

Abstract
Background
Alcohol drinking by adolescents is a major public health concern. Adolescents tend to drink in a chronic, intermittent, that is, “binge,” pattern, and such patterns of ethanol exposure are associated with increased risk of neurotoxicity and the development of alcohol use disorders (Crews et al., 2000; Hunt, 1993). Both adolescent humans and rats are more sensitive to acute ethanol-induced memory impairment than adults (Acheson et al., 1998; Markwiese et al., 1998). Furthermore, in rats, chronic intermittent ethanol (CIE) exposure during adolescence produces a long-lasting, perhaps permanent, maintenance of the adolescent high sensitivity to ethanol’s amnestic effects (White et al., 2000a). We have previously shown that acute ethanol increases tonic inhibitory current mediated by extrasynaptic GABAA receptors more efficaciously in dentate granule cells (DGCs) from adolescent than adult rats (Fleming et al., 2007). In this study, we determined if CIE during adolescence produced long-lasting changes in this tonic current.
Methods
Adolescent rats were subjected to a CIE exposure regimen and allowed to mature to full adulthood. Whole-cell voltage-clamp measurements of tonic inhibitory current and mean phasic current were made in vitro in hippocampal brain slices.
Results
CIE exposure during adolescence increased the ethanol sensitivity of tonic inhibitory current mediated by extrasynaptic GABAA receptors and decreased the ethanol sensitivity of phasic, synaptic GABAA receptor-mediated current in adult DGCs.
Conclusions
CIE exposure during adolescence produces long-lasting changes in the function and ethanol sensitivity of extrasynaptic GABAA receptors in DGCs. These changes appear to “lock-in” and maintain the high adolescent sensitivity to ethanol in these cells. Furthermore, greater ethanol enhancement of tonic inhibition in the hippocampal formation after CIE is consistent with the greater sensitivity to ethanol-induced memory impairment after adolescent CIE. This finding represents the first demonstration of a long-term, memory-related cellular effect of CIE during adolescence, and the “lock-in” of adolescent ethanol sensitivity that these results suggest could represent a conceptual step forward in understanding the vulnerability of the adolescent brain to alcohol.
Keywords: Ethanol, Extrasynaptic GABAA Receptor, Tonic Inhibition, Dentate Gyrus, Adolescence
The use of alcohol by adolescents is a major public health concern with a long-lasting impact on health care in the United States Recent studies have shown that alcohol affects brain function differently during adolescence than adulthood, and we have consistently found that adolescent and adult rats respond differently to acute ethanol exposure. For example, adolescent rats are more sensitive to the memory- impairing effects of ethanol than are adults (Markwiese et al., 1998), but are less sensitive than adults to the effects of ethanol on sedation (Little et al., 1996) and motor impairment (White et al., 2002a, b). In addition, ethanol more efficaciously disrupts both the induction of long-term potentiation (LTP) (Pyapali et al., 1999; Swartzwelder et al., 1995a) and n-methyl-D-aspartate receptor-mediated synaptic activity (Swartzwelder et al., 1995b) in hippocampal slices from adolescents than in those from adults. Acute doses of ethanol have also been shown to impair learning more potently in young adult humans than in slightly older individuals (Acheson et al., 1998).
Clearly, there are substantial developmental differences in the sensitivity of hippocampal neurons and neuronal plasticity to the acute effects of ethanol. However, at present, there has been very little work on the long-lasting effects of chronic ethanol exposure during adolescence. Because most adolescents who drink repeatedly do so in a chronic, intermittent, that is, “binge,” pattern, and such patterns of ethanol exposure are associated with increased risk of neurotoxicity (Crews et al., 2000; Hunt, 1993), we believe that it is critical to thoroughly and systematically examine the long-term effects of chronic intermittent ethanol (CIE) exposure during adolescence. At the behavioral level, we have shown that CIE during adolescence (but not adulthood) alters the subsequent sensitivity of rats to the effects of ethanol on both spatial learning (White et al., 2000b) and motor function (White et al., 2002a). In the case of spatial learning, acute ethanol disrupts spatial learning more powerfully in adolescent rats than in adults (Markwiese et al., 1998), and CIE during adolescence results in the maintenance of high sensitivity to the amnestic effects of ethanol into adulthood (White et al., 2000a). In the case of motor function, adolescents are less sensitive to the acute effects of ethanol (White et al., 2002b), and CIE during adolescence results in the maintenance of this relative insensitivity into adulthood, whereas the same exposure paradigm applied in adulthood did not result in long-lasting alterations of ethanol sensitivity (White et al., 2002a).
These are striking findings because they indicate that CIE during adolescence may “lock-in” adolescent sensitivities to ethanol and sustain them into adulthood. The maintenance of an “adolescent” pattern of behavioral ethanol sensitivity in adulthood suggests that adolescence represents a period of heightened vulnerability to the long-term consequences of repeated ethanol exposures and may reflect a mechanism underlying the increased liability for alcohol abuse disorders in adulthood among those who begin recreational drinking during adolescence. To our knowledge, however, at present, there have been no physiological or cellular studies that have addressed the possibility of long-term alterations in ethanol sensitivity after CIE during adolescence.
We have shown that ethanol promotes GABAA receptor-mediated tonic current more efficaciously in hippocampal neurons from adolescent rats compared with those from adults (Fleming et al., 2007). This tonic inhibitory current is generated by activation of extrasynaptic GABAA receptors, which are known to play a role in inhibiting learning and memory (Chambers et al., 2003; Collinson et al., 2002; Crestani et al., 2002; Martin et al., 2010; Moore et al., 2010; Shen et al., 2010; Wiltgen et al., 2005) and powerfully modulate the function of dentate granule cells (DGCs) (Nusser and Mody, 2002). The fact that ethanol promotes tonic current more efficaciously in DGCs from adolescent than adult animals (Fleming et al., 2007) is fully consistent with its more potent antagonism of learning in adolescent animals (Markwiese et al., 1998). If the adolescent pattern of heightened tonic current ethanol sensitivity was sustained into adulthood, it could have wide-ranging implications for adult CNS functioning in the presence of ethanol. This study was designed to assess the long-term effects of CIE during adolescence on dentate gyrus tonic current and its responsiveness to acute ethanol.
MATERIALS AND METHODS
Animals and Chronic Intermittent Ethanol Exposure
All animal research reported in this study was conducted according to protocols that were approved by the Durham VAMC and Duke University Institutional Animal Care and Use Committees. Male Sprague-Dawley rats (Charles River, Raleigh, NC) were housed 2 animals per cage and provided with ad libitum access to food and water for the duration of the study. Beginning at 30 days of age, adolescent rats were exposed to a CIE exposure regimen that consisted of 10 doses by intragastric gavage of 5 g / kg ethanol (25% w/ v in saline) in a 2-days-on and 2-days-off sequence such that the animals received single-gavage exposures on days 1 and 2, 5 and 6, 9 and 10, 13 and 14, and 17 and 18 of the regimen. Control rats received 20 ml / kg saline by gavage. The rats were weighed daily to track their weight gain. After the last ethanol exposure, the rats were housed in the vivarium for another 22 days, until they had reached adulthood at 70 days of age. The electrophysiology was conducted between 40 and 54 days after the start of the CIE regimen. Thus, all the rats were mature adults of 70 to 84 days old at the time of recording. The exposure group, ethanol or saline, used for each slice preparation was randomized, and the electrophysiologist was blind to the exposure group of the animals during the slice preparation, data acquisition, and analysis phases of the experiment.
Brain Slice Preparation
Rats were anesthetized with halothane or isoflurane vapor and decapitated using a rodent guillotine. The brain was removed, and 300- to 350-μm sagittal slices containing the hippocampal formation were cut using a Vibratome Series 1000 (Vibratome, Bannockburn, IL). The ice-cold cutting solution consisted of (mM) 120 NaCl, 3.3 KCl, 25 NaHCO3, 1.23 NaH2PO4, 15 D-Glucose, 3 Myo-Inositol, 2 Na Pyruvate, 0.4 Na Ascorbate, 0.05 CaCl2, and 12 MgCl equilibrated with a gas mixture of 95% O2 and 5% CO2. The slices were incubated at 35°C for half an hour to 1 hour in artificial cerebrospinal fluid (aCSF) containing (mM) 120 NaCl, 3.3 KCl, 25 NaHCO3, 1.23 NaH2PO4, 15 D-Glucose, 3 Myo-Inositol, 2 Na Pyruvate, 0.4 Na Ascorbate, 2.0 CaCl2, 1.3 MgCl and bubbled with 95% O2 / and 5%CO2. After heating, slices were held at room temperature (RT: 21 to 23°C). Before beginning recording, the slices were allowed to recover for a minimum of 1 hour, which included the duration they were held at 35°C. All drugs were diluted in aCSF and applied in the bath.
Electrophysiology
Individual DGCs were visually identified using a Zeiss Axioskop (Carl Zeiss MicroImaging, LLC, Thornwood, NY) equipped with IR-DIC videomicroscopy and a 40× water immersion objective. While in the recording chamber, the slices were perfused with RT 95 /5% O2 CO2-bubbled aCSF at a rate of 2 to 4 ml / min. Microelectrodes with a tip resistance of 5 to 10 MΩ when filled were pulled from thin-walled borosilicate glass capillaries (World Precision Instruments, Inc., Sarasota, FL, or Garner Glass Co., Claremont, CA) using a Sutter Instrument Co. P-2000 puller (Novato, CA). The electrode solution consisted of (mM) 130 CsCl, 10 4-(2-Hydroxyethyl)piperazine-1-ethanesulfonic acid, N-(2-Hydroxyethyl)-piperazine-N′-(2-ethanesulfonic acid) (HEPES), 4 NaCl, 0.2 ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid (EGTA), 10 Na2CreatinePO4, 4 MgATP, 0.3 TrisGTP, and 6 QX-314; pH 7.2, osm 290. Whole-cell voltage-clamp recordings were performed using an Axopatch 200B amplifier (Molecular Devices, Inc. Sunnyvale, CA). Signals were low-pass filtered at 2 kHz and digitized at 10 kHz using an Axon Instruments Digidata 1440A and Clampex 10.2 (Molecular Devices, Inc.).
Tonic inhibitory currents were measured as previously described (Fleming et al., 2007). Briefly, granule cells were voltage clamped at −80 mV, and the holding current was recorded during 3 periods: (i) baseline, aCSF only; (ii) during bath application of 30 mM ethanol; and (iii) during application of 200 μM picrotoxin. To avoid repeated exposures to ethanol and picrotoxin, only 1 cell was obtained per slice.
Data Analysis and Statistics
Data analysis was performed on data records that had been stripped of exposure and age group information. Tonic and phasic current determination was performed using an in-house function written for MATLAB (MathWorks, Natick, MA) that is based on the method used by Glykys and Mody (2007). For each recording period, all-point histograms were generated for 60 seconds of data, and the Gaussian function f(x) = A exp(−[x − μ]2 / 2σ2) was fitted to the part of each histogram that was not skewed by synaptic events. The center of this distribution (μ) represents the mean holding current, while σ represents the root-mean-square (RMS) noise over the 60-second interval. For each cell, the GABAA receptor-mediated tonic current was determined by calculating the difference between the centers of the fitted Gaussian functions obtained for the baseline and picrotoxin conditions (μpicrotoxin − μbaseline). The tonic noise was determined by calculating the difference in RMS noise between the 2 conditions (σbaseline − σpicrotoxin).
Mean phasic current was determined by subtracting μ from the raw data trace for each interval, which brought the mean of the Gaussian to 0 pA, and computing the cumulative sum of the data, which was then divided by the number of points in the trace. Thus, the baseline noise summed to 0 pA, and the tail generated by the inhibitory postsynaptic currents (IPSCs) summed to yield the mean phasic current (phasic Imean). As for the tonic current measurement, the GABAA receptor-mediated contributions to phasic Imean were computed by subtracting the values obtained under the picrotoxin condition, for example, the baseline mean phasic current equaled Imean / baseline − Imean / picrotoxin.
Inferential statistics included paired t-tests to compare predrug baseline recordings with recordings made under drug conditions. Comparisons across groups were made using 2-way ANOVA or t-test, as appropriate. The criterion for significance was set at p < 0.05; all data are presented as mean ± standard error.
RESULTS
The Effect of Adolescent Ethanol Exposure on Tonic Inhibition in Adulthood
Tonic current was present in DGCs from both CIE and saline-exposed rats (Fig. 1). As shown in Fig. 2A, there was a trend toward a lower level of baseline tonic current in cells from animals in the CIE group: 7.0 ± 1.5 pA, n = 8 cells, 6 rats for the CIE group and 8.7 ± 1.3 pA, n = 9 cells, 5 rats for the saline group, although the trend did not reach statistical significance, t(15) = 0.89, p = 0.20. However, the tonic noise was smaller in CIE animals than in controls: 0.58 ± 0.06 pA and 0.82 ± 0.10 pA, respectively, t(15) = 2.02, p = 0.03 (Fig. 2B), suggesting a decrease in baseline inhibition mediated by extrasynaptic GABAA receptors in DGCs from CIE-treated animals.
Fig. 1.
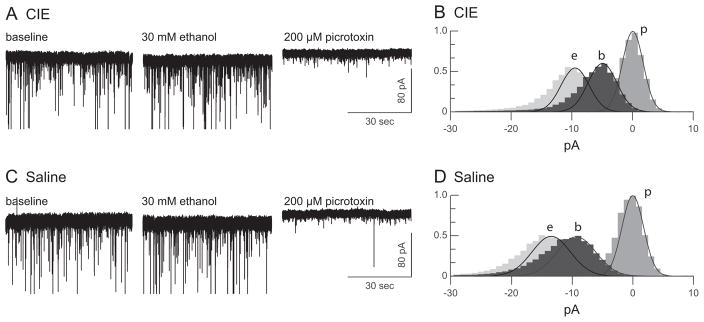
Ethanol increases GABAA receptor-mediated tonic inhibition in dentate granule cells from mature rats exposed to ethanol as adolescents and saline-treated controls. (Left) Representative voltage-clamp traces from a neuron from an adult rat that had been submitted to a chronic intermittent ethanol (CIE) regimen (A) or a saline control regimen (C) during adolescence. Holding current was recorded for 3 periods: a no-drug baseline and during bath application of 30 mM ethanol application and 200 μM picrotoxin. (Right, B, D) All-point histograms and fitted Gaussian functions for the whole-cell, current data shown at left. To facilitate comparison between cells, the data are plotted relative to the center of the distribution for the picrotoxin condition.
Fig. 2.

Chronic intermittent ethanol (CIE) exposure during adolescence increases ethanol enhancement of tonic current in mature dentate granule cells. (A) Tonic current and (B) tonic noise recorded from neurons from adult rats that had been submitted to a CIE regimen or a saline (control) regimen during adolescence. CIE during adolescence reduced tonic noise in adult neurons. (C) CIE during adolescence increases the ethanol (30 mM) enhancement of tonic current in adult neurons, t(15) = 2.269, p = 0.038. *p < 0.05 compared with control.
Ethanol (30 mM) significantly increased the size of the tonic current in both CIE and saline-treated animals, F(1, 15) = 44.34, p ≤ 0.001. With ethanol in the bath, the tonic current, μpicrotoxin − μethanol, was 12.2 ± 1.6 pA in the CIE group and 11.1 ± 1.4 pA in the saline group. For both exposure groups, these ethanol-induced increases in holding current were significant by paired t-test: t(7) = 5.51, p < 0.001 for the CIE group and t(8) = 3.56, p = 0.004 for the saline group. In addition, there was a significant interaction between acute ethanol application and the pretreatment exposure group, F(1, 15) = 16.17, p = 0.03. To control for the variability in the size of the tonic current among cells, ethanol enhancement of tonic current was calculated for each cell as the increase in holding current during bath ethanol application and as a percentage of the tonic current in the absence of ethanol: 100 (μbaseline − μethanol) / (μpicrotoxin − μbaseline). Ethanol enhancement of the tonic current was significantly greater in mature rats that had been exposed to ethanol as adolescents, 98.6 ± 24.9%, compared with rats exposed to saline, 36.3 ± 13.5%, t(15) = 2.269, p = 0.038 (Fig. 2C).
The Effect of Adolescent Ethanol Exposure on Phasic Inhibition in Adulthood
Using a method similar to that reported by Glykys and Mody (2007), we also calculated the mean phasic current (phasic Imean) for each recording interval of each cell. (One cell from the CIE group was excluded from this analysis because of unusual spontaneous IPSC [sIPSC] bursts during the ethanol exposure period.) Baseline phasic Imean did not differ between CIE and saline-exposed rats (Fig. 3A): CIE 3.93 ± 0.49 pA and saline 3.07 ± 0.30 pA, t(14) = 1.564, p = 0.140. In saline controls, 30 mM ethanol significantly increased the phasic Imean to 3.83 ± 0.40 pA, paired t(8) = 4.610, p = 0.002. However, in CIE-exposed rats, the ethanol-induced increase in phasic activity did not occur; in 30 mM ethanol, the phasic Imean for these animals was 3.50 ± 0.64 pA, which was not significantly different from the baseline, paired t(6) = 0.956, p = 0.376. As for the tonic current measurement, to control for the variability in the size of the phasic current among cells, ethanol enhancement of phasic current was calculated for each cell as the increase in phasic Imean during bath ethanol application and as a percentage of the phasic current in the absence of ethanol: 100 (Imean / ethanol − Imean / baseline) /(Imean / baseline − Imean / picrotoxin). In contrast to the effect of CIE exposure on tonic inhibition, ethanol enhancement of the phasic current was significantly smaller in mature rats that had been exposed to ethanol as adolescents, −12.1 ± 9.7%, compared with rats exposed to saline, 25.1 ± 6.1%, t(14) = 3.376, p = 0.005 (Fig. 3B).
Fig. 3.

Chronic intermittent ethanol (CIE) exposure during adolescence abolishes ethanol-induced increases in mean phasic current in mature dentate granule cells (DGCs). (A) Baseline mean phasic current (phasic Imean) in adult DGCs from rats exposed to CIE during adolescence or saline-exposed controls; there is no significant difference between the 2 groups. (B) The % change in phasic Imean during bath application of 30 mM ethanol. Ethanol increases phasic Imean in control but not CIE-exposed rats, t(14) = 3.376, p = 0.005. *p < 0.05 compared with the CIE group.
DISCUSSION
The principal findings of this study are that CIE exposure during adolescence resulted in enhanced ethanol sensitivity of inhibitory tonic currents and decreased ethanol sensitivity of phasic inhibitory current in DGCs during adulthood. Ethanol doubled the amplitude of tonic current in DGCs from animals treated with ethanol during adolescence, compared with an average enhancement of approximately 30% in controls. In addition, the baseline level of “tonic noise,” a reflection of the baseline activity of extrasynaptic GABAA receptors, was diminished in DGCs from adult animals that had been exposed to CIE as adolescents. While tonic noise usually varies along with changes in the size of the tonic current, for example, as in Hanchar and colleagues (2005) and Fleming and colleagues (2007), in this study, we found no significant differences in the average size of the tonic current between groups. In contrast to the effect of CIE on phasic inhibitory currents, ethanol increased synaptic GABAA receptor-mediated phasic current by 25% in control neurons, and this effect was not observed in cells from rats exposed to CIE as adolescents, which suggests that adolescent CIE reduces the ethanol sensitivity of synaptic GABAergic inhibition.
These findings are notable for 2 reasons. First, they indicate that CIE during adolescence results in long-lasting alterations in the functioning and ethanol responsiveness of GABAA receptor-mediated physiological activity in the hippocampal formation. This is the first demonstration of such a long-lasting effect of adolescent ethanol exposure on hippocampal GABAA receptor-mediated activity at the cellular neurophysiological level. As such, it could have mechanistic implications for understanding the effects of adolescent alcohol drinking on several neurological processes, including learning and seizure susceptibility.
Second, and perhaps more importantly, when viewed in the context of previous studies, these data suggest that CIE during adolescence results in the persistence of adolescent ethanol sensitivity into adulthood. We did not make a direct comparison of CIE during adolescence with CIE during adulthood in the present study. However, we have previously shown that tonic currents recorded in DGCs from adolescent rats are more sensitive to ethanol than are those recorded in DGCs from adults (Fleming et al., 2007). Indeed, the magnitude of tonic current enhancement by 30 mM ethanol in DGCs from naïve adolescent rats in that study (108%, compared with 47% in DGCs from adults) is quite consistent with the enhancement that we observed among adults who had been exposed to CIE during adolescents in the present study (99%). Such a “lock-in” of the adolescent pattern of ethanol sensitivity in hippocampal GABA function could be of mechanistic significance for understanding some of the effects that ethanol has on learning in adolescents and after repeated adolescent ethanol exposure. For example, we have shown that ethanol impairs spatial learning in adolescent rats more potently than in adults (Markwiese et al., 1998) and that adult rats exposed to CIE during adolescence are more sensitive to ethanol-induced impairments of spatial working memory (White et al., 2000a). The fact that ethanol promotes GABAA receptor-mediated tonic current more efficaciously in hippocampal DGCs from adolescent rats than in those from adults (Fleming et al., 2007) is consistent with ethanol impairing spatial learning more potently in adolescents, compared with adults (Markwiese et al., 1998). Similarly, the present finding that CIE in adolescence results in greater ethanol sensitivity of hippocampal tonic current in adulthood is consistent with greater behavioral sensitivity to learning impairment in adulthood after CIE during adolescence (White et al., 2000a).
Activation of extrasynaptic GABAA receptors produces shunting inhibition that decreases neuronal excitability and decreases the likelihood of action potential firing, and could thereby influence memory mechanisms such as LTP (Semyanov et al., 2004). Indeed, extrasynaptic GABAA receptors have been shown to play a role in inhibiting learning and memory. For example, GABAA receptor α5 subunits are expressed extrasynaptically in the hippocampus, and reduction or loss of the α5 subunit in mice improves performance in trace fear conditioning and spatial learning tasks, which are known to be highly dependent on hippocampal synaptic plasticity (Collinson et al., 2002; Crestani et al., 2002; Fritschy and Brunig, 2003). Similarly, α5 selective inverse agonists improve spatial learning in the Morris water maze (Chambers et al., 2003).
In hippocampal DGCs, ethanol-sensitive δ-type GABAA receptors mediate tonic current that strongly influences their function (Nusser and Mody, 2002). Memory-related synaptic information entering the hippocampal formation arrives first in the dentate gyrus, and DGCs are the principal neurons that project from the dentate onto CA3 pyramidal cells, the next stage in hippocampal processing, which then relay activity forward to area CA1 (Johnston and Amaral, 2003). Therefore, alcohol modulation of tonic inhibition in DGCs could function as a “gate” that affects not only the excitability of the DGCs but also the function of the entire hippocampal network. Indeed, knockout of the δ subunit enhances trace fear conditioning in mice, providing further evidence that tonic inhibition mediated by extrasynaptic GABAA receptors in the dentate gyrus suppresses hippocampus-mediated learning (Wiltgen et al., 2005).
Long-term alterations of extrasynaptic GABAA receptor-mediated currents after CIE have been observed previously when the ethanol exposure occurred during adulthood (Liang et al., 2007). In that study, there was a long-lasting reduction of extrasynaptic GABAA receptor-mediated current in CA1 neurons after a CIE paradigm that involved 60 doses across 120 days, beginning at about postnatal day 50. That study also reported the long-lasting down-regulation of GABA receptor δ subunit protein in hippocampus after CIE. Such changes may be related to both behavioral tolerance and changes in neuronal excitability after CIE in adulthood. These results are, in part, consistent with the present observations because they demonstrate long-term changes in hippocampal extrasynaptic receptor activity after CIE. However, there are several important differences between that study and the present one. Based on the body weights reported, it is likely that the animals were about 50 to 54 days of age, when CIE was initiated. Thus, even at the beginning of CIE, they would fall outside the age range typically considered adolescent in the male Sprague-Dawley rat (Spear, 2000). In addition, the duration of CIE was quite long, extended well into adulthood, and involved 60 ethanol doses. In contrast, our paradigm involved only 10 total ethanol doses, presented between postnatal days 30 and 48, a range widely considered to be within the window of adolescence in the male rat. Furthermore, the decrease in expression of the δ subunit shown by Liang and colleagues (2007) seems to be inconsistent with the increase in ethanol sensitivity of tonic inhibition shown here. Our previous work on differences in ethanol sensitivity of tonic current in ethanol-naïve adolescent and adult rats has suggested that the regulation of ethanol’s effect on tonic inhibition is more complex than a simple change in GABAA receptor subunit composition (Fleming et al., 2007). Changes in ambient GABA concentration, intracellular signaling, and Cl− reversal potential all could play roles in the effects of CIE on tonic inhibition (Fleming et al., 2011; Smith et al., 2009; Weiner and Valenzuela, 2006; Yamashita et al., 2006).
In mice, spillover from sIPSC events is the main source of ambient GABA that produces tonic inhibition in CA1 pyramidal cells and DG interneurons (Glykys and Mody, 2007). Furthermore, increases in GABA release may play a role in the ethanol enhancement of tonic inhibitory current that is observed in cerebellar granule cells (Carta et al., 2004), although that finding is controversial (Hanchar et al., 2005). Therefore, changes in GABA spillover from synaptic events could contribute to the changes in tonic inhibition that we observed in this study. Therefore, we used the mean phasic current calculation to detect changes in synaptic GABA release over the same recording intervals used for tonic current determination. The Imean method has been shown to be a highly accurate method that detects small changes in sIPSCs better than traditional methods, which are based on detecting and averaging individual events (Glykys and Mody, 2007). We found no significant differences in baseline phasic Imean between CIE-exposed rats and saline controls. Ethanol increased the phasic Imean only in control animals; this effect was not observed in CIE-exposed rats. Previously, we and others have shown no effect of ethanol on sIPSCs in DGCs from adult animals, but those studies used less-sensitive traditional sIPSC analysis methods (Fleming et al., 2007; Wei et al., 2004). The fact that ethanol did not increase mean phasic current in CIE-exposed rats suggests that the enhanced effects of ethanol on tonic current in these animals are not because of an increase in GABA release. Instead, changes in GABA uptake or the postsynaptic mechanisms discussed earlier may underlie the changes in ethanol sensitivity observed in this study, and more work is needed to determine how CIE alters tonic currents.
The present finding that CIE during adolescence results in enhanced sensitivity of DGC tonic current to ethanol-induced enhancement in adulthood could be of mechanistic significance for explaining previous studies that have shown enhanced sensitivity to the memory-impairing effects of ethanol during adulthood after CIE in adolescence (White et al., 2000a). Beyond their mechanistic implications, the present findings may also be of considerable value for health education efforts. They represent the first demonstration of a long-term, memory-related cellular effect of CIE during adolescence, and the “lock-in” of adolescent ethanol sensitivity that these results suggest could represent a conceptual step forward in understanding the vulnerability of the adolescent brain to alcohol.
Acknowledgments
This research was supported by a grant from the Institute for Medical Research and a VA Career Development Award to RLF, VA Merit Review grants to SDM and HSS, NIH (NIAAA) grant # 1U01AA019925-01 (NADIA) to HSS, and by VA Senior Research Career Scientist Awards to HSS and WAW.
References
- Acheson SK, Stein RM, Swartzwelder HS. Impairment of semantic and figural memory by acute ethanol: age-dependent effects. Alcohol Clin Exp Res. 1998;22:1437–1442. doi: 10.1111/j.1530-0277.1998.tb03932.x. [DOI] [PubMed] [Google Scholar]
- Carta M, Mameli M, Valenzuela CF. Alcohol enhances GABAergic transmission to cerebellar granule cells via an increase in Golgi cell excitability. J Neurosci. 2004;24:3746–3751. doi: 10.1523/JNEUROSCI.0067-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers MS, Atack JR, Broughton HB, Collinson N, Cook S, Dawson GR, Hobbs SC, Marshall G, Maubach KA, Pillai GV, Reeve AJ, MacLeod AM. Identification of a novel, selective GABA(A) alpha5 receptor inverse agonist which enhances cognition. J Med Chem. 2003;46:2227–2240. doi: 10.1021/jm020582q. [DOI] [PubMed] [Google Scholar]
- Collinson N, Kuenzi FM, Jarolimek W, Maubach KA, Cothliff R, Sur C, Smith A, Otu FM, Howell O, Atack JR, Mckernan RM, Seabrook GR, Dawson GR, Whiting PJ, Rosahl TW. Enhanced learning and memory and altered GABAergic synaptic transmission in mice lacking the alpha 5 subunit of the GABAA receptor. J Neurosci. 2002;22:5572–5580. doi: 10.1523/JNEUROSCI.22-13-05572.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crestani F, Keist R, Fritschy JM, Benke D, Vogt K, Prut L, Bluthmann H, Mohler H, Rudolph U. Trace fear conditioning involves hippocampal alpha5 GABA(A) receptors. Proc Natl Acad Sci USA. 2002;99:8980–8985. doi: 10.1073/pnas.142288699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews FT, Braun CJ, Hoplight B, Switzer RC, III, Knapp DJ. Binge ethanol consumption causes differential brain damage in young adolescent rats compared with adult rats. Alcohol Clin Exp Res. 2000;24:1712–1723. [PubMed] [Google Scholar]
- Fleming RL, Acheson SK, Moore SD, Wilson WA, Swartzwelder HS. GABA transport modulates the ethanol sensitivity of tonic inhibition in the rat dentate gyrus. Alcohol. 2011;45:577–583. doi: 10.1016/j.alcohol.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming RL, Wilson WA, Swartzwelder HS. Magnitude and ethanol sensitivity of tonic GABAA receptor-mediated inhibition in dentate gyrus changes from adolescence to adulthood. J Neurophysiol. 2007;97:3806–3811. doi: 10.1152/jn.00101.2007. [DOI] [PubMed] [Google Scholar]
- Fritschy JM, Brunig I. Formation and plasticity of GABAergic synapses: physiological mechanisms and pathophysiological implications. Pharmacol Ther. 2003;98:299–323. doi: 10.1016/s0163-7258(03)00037-8. [DOI] [PubMed] [Google Scholar]
- Glykys J, Mody I. The main source of ambient GABA responsible for tonic inhibition in the mouse hippocampus. J Physiol. 2007;582:1163–1178. doi: 10.1113/jphysiol.2007.134460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanchar HJ, Dodson PD, Olsen RW, Otis TS, Wallner M. Alcohol-induced motor impairment caused by increased extrasynaptic GABA(A) receptor activity. Nat Neurosci. 2005;8:339–345. doi: 10.1038/nn1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt WA. Are binge drinkers more at risk of developing brain damage? Alcohol. 1993;10:559–561. doi: 10.1016/0741-8329(93)90083-z. [DOI] [PubMed] [Google Scholar]
- Johnston D, Amaral D. Hippocampus. In: Shepherd G, editor. The Synaptic Organization of the Brain. Oxford University Press; New York: 2003. pp. 455–498. [Google Scholar]
- Liang J, Suryanarayanan A, Abriam A, Snyder B, Olsen RW, Spigelman I. Mechanisms of reversible GABAA receptor plasticity after ethanol intoxication. J Neurosci. 2007;27:12367–12377. doi: 10.1523/JNEUROSCI.2786-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little PJ, Kuhn CM, Wilson WA, Swartzwelder HS. Differential effects of ethanol in adolescent and adult rats. Alcohol Clin Exp Res. 1996;20:1346–1351. doi: 10.1111/j.1530-0277.1996.tb01133.x. [DOI] [PubMed] [Google Scholar]
- Markwiese BJ, Acheson SK, Levin ED, Wilson WA, Swartzwelder HS. Differential effects of ethanol on memory in adolescent and adult rats. Alcohol Clin Exp Res. 1998;22:416–421. [PubMed] [Google Scholar]
- Martin LJ, Zurek AA, MacDonald JF, Roder JC, Jackson MF, Orser BA. Alpha5GABAA receptor activity sets the threshold for long-term potentiation and constrains hippocampus-dependent memory. J Neurosci. 2010;30:5269–5282. doi: 10.1523/JNEUROSCI.4209-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore MD, Cushman J, Chandra D, Homanics GE, Olsen RW, Fanselow MS. Trace and contextual fear conditioning is enhanced in mice lacking the alpha4 subunit of the GABA(A) receptor. Neurobiol Learn Mem. 2010;93:383–387. doi: 10.1016/j.nlm.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusser Z, Mody I. Selective modulation of tonic and phasic inhibitions in dentate gyrus granule cells. J Neurophysiol. 2002;87:2624–2628. doi: 10.1152/jn.2002.87.5.2624. [DOI] [PubMed] [Google Scholar]
- Pyapali GK, Turner DA, Wilson WA, Swartzwelder HS. Age and dose-dependent effects of ethanol on the induction of hippocampal long-term potentiation. Alcohol. 1999;19:107–111. doi: 10.1016/s0741-8329(99)00021-x. [DOI] [PubMed] [Google Scholar]
- Semyanov A, Walker MC, Kullmann DM, Silver RA. Tonically active GABA A receptors: modulating gain and maintaining the tone. Trends Neurosci. 2004;27:262–269. doi: 10.1016/j.tins.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Shen H, Sabaliauskas N, Sherpa A, Fenton AA, Stelzer A, Aoki C, Smith SS. A critical role for alpha4betadelta GABAA receptors in shaping learning deficits at puberty in mice. Science. 2010;327:1515–1518. doi: 10.1126/science.1184245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SS, Aoki C, Shen H. Puberty, steroids and GABA(A) receptor plasticity. Psychoneuroendocrinology. 2009;34(Suppl 1):S91–S103. doi: 10.1016/j.psyneuen.2009.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spear L. Modeling adolescent development and alcohol use in animals. Alcohol Res Health. 2000;24:115–123. [PMC free article] [PubMed] [Google Scholar]
- Swartzwelder HS, Wilson WA, Tayyeb MI. Age-dependent inhibition of long-term potentiation by ethanol in immature versus mature hippocampus. Alcohol Clin Exp Res. 1995a;19:1480–1485. doi: 10.1111/j.1530-0277.1995.tb01011.x. [DOI] [PubMed] [Google Scholar]
- Swartzwelder HS, Wilson WA, Tayyeb MI. Differential sensitivity of NMDA receptor-mediated synaptic potentials to ethanol in immature versus mature hippocampus. Alcohol Clin Exp Res. 1995b;19:320–323. doi: 10.1111/j.1530-0277.1995.tb01509.x. [DOI] [PubMed] [Google Scholar]
- Wei W, Faria LC, Mody I. Low ethanol concentrations selectively augment the tonic inhibition mediated by delta subunit-containing GABAA receptors in hippocampal neurons. J Neurosci. 2004;24:8379–8382. doi: 10.1523/JNEUROSCI.2040-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner JL, Valenzuela CF. Ethanol modulation of GABAergic transmission: the view from the slice. Pharmacol Ther. 2006;111:533–554. doi: 10.1016/j.pharmthera.2005.11.002. [DOI] [PubMed] [Google Scholar]
- White AM, Bae JG, Truesdale MC, Ahmad S, Wilson WA, Swartzwelder HS. Chronic-intermittent ethanol exposure during adolescence prevents normal developmental changes in sensitivity to ethanol-induced motor impairments. Alcohol Clin Exp Res. 2002a;26:960–968. doi: 10.1097/01.ALC.0000021334.47130.F9. [DOI] [PubMed] [Google Scholar]
- White AM, Ghia AJ, Levin ED, Swartzwelder HS. Binge pattern ethanol exposure in adolescent and adult rats: differential impact on subsequent responsiveness to ethanol. Alcohol Clin Exp Res. 2000a;24:1251–1256. [PubMed] [Google Scholar]
- White AM, Matthews DB, Best PJ. Ethanol, memory, and hippocampal function: a review of recent findings. Hippocampus. 2000b;10:88–93. doi: 10.1002/(SICI)1098-1063(2000)10:1<88::AID-HIPO10>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- White AM, Truesdale MC, Bae JG, Ahmad S, Wilson WA, Best PJ, Swartzwelder HS. Differential effects of ethanol on motor coordination in adolescent and adult rats. Pharmacol Biochem Behav. 2002b;73:673–677. doi: 10.1016/s0091-3057(02)00860-2. [DOI] [PubMed] [Google Scholar]
- Wiltgen BJ, Sanders MJ, Ferguson C, Homanics GE, Fanselow MS. Trace fear conditioning is enhanced in mice lacking the delta subunit of the GABAA receptor. Learn Mem. 2005;12:327–333. doi: 10.1101/lm.89705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita M, Marszalec W, Yeh JZ, Narahashi T. Effects of ethanol on tonic GABA currents in cerebellar granule cells and mammalian cells recombinantly expressing GABA(A) receptors. J Pharmacol Exp Ther. 2006;319:431–438. doi: 10.1124/jpet.106.106260. [DOI] [PubMed] [Google Scholar]