

Abstract
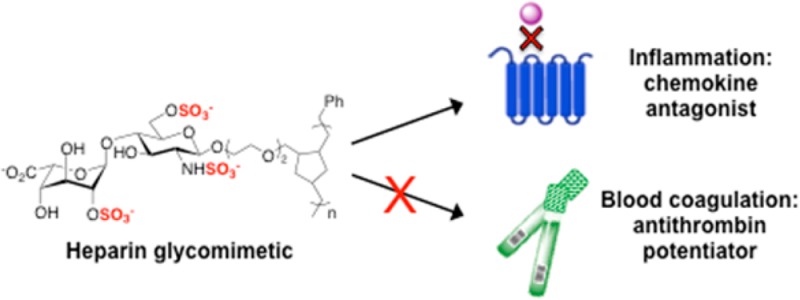
Heparan sulfate (HS) glycosaminoglycans participate in critical biological processes by modulating the activity of a diverse set of protein binding partners. Such proteins include all known members of the chemokine superfamily, which are thought to guide the migration of immune cells through their interactions with HS. Here, we describe an expedient, divergent synthesis to prepare defined HS glycomimetics that recapitulate the overall structure and activity of HS glycosaminoglycans. Our approach uses a core disaccharide precursor to produce a variety of differentially sulfated glycopolymers. We demonstrate that a specific trisulfated mimetic antagonizes the chemotactic activity of the proinflammatory chemokine RANTES with potency similar to that of heparin, without inhibiting serine proteases in the blood coagulation cascade. Our work provides a general strategy for modulating chemokine activity and dissecting the pleiotropic functions of HS/heparin through the presentation of defined sulfation motifs within polymeric scaffolds.
Heparan sulfate (HS) glycosaminoglycans are a ubiquitous class of sulfated polysaccharides that are involved in diverse physiological processes, such as development, wound healing, angiogenesis, and inflammation.1 Assembled from disaccharide subunits, HS polysaccharides exhibit subtle variations in stereochemistry, length, and patterns of sulfation. This structural diversity enables HS to modulate the activity of >400 different proteins, including ∼50 members of the chemokine superfamily.2 Chemokines are chemoattractant proteins that control the activation and migration of specific leukocyte populations toward sites of injury, inflammation, and atherosclerosis. Accumulation of pro-inflammatory chemokines into epithelial spaces contributes to the pathogenesis of allergy, arthritis, psoriasis, and other inflammatory disorders.3 Although HS proteoglycans are required for establishing chemokine gradients in vivo,4 the precise carbohydrate structural determinants involved have yet to be elucidated, limiting the development of HS-based strategies for targeting chemokine activity.
A major challenge to understanding the structure–activity relationships of HS and developing HS-based therapeutic approaches has been the chemical complexity and heterogeneity of HS in vivo. Heparin, a close structural relative of HS, displays less heterogeneity and is used clinically as an anti-coagulant drug for the prevention and treatment of thrombosis.5 Elegant studies have demonstrated that a unique sulfated sequence found within heparin is primarily responsible for its anti-coagulant activity.6 Heparin has also been shown to have potent anti-inflammatory activity in models of asthma, chronic dermatitis, and ulcerative colitis, but it is not recommended as an anti-inflammatory agent in clinical practice due to its anti-coagulant activity.7 Moreover, heparin isolated from natural sources can induce other undesirable physiological effects due to its structural heterogeneity.6a The development of chemically defined HS/heparin mimetics that lack anti-coagulant activity offers a potential solution to these challenges. Although a few studies have modified natural polysaccharides or undertaken the semi-synthesis of glucan sulfate mimetics,8 such approaches lack the precision of chemical synthesis9 in controlling the sulfation sequence. To our knowledge, only one study9a exploited chemical synthesis to generate complex, HS-based oligosaccharides that modulate pro-inflammatory chemokines. Here, we describe a new class of simplified HS/heparin glycomimetics that have a highly tunable structure, controllable lengths, and defined sulfation motifs. Importantly, these molecules possess the ability to inhibit pro-inflammatory chemokines with similar efficiency to natural heparin, yet they do not interact with key factors in the coagulation cascade.
Most chemokines have a primary HS-binding site occupied by at least two sugar residues.10 Based on our prior work with chondroitin sulfate glycosaminoglycans,11 we envisaged that HS disaccharide epitopes appended onto a multivalent polymer backbone might be sufficient to target this binding site, provided that their binding affinity could be enhanced through avidity (Scheme 1a). A polynorbornene backbone was selected to allow for maximal control over chain length and low polydispersity through ring-opening metathesis polymerization chemistry (Scheme 1b).12 Reduction of the unsaturated backbone was expected to emulate the conformational plasticity found in native HS/heparin and facilitate its interaction with proteins.13 Furthermore, by controlling the sulfation pattern prior to polymerization, we sought to increase the specificity of the polymers for particular HS-binding proteins. Notably, these structures would represent the first example of high-molecular-weight polymers prepared from minimal HS disaccharide units.
Scheme 1. (a) Synthetic Mimetic of Heparin/HS Polysaccharides and (b) Retrosynthesis of Glycopolymers 1–4.
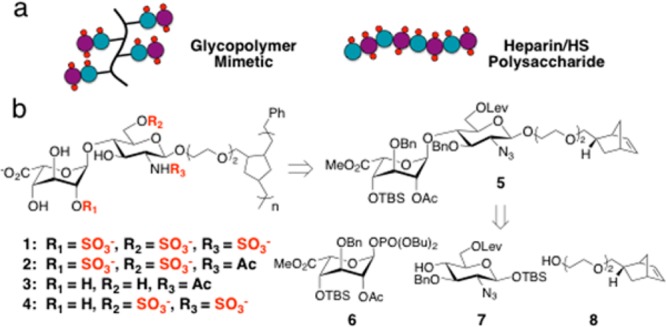
A library of differentially sulfated glycopolymers 1–4 was derived from a single synthetic precursor, 5 (Scheme 1b). The orthogonal protecting group strategy for 5 was designed to allow multiple sulfation patterns to be accessed using a minimum number of steps. We chose to install a tert-butyldimethylsilyl (TBS) group at the C4′-hydroxyl and benzyl ethers (Bn) at the non-sulfated positions C3′ and C3 to enhance the solubility of the sulfated monomers during the polymerization step. Acetyl (Ac) and levulinoyl (Lev) ester groups were selected for sites that would ultimately carry the O-sulfonate groups (C2′ and C6, respectively); the former group would also provide anchimeric assistance in generating the 1,2-trans glycosidic linkage.
Disaccharide 5 was generated from iduronic acid (IdoA) phosphate donor 6, glucosamine (GlcN) acceptor 7,14a and norbornyl linker 8.12c An efficient route to donor 6 and its subsequent transformation to disaccharide 5 is shown in Scheme 2. First, the 1,2,4-triol 9 was synthesized from commercially available diacetone glucose in six steps14b and treated with AcCl and DMAP at −40 °C15a to afford the triacetylated β-isomer 10 in quantitative yield. Conditions using Ac2O or higher temperatures15b were avoided as they led to a mixture of α/β-pyranose and α/β-furanose isomers. Anomeric bromination of 10 using TiBr415a formed a glycosyl halide intermediate, which was directly transformed into 1,2-orthoester 11 using 2,4,6-collidine as the base. The 1,2-orthoester moiety locked the pyranose ring into the 4C1 conformation and allowed for selective cleavage of the remaining acetyl group under Zemplén conditions. After several unsuccessful attempts using standard conditions, we found that TBS protection of the C4′ hydroxyl of 12 required excess TBSOTf in neat pyridine to compensate for its poor nucleophilicity.
Scheme 2. Synthesis of Disaccharide 5.

Reagents and conditions: (a) AcCl, Py, DMAP, CH2Cl2, −40 °C, quant.; (b) (i) TiBr4, CH2Cl2, (ii) 2,4,6-collidine, MeOH, CH2Cl2, 75%; (c) NaOMe, MeOH, −10 °C, 80%; (d) TBSOTf, Py, 0 °C, 92%; (e) HOPO(OBu)2, CH2Cl2, 4 Å MS, quant.; (f) TMSOTf, 7, CH2Cl2, 4 Å MS, 93%; (g) (i) TBAF, AcOH, THF, 0 °C, (ii) Cl3CCN, DBU, CH2Cl2, 0 °C, 89%; (h) BF3·OEt2, 8, CH2Cl2, 4 Å MS, 80%.
Direct reaction of orthoester 13 with acceptor 7 gave low yields of the desired IdoA-GlcN disaccharide 14. We therefore chose to explore the reactivity of the corresponding IdoA phosphate donor, which could be accessed from 13 using HOPO(OBu)217 in a single quantitative step. Fortuitously, phosphate donor 6 reacted with acceptor 7 and stoichiometric TMSOTf17 to deliver the α-linked disaccharide 14 in 93% yield. To tether the HS disaccharide to norbornyl linker 8, we converted 14 into glycosyl imidate 15 by deprotecting the anomeric TBS and treating the resulting hemiacetal with trichloroacetonitrile and DBU. Glycosidic coupling of 15 and 8 with catalytic TMSOTf resulted in a 1:1 mixture of α/β isomers; however, treatment with BF3·OEt2 (0.34 equiv) gave exclusively the β-isomer in 80% yield (δ = 4.34 ppm, J12 = 8 Hz).
Disaccharide 5 was then diversified into four differentially sulfated polymers (1–4; Scheme 3). The azide was reduced using 1,3-propanedithiol,9b and the resulting intermediate was treated either with K2CO3 to deprotect both ester groups or with hydrazine acetate9c to selectively cleave the Lev ester. The resulting compounds 16 and 17 were regioselectively N-sulfated, O-sulfated, and N-acetylated at the desired positions. Conveniently, we found that simultaneous sulfation at the O- and N-positions could be achieved using SO3·Py and mild heating conditions (55 °C). Polymerization of the sulfated monomers with 5 mol % of Grubbs’ fast-initiating catalyst [(H2IMes)(Py)2(Cl)2Ru=CHPh]19 led to rapid conversion within minutes to the desired glycopolymers (22–25). Late-stage deprotection conditions were first established using monomer 18 to prepare a monovalent trisulfated disaccharide bearing the norbornyl linker at the reducing end (26, see SI) and then applied to 22–25. We were pleased to find that TMSOK-mediated saponification of the methyl ester also deprotected the TBS group at C4′, likely due to its proximity to the resulting C5′ carboxylate anion. Finally, catalytic hydrogenation using Pd(OH)2/charcoal in a co-solvent of phosphate buffer and methanol9b delivered 26 and glycopolymers 1–4, which were characterized by 1H NMR spectroscopy and size exclusion chromatography multi-angle light scattering. Comparison of the 1H–1H gCOSY NMR spectrum of trisulfated monomer 26 to that of glycopolymer 1 confirmed that the sulfation pattern remained intact during the post-polymerization deprotection steps. By varying the amount of catalyst, we could predictably control the glycopolymer lengths within a relatively narrow polydispersity range, affording polymers with lengths comparable to the length of commercially available heparin (Table S1).
Scheme 3. Synthesis of Glycopolymers 1–4.
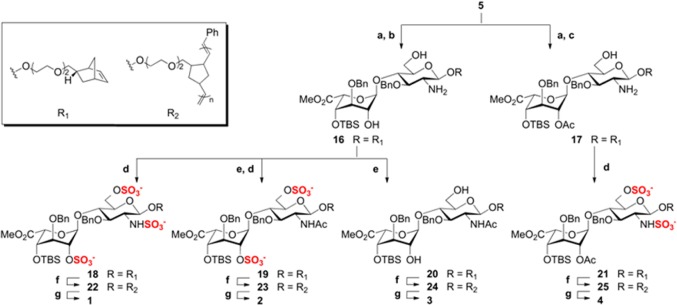
Reagents and conditions: (a) 1,3-propanedithiol, DIPEA, MeOH, 87%; (b) K2CO3, MeOH, 78%; (c) H2NNH2·H2O, AcOH, Py, quant.; (d) SO3·Py, NEt3, Py, 55 °C, 78–85%; (e) Ac2O, NEt3, MeOH, quant.; (f) (H2IMes)(Py)2(Cl)2Ru=CHPh, DCE, MeOH, quant.; (g) (i) TMSOK, TBAI, THF, 80–88%, (ii) Pd(OH)2/charcoal, H2 (1 atm), phosphate buffer (pH 7.4), MeOH, 37–55%.
With the library of glycopolymers in hand, we characterized their binding specificities for RANTES, a basic chemokine that induces the migration of T cells, monocytes, natural killer cells, and other classes of leukocytes.20 Although RANTES preferentially recognizes trisulfated heparin (Kd = 32.1 nM) over other classes of glycosaminoglycans,21 the effects of HS sulfation on RANTES binding have not been examined using homogeneously sulfated molecules. We compared the relative ability of each glycopolymer to block RANTES binding to heparin (EC50 = 12.2 nM; Figure S1) using a competitive enzyme-linked immunosorbent assay. Trisulfated glycopolymer 1 bound strongly to RANTES (IC50 = 9.3 ± 1.1 μg/mL (334 ± 39 nM)), albeit with reduced affinity compared to heparin of similar chain length (IC50 = 0.90 ± 0.03 μg/mL (45.0 ± 1.5 nM); Figure 1 and Table S2). However, the glycopolymer competed more effectively for RANTES binding compared to heparin at its maximum inhibitory concentration. Whereas heparin exhibited a maximum inhibition of 58.4%, 1 inhibited RANTES binding by up to 90.8% under the same assay conditions, which could be attributed to differences in their interactions with oligomeric forms of RANTES.4 As expected, monovalent disaccharide 26 failed to compete with heparin for RANTES binding (Figure 1). These results provide the first demonstration that an HS disaccharide epitope can be sufficient for chemokine binding when presented in a multivalent framework and underscore the importance of avidity in the recognition of glycosaminoglycan structures.
Figure 1.
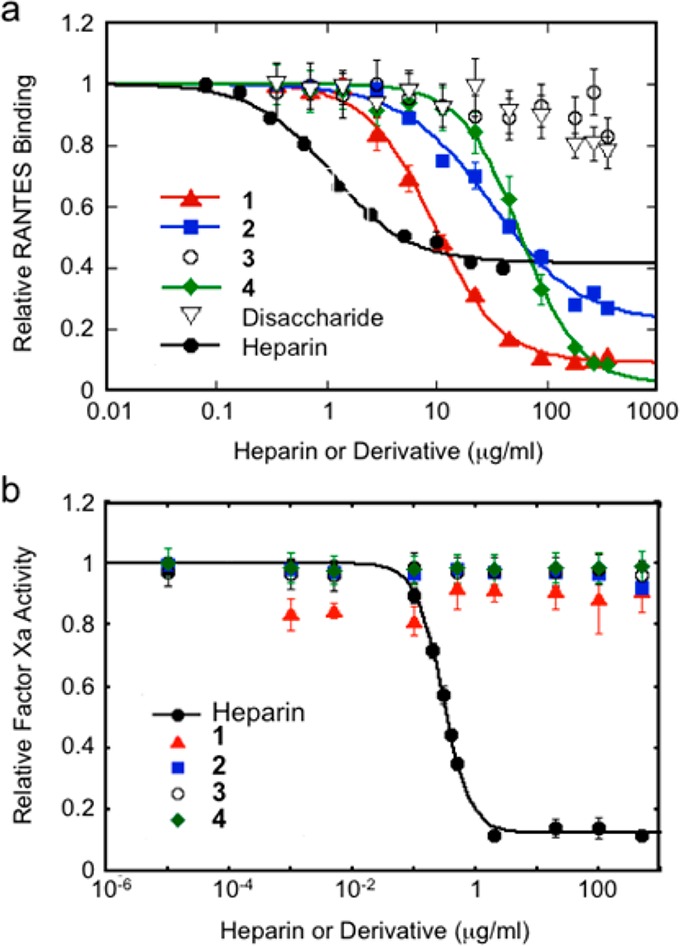
(a) Comparison of the ability of 1–4 to compete with heparin for binding to RANTES. P < 0.05 for IC50 values of 2 and 4 compared to 1; P < 0.05 for IC50 value of 2 compared to 4. IC50 values corrected for ligand valence are reported in Table S2. (b) 1–4 do not potentiate the anti-coagulant activity of antithrombin III, as determined by their ability to inhibit factor Xa substrates in a chromogenic assay. Data represent the mean ± standard error for quadruplicate assays in (a) and (b).
Next, we tested whether site-defined modifications to the sulfation pattern of 1 would alter its affinity for RANTES. Removal of either the N-sulfate group of GlcN (2) or the 2-O-sulfate group of IdoA (4) decreased binding to RANTES (IC50 = 31.1 ± 6.2 μg/mL (852 ± 170 nM) and 58.0 ± 5.7 μg/mL (1760 ± 170 nM), respectively), and unsulfated 3 had no appreciable activity (Figure 1a). The observation that removal of 2-O-sulfation has a greater effect than removal of N-sulfation suggests that precise positioning of the sulfate groups (in addition to overall charge) is important for determining the affinity of the glycopolymers for RANTES. Importantly, none of the glycopolymers possessed anti-coagulant activity, as demonstrated by their inability to potentiate the inhibition of factor Xa and thrombin substrates by antithrombin III (Figures 1b and S2). Thus, controlling the positioning of sulfate groups within the glycopolymer enables the anti-inflammatory function of HS/heparin to be dissected from its anti-coagulant function. Furthermore, modifications to the sulfation pattern can be exploited to adjust the affinity of the glycopolymers for different HS-binding proteins and may facilitate the development of glycosaminoglycan-based therapeutic agents with fewer off-target side effects.
The chemotactic activity of RANTES, which is essential to the pathogenesis of allergic inflammatory responses such as asthma, is mediated in part by the G-protein coupled receptor CCR3.22 To test whether 1 can interfere with RANTES-induced chemotaxis via CCR3, we probed the migration of murine L1.2 pre-B cells that were stably transfected with the CCR3 receptor.23 Using a modified Boyden chamber, we observed that the directional migration of L1.2-CCR3 cells (but not wild-type L1.2 cells lacking CCR3) was dependent on the RANTES concentration and elicited a maximal response at 10 nM (Figure S3). Pre-incubation of RANTES (10 nM) with either 1 or heparin diminished the chemotactic activity of RANTES in a dose-dependent manner (Figure 2). Further corroborating these results, lower levels of RANTES were detected on the surface of CCR3-expressing cells after the chemokine (100 nM) was pretreated with 1 or heparin, as determined by flow cytometry analysis (Figure S4). Notably, 1 and heparin failed to block the migration of L1.2 cells transfected with CCR5, an alternative receptor for RANTES (Figure S5). Consistent with these findings, the reported binding sites for HS and RANTES overlap on CCR3, whereas the two binding sites share no overlap on CCR5.22 Together, our results show that 1 effectively antagonizes the CCR3-dependent chemotactic activity of RANTES in cells, with comparable potency as heparin. Moreover, we demonstrate the ability of these HS glycomimetics to selectively target specific chemokine–receptor interactions.
Figure 2.
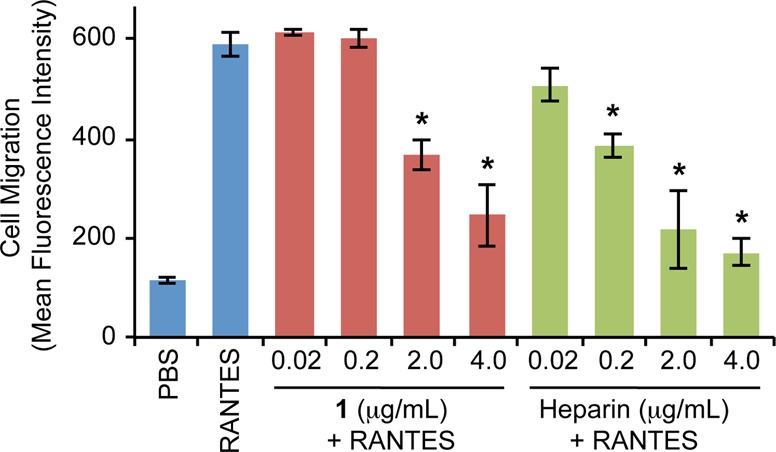
Heparin or 1 antagonizes the RANTES-induced migration of CCR3-expressing cells. Data represent the mean ± standard error for three independent experiments, each conducted in quadruplicate (*, P < 0.01).
In conclusion, we have developed a new class of HS glycomimetics that are synthetically accessible and highly tunable in structure and sulfation sequence. By controlling the sulfation sequence and exploiting the principles of multivalency to enhance glycan recognition, the binding affinity of HS disaccharides for protein binding partners can be amplified to target chemokines and their receptor interactions. We demonstrate that a trisulfated HS glycopolymer binds to RANTES with nanomolar affinity and inhibits the CCR3-dependent cellular response to this therapeutically important chemokine, without affecting components of the blood coagulation cascade. We envision that variations of these glycomimetics can be synthetically tailored to antagonize a wide range of HS-binding proteins with clinical relevance to atherosclerosis, cancer, and autoimmune disorders.
Acknowledgments
We thank Rochelle Diamond at the Flow Cytometry Cell Sorting Facility, Dr. Mona Shahgholi in the Mass Spectrometry Facility, and Dr. David VanderVelde at the High Resolution NMR Facility at Caltech. We also thank Dr. Osamu Yoshie (Kinki University, School of Medicine, Japan) for generously providing murine L1.2 cells stably expressing CCR3 and CCR5. This work was supported by the NIH (R01 GM093627).
Supporting Information Available
Supporting figures, experimental procedures, synthesis, and characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Bishop J. R.; Schuksz M.; Esko J. D. Nature 2007, 446, 1030. [DOI] [PubMed] [Google Scholar]
- a Ori A.; Wilkinson M. C.; Fernig D. G. J. Biol. Chem. 2011, 286, 19892. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lortat-Jacob H.; Grosdidier A.; Imberty A. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerard C.; Rollins B. J. Nature Immunol. 2001, 2, 108. [DOI] [PubMed] [Google Scholar]
- Proudfoot A. E.; Handel T. M.; Johnson Z.; Lau E. K.; LiWang P.; Clark-Lewis I.; Borlat F.; Wells T. N.; Kosco-Vilbois M. H. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackman N. Nature 2008, 451, 914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Petitou M.; Hérault J. P.; Bernat A.; Driguez P. A.; Duchaussoy P.; Lormeau J. C.; Herbert J. M. Nature 1999, 398, 417. [DOI] [PubMed] [Google Scholar]; b Choay J.; Petitou M.; Lormeau J. C.; Sinay P.; Casu B.; Gatti G. Biochem. Biophys. Res. Commun. 1983, 116, 492. [DOI] [PubMed] [Google Scholar]; c Xu Y.; Masuko S.; Takieddin M.; Xu H.; Liu R.; Jing J.; Mousa S. A.; Linhardt R. J.; Liu J. Science 2011, 334, 498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young E. Thromb. Res. 2008, 122, 743. [DOI] [PubMed] [Google Scholar]
- a Wang L. C.; Brown J. R.; Varki A.; Esko J. D. J. Clin. Invest. 2002, 110, 127. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Wang J.-G.; Mu J.-S.; Zhu H.-S.; Geng J.-G. Inflamm. Res. 2002, 51, 435. [DOI] [PubMed] [Google Scholar]; c Alban S.; Ludwig R. J.; Bendas G.; Schön M. P.; Oostingh G. J.; Radeke H. H.; Fritzsche J.; Pfeilschifter J.; Kaufmann R.; Boehncke W.-H. J. Invest. Derm. 2009, 129, 1192. [DOI] [PubMed] [Google Scholar]; d Powell A. L.; Ahmed Y. A.; Yates E. A.; Turnbull J. E. Nat. Protoc. 2010, 5, 821. [DOI] [PubMed] [Google Scholar]
- a de Paz J. L.; Moseman E. A.; Noti C.; Polito L.; von Andrian U. H.; Seeberger P. H. ACS Chem. Biol. 2007, 2, 735. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Hu Y.-P.; Zhong Y.-Q.; Chen Z.-G.; Chen C.-Y.; Shi Z.; Zulueta M. M.; Ku C.-C.; Lee P.-Y.; Wang C.-C.; Hung S.-C. J. Am. Chem. Soc. 2012, 134, 20722. [DOI] [PubMed] [Google Scholar]; c Fan R.-H.; Achkar J.; Hernández-Torres J. M.; Wei A. Org. Lett. 2005, 7, 5095. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Arungundram S.; Al-Mafraji K.; Asong J.; Leach F. E. III; Amster I. J.; Venot A.; Turnbull J. E.; Boons G.-J. J. Am. Chem. Soc. 2009, 131, 17394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Goger B.; Halden Y.; Rek A.; Mösl R.; Pye D.; Gallagher J.; Kungl A. J. Biochemistry 2002, 41, 1640. [DOI] [PubMed] [Google Scholar]; b Shaw J. P.; Johnson Z.; Borlat F.; Zwahlen C.; Kungl A.; Roulin K.; Harrenga A.; Wells T. N.; Proudfoot A. E. Structure 2004, 12, 2081. [DOI] [PubMed] [Google Scholar]
- Rawat M.; Gama C. I.; Matson J. B.; Hsieh-Wilson L. C. J. Am. Chem. Soc. 2008, 130, 2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Grubbs R. H.Handbook of Metathesis, 1st ed.; Wiley-VCH: Weinheim, Germany, 2003. [Google Scholar]; b Kanai M.; Mortell K. H.; Kiessling L. L. J. Am. Chem. Soc. 1997, 119, 9931. [Google Scholar]; c Lee S.-G.; Brown J. M.; Rogers C. J.; Matson J. B.; Krishnamurthy C.; Rawat M.; Hsieh-Wilson L. C. Chem. Sci. 2010, 1, 322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canales A.; Angulo J.; Ojeda R.; Bruix M.; Fayos R.; Lozano R.; Giménez-Gallego G.; Martín-Lomas M.; Nieto P. M.; Jiménez-Barbero J. J. Am. Chem. Soc. 2005, 127, 5778. [DOI] [PubMed] [Google Scholar]
- a Orgueira H. A.; Bartolozzi A.; Schell P.; Litjens R. E.; Palmacci E. R.; Seeberger P. H. Chem.—Eur. J. 2003, 9, 140. [DOI] [PubMed] [Google Scholar]; b Lohman G. J; Hunt D. K.; Högermeier J. A.; Seeberger P. H. J. Org. Chem. 2003, 68, 7559. [DOI] [PubMed] [Google Scholar]
- a Gavard O.; Hersant Y.; Alais J.; Duverger V.; Dilhas A.; Bascou A.; Bonnaffé D. Eur. J. Org. Chem. 2003, 2003, 3603. [Google Scholar]; b Dilhas A.; Bonnaffé D. Carbohydr. Res. 2003, 338, 681. [DOI] [PubMed] [Google Scholar]
- Ravidà A.; Liu X.; Kovacs L.; Seeberger P. H. Org. Lett. 2006, 8, 1815. [DOI] [PubMed] [Google Scholar]
- Scholl M.; Ding S.; Lee C. W.; Grubbs R. H. Org. Lett. 1999, 1, 953. [DOI] [PubMed] [Google Scholar]
- Qin S.; Rottman J. B.; Myers P.; Kassam N.; Weinblatt M.; Loetscher M.; Koch A. E.; Moser B.; Mackay C. R. J. Clin. Invest. 1998, 101, 746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin L.; Blanpain C.; Garnier P.; Wittamer V.; Parmentier M.; Vita C. Biochemistry 2001, 40, 6303. [DOI] [PubMed] [Google Scholar]
- a Kitaura M.; Nakajima T.; Imai T.; Harada S.; Combadiere C.; Tiffany H. L.; Murphy P. M.; Yoshie O. J. Biol. Chem. 1996, 271, 7725. [DOI] [PubMed] [Google Scholar]; b Borish L. C.; Steinke J. W. J. Allergy Clin. Immunol. 2003, 111, S460. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.