19-bispecific antibody, has been shown in a Phase I/II trial to induce objective responses in non-Hodgkin lymphoma patients.32 Unfortunately, the wider scale therapeutic use of bispecific antibodies has faced obstacles related to manufacturing feasibility and limitations related to pharmacokinetics and stability.33 Tandem-scFv, diabodies, two-in-one antibodies, and dual variable domain antibodies were introduced as novel designs that were able to overcome some of these limitations but still suffer from residual shortcomings of antibody-based approaches.33,34,35,36 Antibodies, unlike T cells, do not actively migrate through microvascular-walls or penetrate the core of solid tumors to exert their antitumor activity and usually have no access to the neuraxis, a common sanctuary for cancer cells.37,38 Furthermore, in the context of modest expression of target antigens, antibodies exhibit limited efficacy.31,39 T cells expressing antibody-derived CARs have been shown to overcome such limitations.31,40
Our group previously redirected the specificity of T cells to two distinct entities by grafting tumor-restricted antigen-specific CARs onto T cells whose native receptor was specific for latent-virus antigens. Thus, we grafted EBV-specific cytotoxic T cells (EBV-CTLs) with a GD2-specific CAR to treat Neuroblastoma, whereas CMV.CTLs were grafted with HER2-specific CARs to treat Glioblastoma.7 The intent was to enable CAR expressing CTLs to receive optimal costimulation after native-receptor engagement of viral antigens on professional antigen presenting cells, and thereby enhance their in vivo survival in these first-in-man studies.7,41
This work describes the first rationally designed artificial molecule to render a T cell bispecific. We demonstrate synergistic killing which should further enhance function and resistance against antigen loss. A combination of these approaches may also be feasible by grafting a TanCAR to a latent-virus–specific CTL, thereby providing a trispecific T cell with enhanced in vivo survival. The TanCAR could be further modified to incorporate additional costimulatory endodomains to enhance the degree of T cell activation and persistence.6,10
Though much progress has been made, designing novel protein molecules with correct protein folds is still very challenging.13,14 Indeed, Patel et al. described, in 2000, T cell products expressing four empiric designs of anti-tumoral antigen bispecific chimeric immune receptors one of which lysed both CEA- and TAG-72-expressing targets and did not kill antigen-negative targets or target cells expressing other members of the CEA family.42 However, the recent advances in computational modeling and protein–protein docking make structure design and analysis of a novel protein, such as the aforementioned tandem CAR, more feasible.15,16 We used such tools, in a staged methodology, to profile the energy landscapes as well as the dynamics of the polypeptide chains in all stages of the folding process, in an attempt to predict functionality of this molecule. We reasoned that although CD19 and HER2 are not naturally coexpressed on normal or malignant cells, using them as targets would allow us to directly test the effects of binding one or both target antigens and the consequences of such binding on T cell activation and target cell killing. Both molecules are well characterized as targets and are in multiple clinical trials using CAR T cells. Furthermore, a crystal structure of HER2 was available.21 Although the structure of CD19 is unknown, several potential structural templates, with relatively high sequence similarity, are available for constructing a CD19 homolog model. Coupled with a model of TanCAR, it is possible to reliably assess its docking potential to these target molecules.
The TanCAR model was produced from several closely related structures (>50% identity between the sequences of interest and the template structures), demonstrating that computational modeling is a useful tool in the construction of such complex molecules by interrogating their functional structure and predicting their functionality. However, in the proposed model, the Gly-Ser linker has no structural template; therefore, the exact structure of this loop and the orientations of the two TanCAR domains with respect to each other most likely exhibit some degree of variation. It is conceivable that, whereas unbound, TanCAR remains as a soluble, compact, globular protein. When bound to HER2 and CD19, steric forces may cause the two domains to separate, remaining tethered to each other through an extended conformation of the Gly-Ser linker. In an extended conformation, this loop could help resolve some of the differences in “height” of the two target molecules on the surface of the cell. Regardless of the conformation of the loop, the interface proposed by our docking is compatible with either a compact or extended form of the linker. In this respect, the achievement of a definite low energy model that1 incorporates docking routines of both scFv's to their respective molecules, simultaneously, while2 maintaining the polarity and distance of the target versus effector ends of all three molecules was an indicator of the correct orientation of the proposed design. Using alternative positions of the scFv's or different lengths of the linker segment failed to achieve these goals and thus a collective docking model was not generable. This TanCAR design with the FRP5 in the juxta-membrane position and a 20 amino acid Gly-Ser linker was then expressed in T cells and analyzed for its biological activity.
Indeed, when the hypothetical molecule was physically generated and tested, our results corroborated the predictive modeling. The surface expression of the TanCAR exodomain in its entirety was validated by detection of the juxta-membrane HER2-scFv. The proper folding and retention of the VL and VH stereo-orientation for HER2-scFv and CD19-scFv was evident in the specific recognition and distinct lysis by TanCAR T cells of tumor cells expressing these target molecules and the blockade of this lysis with the respective competitors. Failure to block the cytolytic effect against tumor cells expressing both targets using soluble HER2 indicated that the dual functionality of TanCAR T cells allowed for persistence of their effector functions despite antigen loss. This redundancy in function would be favorable in such a scenario. Lastly, the enhancement of cytolysis upon induction of CD19 in HER2-expressing targets indicated synergistic dynamics that further translated into improved tumor control in an animal model using a relatively small dose of T cells. These findings point to the potential of TanCAR T cells for therapeutic application in human disease.
With the enhanced activation of T cells comes the risk of adverse events related to recognition of modestly expressed antigens resulting in off-target effects. Although our group has recently shown that it is possible to quickly eliminate adoptively transferred T cells in case of adverse events by inducing apoptosis through a suicide gene43, TanCAR molecules could be conceptually used to “bar code” tumor cells—wherein only dual-antigen expressers are killed. Similarly, other groups have used a functionally bipartite CAR molecule that conditionally triggers sufficient T cell activation only on simultaneous engagement of two distinct tumor antigens.44 Alternatively, TanCARs themselves could also be engineered to include antibody recognizable moieties that, in the advent of adverse events, would allow for the rapid elimination of TanCAR T cells.
In summary, we here describe a novel bispecific CAR molecule—termed TanCAR. TanCAR induced bispecific activation of T cells and exhibited potent effector functions against individual target antigens as well as synergistic enhancement of functionality upon simultaneous engagement of both. Preclinical studies of TanCAR T cells in an animal tumor model demonstrated its potential for therapeutic application in human disease.
Materials and methods
Blood donors and tumor cell lines. Studies were performed on Baylor College of Medicine (BCM) IRB-approved protocols and informed consent was obtained from all blood donors. The tumor lines Daoy, Raji, and MDA-MB-468 were purchased from ATCC (Manassas, VA). All cell lines were grown in DMEM (Invitrogen, Carlsbad, CA) with 10% fetal calf serum (HyClone, Logan, UT), with 2 mmol/l GlutaMAX-I, 1.5 g/l sodium bicarbonate and 1.0 mmol/l sodium pyruvate (Invitrogen). T cells derived from peripheral blood mononuclear cells were activated on CD3/CD28 antibody-coated plates and were expanded in IL-2 (100 U/ml)-containing RPMI1640 with 10% fetal calf serum and 2 mmol/l GlutaMAX-I.
Protein structure modeling and docking. A model for the bispecific CAR was constructed using ModWeb, an automated web server for protein-structure modeling.20 The full-length TanCAR sequence was submitted to the ModWeb server, which identified the two antibody-like fragment domains. 1OP345 was identified as a candidate structural template for residues 40–146 (59.81% sequence identity) and 1F3R46 as a candidate structural template for residues 167–329 (58.90% sequence identity). Additional structural templates, 3ESV (64.76% sequence identity) and 2KH2 (59.51% sequence identity),47,48 were also identified covering residues 38–285. From these templates, a homology model was constructed spanning residues 39–329, including the 20 amino acid long Gly-Ser linker. The model was truncated at residue 285 as residues 286–329 were poorly modeled. The structure for HER2 was available (PDB-ID: 1N8Z).21 As a note, the 1N8Z-structure contains residues 23–629, resulting in a difference in numbering between 1N8Z and the HER2 precursor protein sequence (uniprot P04626). A homologous structure for CD19 was identified using BioInfoBank metaserver.49 A model for residues 1–272 of CD19 was constructed with Modeller V9.1 using 3MJG (12.59% sequence identity) as a structural template.22
Initially, pairwise docking was performed with PatchDock23 using the individual TanCAR domains and the corresponding receptor; residues 39–155 corresponded to the HER2 binding portion of TanCAR, whereas residues 156–285 were assigned to the CD19 binding portion of TanCAR. Fits were evaluated visually and based on their PatchDock score. For the TanCAR-HER2 docking, results were additionally filtered based on peptide spotting experiments that had suggested binding residues (Supplementary Figure S1). Further refinement of the individual candidate dockings was done using FireDock.24 Candidate dockings from both CAR-HER2 and CAR-CD19 were then combined in UCSF Chimera29 and evaluated for steric clashes. The final model for the CD19-CAR-HER2 docking was selected based on lowest global energy in each of the pairwise dockings from FireDock and steric constraints in the entire assembly.
Construction, delivery, and expression of the TanCAR-encoding transgene. The CD19-specific scFv was provided by Heddy Zola (Women's and Children's Hospital, Adelaide, Australia).50 The HER2-specific scFv, FRP5, was previously described by Wels and colleagues.51 The modeled bispecific extracellular domain was assembled on Clone Manager (Sci-Ed Software, Cary, NC). The designed transgene DNA sequence was modified to include restriction enzyme sites at the cloning sites and exclude any inadvertently inserted sites within the translation elements, then optimized using the GeneOptimizer software for maximum protein production.26 The TanCAR extracellular domain was then synthesized by GeneArt Inc. using oligonucleotides, cloned into the Gateway entry vector pDONR221, and sequence verified. This antigen recognition domain was then subcloned in frame into an SFG retroviral vector containing a short hinge, and the transmembrane and signaling domain of the costimulatory molecule, CD28 and the signaling domain of the T cell receptor ζ-chain (Figure 3a). The structure of the construct was confirmed using restriction digests. The 5′-3′ as well as the 3′-5′ sequence of the whole construct was confirmed using single base pair pyro-sequencing (SeqWright DNA-Technology, Houston, TX) with >97% homology with the optimized construct map.
Retrovirus production and transduction of T cells. To produce retroviral supernatant, human embryonic kidney (HEK) 293T cells were cotransfected with the TanCAR-encoding retroviral transfer plasmid, Peg-Pam-e plasmid encoding MoMLV gag-pol, and plasmid pMEVSVg containing the sequence for VSV-G envelope,28 using GeneJuice transfection reagent (EMD Biosciences, San Diego, CA).28 Supernatants containing retroviral vector were collected 48 and 72 hours later.
Anti-CD3(OKT3)/anti-CD28 activated T cells were transduced with retroviral vectors as described.19 Briefly, peripheral blood mononuclear cells were isolated by Lymphoprep (Bio-One, Monroe, NC) then activated with OKT3 (OrthoBiotech, Raritan, NJ) and CD28 monoclonal antibodies (BD Biosciences, Palo Alto, CA) at a final concentration of 1 μg/ml. On day 2, recombinant human IL-2 (Chiron, Emeryville, CA) was added at a final concentration of 100 U/ml, and 2 days later, cells were harvested for retroviral transduction over recombinant fibronectin fragment (Takara-Bio-USA, Madison, WI) pre-coated plates. Subsequently, 3 × 105 T cells per well were transduced with retrovirus in the presence of 100 U/ml IL-2. After 48–72 hours cells were removed and expanded in the presence of 50–100 U/ml IL-2 for 10–15 days before use.
Cytotoxicity assays. Cytotoxicity assays were performed as previously described.19 Briefly, 1 × 106 target cells were labeled with 0.1 mCi (3.7 MBq) 51Cr and mixed with decreasing numbers of effector cells to give effector to target ratios of 40:1, 20:1, 10:1, and 5:1. Target cells incubated in complete medium alone or in 1% Triton X-100 were used to determine spontaneous and maximum 51Cr release, respectively. After 4 hours, we collected supernatants and measured radioactivity in a gamma counter (Cobra Quantum; PerkinElmer; Wellesley, MA). The mean percentage of specific lysis of triplicate wells was calculated according to the following formula: (test release − spontaneous release)/(maximal release − spontaneous release) × 100.
Analysis of cytokine production and T cell expansion. Effector T cells (TanCAR-expressing T cells or non-transduced T cells) from healthy volunteers were cocultured with tumor cells in short-term culture, HER2-positive and HER2-negative cell lines, at various effector to target ratios in a 24 well plate. After 24 to 48 hours incubation, culture supernatants were harvested and the presence of IFN-γ and IL-2 was determined by ELISA as per the manufacturer's instructions (R&D Systems, Minneapolis, MN). T cell expansion was determined by counting viable cells (trypan blue exclusion) 7 days after stimulation.
Tetracycline-inducible system. To express the truncated CD19 (thereafter referred to as CD19) on Daoy cells, we used the Tet-On 3G Tetracycline-Inducible Expression System (CloneTech, Mountainview, CA). The CD19 encoding DNA fragment was subcloned downstream of the inducible promoter PTRE3G using PCR amplification. Daoy cells expressed CD19, but only when cultured in the presence of doxycycline (DOX+), a tetracycline analog. When bound by doxycycline, the Tet-On 3G protein undergoes a conformational change that allows it to bind to tet operator sequences located in the inducible promoter PTRE3G. The addition of doxycycline also initiated a proportionate expression of the reporter gene mCherry.
Flow cytometry. A FACScalibur instrument (Becton Dickinson, Mountain View, CA) and CellQuest software (BD Biosciences) were used. Analysis was done on >10,000 events. Negative controls included isotype antibodies. Cells were incubated 15–30 minutes with antibodies at 4 °C in the dark then washed and fixed in 0.5% paraformaldehyde before analysis. Surface expression of the TanCAR exodomain was assessed using a HER2-scFv (FRP5)-specific method by incubation with a soluble HER2.Fc fragment followed by Fc-specific FITC-labeled antibody. Alternatively, APC-conjugated Fab-specific antibody was used to detect either HER2-scFv or CD19-scFv.
Animal studies. All animal experiments were conducted on a protocol approved by the BCM Institutional Animal Care and Use Committee. Male recipient NOD-SCID mice of 8–10 weeks old (FOX CHASE CB-17 SCID ICR; Taconic, Hudson, NY) were anesthetized with isofluorane (Abbott Laboratories, Abbott Park, IL) and received a subcutaneous injection of 1 × 106 DAOY.TET.CD19 cells (in 100 µl PBS) per flank per mouse. Tumors were then allowed ~3 weeks to fully engraft. Mice with established tumors (mean tumor volume 94 µl: SD 28 µl) were randomly assigned to four groups (n = 5 tumors/group). Tumor volume was calculated from the product of the bidimensional area measured with an electronic caliper. All four groups of animals had similar tumor volumes after randomization. To induce the expression of CD19 (DOX+) two groups received intraperitoneally 80 µg/mouse daily for 1 week followed thrice-weekly doses thereafter. The other half received an equal volume of PBS (DOX−) on the same dosing schedule. On the third day of doxycycline/PBS administration, within the DOX+ and DOX− groups, 5 animals each received one intratumoral injection of 10,000 TanCAR T cells per µl tumor volume. All mice were then blindly assessed for fold changes in tumor volume.
Immunofluorescence. Staining for CD19 was performed on Daoy.DOX.CD19 explants in the presence or absence doxycycline. Sections were deparaffinized in xylene and rehydrated in decreasing concentrations of ethanol. For antigen retrieval, slides were immersed in DAKO Citrate Buffer in a pressure cooker for 45 minutes. After blocking with 5% donkey serum, tissues were incubated with CD19 primary antibody at 4°C overnight in a humidified chamber and with secondary anti-rabbit antibody at room temperature for 1 hour. Samples were counterstained with DAPI and examined under a fluorescent microscope.
Statistical analysis. The two-tailed Student's t-test was used to test for significance in each set of values, assuming equal variance. Mean values ±SD are given unless otherwise stated. Kaplan–Meier analysis was used to assess survival of experimental animal groups.
SUPPLEMENTARY MATERIAL Figure S1. Binding of anti-HER2 antibody FRP5 to peptide arrays. Figure S2. Surface expression of the TanCAR was tested using two different methods. Figure S3. We used a tetracycline-inducible system to conditionally express a truncated non-signaling CD19 molecule on Daoy cells (Daoy.TET.CD19).30
Acknowledgments
We thank Malcolm K Brenner for helpful discussions and advice, and for critical review of the manuscript and Carl E Allen for kindly reviewing the final version. The authors were supported by The Alliance for Cancer Gene Therapy (P30CA125123). The V-Foundation for Cancer Research, The Dana Foundation, The Larry and Helen Hoag Foundation, and The American Brain Tumor Association. M.L.B. is supported by grants from NIH through the National Center for Research Resources (P41RR002250) and the National Institute of General Medical Science (R01GM079429 and R01GM080139), T.B. and D.R.S. by T32 (HL092332), and H.E.H. by a Dan L Duncan Chair. The authors declared no conflict of interest.
Supplementary Material
Binding of anti-HER2 antibody FRP5 to peptide arrays.
Surface expression of the TanCAR was tested using two different methods.
We used a tetracycline-inducible system to conditionally express a truncated non-signaling CD19 molecule on Daoy cells (Daoy.TET.CD19).30
References
- Kochenderfer JN, Rosenberg SA. Chimeric antigen receptor-modified T cells in CLL. N Engl J Med. 2011;365:1937–8; author reply 1938. doi: 10.1056/NEJMc1111004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci USA. 1993;90:720–724. doi: 10.1073/pnas.90.2.720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadelain M, Rivière I, Brentjens R. Targeting tumours with genetically enhanced T lymphocytes. Nat Rev Cancer. 2003;3:35–45. doi: 10.1038/nrc971. [DOI] [PubMed] [Google Scholar]
- Brentjens RJ, Latouche JB, Santos E, Marti F, Gong MC, Lyddane C, et al. Eradication of systemic B-cell tumors by genetically targeted human T lymphocytes co-stimulated by CD80 and interleukin-15. Nat Med. 2003;9:279–286. doi: 10.1038/nm827. [DOI] [PubMed] [Google Scholar]
- Pule M, Straathof K, Dotti G, Heslop HE, Rooney CM, Brenner MK. Three-module signaling endo-domain artificial T-cell receptor which transmits CD28, OX40 and CD3-zeta signals enhances IL-2 release and proliferative response in transduced primary T-cells. Blood. 2004;104:485a. [Google Scholar]
- Savoldo B, Ramos CA, Liu E, Mims MP, Keating MJ, Carrum G, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest. 2011;121:1822–1826. doi: 10.1172/JCI46110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med. 2008;14:1264–1270. doi: 10.1038/nm.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119:2709–2720. doi: 10.1182/blood-2011-10-384388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen MC, Popplewell L, Cooper LJ, DiGiusto D, Kalos M, Ostberg JR, et al. Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T cells in humans. Biol Blood Marrow Transplant. 2010;16:1245–1256. doi: 10.1016/j.bbmt.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol. 2004;22:329–360. doi: 10.1146/annurev.immunol.22.012703.104803. [DOI] [PubMed] [Google Scholar]
- Weijtens ME, Hart EH, Bolhuis RL. Functional balance between T cell chimeric receptor density and tumor associated antigen density: CTL mediated cytolysis and lymphokine production. Gene Ther. 2000;7:35–42. doi: 10.1038/sj.gt.3301051. [DOI] [PubMed] [Google Scholar]
- Buchner GS, Murphy RD, Buchete NV, Kubelka J. Dynamics of protein folding: probing the kinetic network of folding-unfolding transitions with experiment and theory. Biochim Biophys Acta. 2011;1814:1001–1020. doi: 10.1016/j.bbapap.2010.09.013. [DOI] [PubMed] [Google Scholar]
- Kuhlman B, Baker D. Exploring folding free energy landscapes using computational protein design. Curr Opin Struct Biol. 2004;14:89–95. doi: 10.1016/j.sbi.2004.01.002. [DOI] [PubMed] [Google Scholar]
- Perez-Aguilar JM, Saven JG. Computational design of membrane proteins. Structure. 2012;20:5–14. doi: 10.1016/j.str.2011.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samish I, MacDermaid CM, Perez-Aguilar JM, Saven JG. Theoretical and computational protein design. Annu Rev Phys Chem. 2011;62:129–149. doi: 10.1146/annurev-physchem-032210-103509. [DOI] [PubMed] [Google Scholar]
- Kiel C, Beltrao P, Serrano L. Analyzing protein interaction networks using structural information. Annu Rev Biochem. 2008;77:415–441. doi: 10.1146/annurev.biochem.77.062706.133317. [DOI] [PubMed] [Google Scholar]
- Matsushima N, Yoshida H, Kumaki Y, Kamiya M, Tanaka T, Izumi Y, et al. Flexible structures and ligand interactions of tandem repeats consisting of proline, glycine, asparagine, serine, and/or threonine rich oligopeptides in proteins. Curr Protein Pept Sci. 2008;9:591–610. doi: 10.2174/138920308786733886. [DOI] [PubMed] [Google Scholar]
- Ahmed N, Salsman VS, Kew Y, Shaffer D, Powell S, Zhang YJ, et al. HER2-specific T cells target primary glioblastoma stem cells and induce regression of autologous experimental tumors. Clin Cancer Res. 2010;16:474–485. doi: 10.1158/1078-0432.CCR-09-1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieper U, Webb BM, Barkan DT, Schneidman-Duhovny D, Schlessinger A, Braberg H, et al. ModBase, a database of annotated comparative protein structure models, and associated resources. Nucleic Acids Res. 2011;39 Database issue:D465–D474. doi: 10.1093/nar/gkq1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho HS, Mason K, Ramyar KX, Stanley AM, Gabelli SB, Denney DW.et al. (2003Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature 421756–760. [DOI] [PubMed] [Google Scholar]
- Shim AH, Liu H, Focia PJ, Chen X, Lin PC, He X. Structures of a platelet-derived growth factor/propeptide complex and a platelet-derived growth factor/receptor complex. Proc Natl Acad Sci USA. 2010;107:11307–11312. doi: 10.1073/pnas.1000806107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneidman-Duhovny D, Inbar Y, Nussinov R, Wolfson HJ. PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res. 2005;33 Web Server issue:W363–W367. doi: 10.1093/nar/gki481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrusier N, Nussinov R, Wolfson HJ. FireDock: fast interaction refinement in molecular docking. Proteins. 2007;69:139–159. doi: 10.1002/prot.21495. [DOI] [PubMed] [Google Scholar]
- Gerstmayer B, Altenschmidt U, Hoffmann M, Wels W. Costimulation of T cell proliferation by a chimeric B7-2 antibody fusion protein specifically targeted to cells expressing the erbB2 proto-oncogene. J Immunol. 1997;158:4584–4590. [PubMed] [Google Scholar]
- Raab D, Graf M, Notka F, Schödl T, Wagner R. The GeneOptimizer Algorithm: using a sliding window approach to cope with the vast sequence space in multiparameter DNA sequence optimization. Syst Synth Biol. 2010;4:215–225. doi: 10.1007/s11693-010-9062-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossig C, Bollard CM, Nuchtern JG, Merchant DA, Brenner MK. Targeting of G(D2)-positive tumor cells by human T lymphocytes engineered to express chimeric T-cell receptor genes. Int J Cancer. 2001;94:228–236. doi: 10.1002/ijc.1457. [DOI] [PubMed] [Google Scholar]
- Ahmed N, Ratnayake M, Savoldo B, Perlaky L, Dotti G, Wels WS, et al. Regression of experimental medulloblastoma following transfer of HER2-specific T cells. Cancer Res. 2007;67:5957–5964. doi: 10.1158/0008-5472.CAN-06-4309. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Zhou X, Vink M, Klaver B, Berkhout B, Das AT. Optimization of the Tet-On system for regulated gene expression through viral evolution. Gene Ther. 2006;13:1382–1390. doi: 10.1038/sj.gt.3302780. [DOI] [PubMed] [Google Scholar]
- Ahmed N, Salsman VS, Yvon E, Louis CU, Perlaky L, Wels WS, et al. Immunotherapy for osteosarcoma: genetic modification of T cells overcomes low levels of tumor antigen expression. Mol Ther. 2009;17:1779–1787. doi: 10.1038/mt.2009.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bargou R, Leo E, Zugmaier G, Klinger M, Goebeler M, Knop S, et al. Tumor regression in cancer patients by very low doses of a T cell-engaging antibody. Science. 2008;321:974–977. doi: 10.1126/science.1158545. [DOI] [PubMed] [Google Scholar]
- Hagemeyer CE, von Zur Muhlen C, von Elverfeldt D, Peter K. Single-chain antibodies as diagnostic tools and therapeutic agents. Thromb Haemost. 2009;101:1012–1019. [PubMed] [Google Scholar]
- Wu C, Ying H, Grinnell C, Bryant S, Miller R, Clabbers A, et al. Simultaneous targeting of multiple disease mediators by a dual-variable-domain immunoglobulin. Nat Biotechnol. 2007;25:1290–1297. doi: 10.1038/nbt1345. [DOI] [PubMed] [Google Scholar]
- Gu J, Ghayur T. Generation of dual-variable-domain immunoglobulin molecules for dual-specific targeting. Meth Enzymol. 2012;502:25–41. doi: 10.1016/B978-0-12-416039-2.00002-1. [DOI] [PubMed] [Google Scholar]
- Doppalapudi VR, Huang J, Liu D, Jin P, Liu B, Li L, et al. Chemical generation of bispecific antibodies. Proc Natl Acad Sci USA. 2010;107:22611–22616. doi: 10.1073/pnas.1016478108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchnowska R, Szczylik C. Central nervous system metastases in breast cancer patients administered trastuzumab. Cancer Treat Rev. 2005;31:312–318. doi: 10.1016/j.ctrv.2005.04.008. [DOI] [PubMed] [Google Scholar]
- Shmueli E, Wigler N, Inbar M. Central nervous system progression among patients with metastatic breast cancer responding to trastuzumab treatment. Eur J Cancer. 2004;40:379–382. doi: 10.1016/j.ejca.2003.09.018. [DOI] [PubMed] [Google Scholar]
- Verneris MR, Arshi A, Edinger M, Kornacker M, Natkunam Y, Karami M, et al. Low levels of Her2/neu expressed by Ewing's family tumor cell lines can redirect cytokine-induced killer cells. Clin Cancer Res. 2005;11:4561–4570. doi: 10.1158/1078-0432.CCR-05-0157. [DOI] [PubMed] [Google Scholar]
- Hong JJ, Rosenberg SA, Dudley ME, Yang JC, White DE, Butman JA, et al. Successful treatment of melanoma brain metastases with adoptive cell therapy. Clin Cancer Res. 2010;16:4892–4898. doi: 10.1158/1078-0432.CCR-10-1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Study of administration of CMV-specific cytotoxic T lymphocytes expressing CAR targeting HER2 in patients with GBM (HERT-GBM). 2011 ) < http://clinicaltrials.gov/ct2/show/NCT01109095?term=HERT-GBM&rank=1 >.
- Patel SD, Moskalenko M, Tian T, Smith D, McGuinness R, Chen L, et al. T-cell killing of heterogenous tumor or viral targets with bispecific chimeric immune receptors. Cancer Gene Ther. 2000;7:1127–1134. doi: 10.1038/sj.cgt.7700213. [DOI] [PubMed] [Google Scholar]
- Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med. 2011;365:1673–1683. doi: 10.1056/NEJMoa1106152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkie S, van Schalkwyk MC, Hobbs S, Davies DM, van der Stegen SJ, Pereira AC, et al. Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J Clin Immunol. 2012;32:1059–1070. doi: 10.1007/s10875-012-9689-9. [DOI] [PubMed] [Google Scholar]
- Calarese DA, Scanlan CN, Zwick MB, Deechongkit S, Mimura Y, Kunert R, et al. Antibody domain exchange is an immunological solution to carbohydrate cluster recognition. Science. 2003;300:2065–2071. doi: 10.1126/science.1083182. [DOI] [PubMed] [Google Scholar]
- Kleinjung J, Petit MC, Orlewski P, Mamalaki A, Tzartos SJ, Tsikaris V, et al. The third-dimensional structure of the complex between an Fv antibody fragment and an analogue of the main immunogenic region of the acetylcholine receptor: a combined two-dimensional NMR, homology, and molecular modeling approach. Biopolymers. 2000;53:113–128. doi: 10.1002/(SICI)1097-0282(200002)53:2<113::AID-BIP1>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Leysath CE, Monzingo AF, Maynard JA, Barnett J, Georgiou G, Iverson BL, et al. Crystal structure of the engineered neutralizing antibody M18 complexed to domain 4 of the anthrax protective antigen. J Mol Biol. 2009;387:680–693. doi: 10.1016/j.jmb.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson IC, Hall CJ, Veverka V, Shi JY, Muskett FW, Stephens PE, et al. High resolution NMR-based model for the structure of a scFv-IL-1beta complex: potential for NMR as a key tool in therapeutic antibody design and development. J Biol Chem. 2009;284:31928–31935. doi: 10.1074/jbc.M109.025304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginalski K, Elofsson A, Fischer D, Rychlewski L. 3D-Jury: a simple approach to improve protein structure predictions. Bioinformatics. 2003;19:1015–1018. doi: 10.1093/bioinformatics/btg124. [DOI] [PubMed] [Google Scholar]
- Zola H, MacArdle PJ, Bradford T, Weedon H, Yasui H, Kurosawa Y. Preparation and characterization of a chimeric CD19 monoclonal antibody. Immunol Cell Biol. 1991;69 (Pt 6):411–422. doi: 10.1038/icb.1991.58. [DOI] [PubMed] [Google Scholar]
- Wels W, Harwerth IM, Zwickl M, Hardman N, Groner B, Hynes NE. Construction, bacterial expression and characterization of a bifunctional single-chain antibody-phosphatase fusion protein targeted to the human erbB-2 receptor. Biotechnology (NY) 1992;10:1128–1132. doi: 10.1038/nbt1092-1128. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Binding of anti-HER2 antibody FRP5 to peptide arrays.
Surface expression of the TanCAR was tested using two different methods.
We used a tetracycline-inducible system to conditionally express a truncated non-signaling CD19 molecule on Daoy cells (Daoy.TET.CD19).30