

Abstract
The ganglionic eminence contributes cells to several forebrain structures including the cerebral cortex, for which it provides GABAergic interneurons. Migration of neuronal precursors from the retinoic-acid rich embryonic ganglionic eminence to the cerebral cortex is known to be regulated by several factors, but retinoic acid has not been previously implicated. We found retinoic acid to potently inhibit cell migration in slice preparations of embryonic mouse forebrains, which was reversed by an antagonist of the dopamine-D2 receptor, whose gene is transcriptionally regulated by retinoic acid. Histonedeacetylase inhibitors, which amplify nuclear receptor-mediated transcription, potentiated the inhibitory effect of retinoic acid. Surprisingly, when retinoic acid signalling was completely blocked with a pan-retinoic acid receptor antagonist, this also decreased cell migration into the cortex, implying that a minimal level of endogenous retinoic acid is necessary for tangential migration. Given these opposing effects of retinoic acid in vitro, the in vivo contribution of retinoic acid to migration was tested by counting GABAergic interneurons in cortices of adult mice with experimental reductions in retinoic acid signalling: a range of perturbations resulted in significant reductions in the numerical density of some GABAergic interneuron subpopulations. These observations suggest functions of retinoic acid in interneuron diversity and organization of cortical excitatory–inhibitory balance.
Keywords: cortical homeostasis, dopamine D2 receptor, histone deacetylase inhibitor, interneurons, tangential migration, telencephalon
Retinol (vitamin A) is an important nutrient with indispensable input into the control of embryonic development and homeostasis (Ross et al. 2000; Clagett-Dame and DeLuca 2002). Retinol is converted to retinoic acid (RA) through sequential oxidative steps: the reversible oxidation of retinol to retinaldehyde can be catalysed by a multitude of retinol dehydrogenases, several of which are widely expressed in the forebrain. The irreversible oxidation of retinaldehyde to RA, however, is primarily mediated by three retinaldehyde dehydrogenases (RALDHs), RALDH1, RALDH2 and RALDH3, whose expressions are restricted to a few sites (Smith et al. 2001; Duester 2008). RA treatment is estimated to regulate expression of several thousand genes by two-fold or more based on in vitro studies on human cell lines (Cawley et al. 2004), but where and when endogenous RA influences gene expression in vivo depends on the cellular and developmental context and is less understood. The best predictors for sites of RA actions in vivo are local peaks in RA levels, as they are generated at local RALDH expression sites (McCaffery and Dräger 1994). Being a small amphipathic lipid, RA can rapidly diffuse out of RALDH-positive cells and travel extended distances through tissues (McCaffery et al. 2006; Duester 2008). Because for technical reasons RA cannot be directly visualized in the tissue, its concentration at a particular site can only be inferred from the known topographies of RALDHs in the neighbourhood, as well as from RA measurements in dissected tissue regions.
Whereas at some embryonic sites, including the eye and face, RA synthesis is very high, RALDH expression in the forebrain is exceptionally sparse, and almost 90% of total RA content in the adult brain is supplied by the circulation (Kurlandsky et al. 1995; Luo et al. 2004). The meninges surrounding the brain express RALDH2, which generates high RA levels at the brain surface, and a unique region of very high neuronal RA synthesis is a transient neuroepithelial structure in the ventral telencephalon of the embryo, the ganglionic eminence (GE), and its descendant in the mature brain, the basal ganglia of the forebrain (McCaffery and Dräger 1994). The GE is the birthplace of GABAergic interneurons for the cerebral cortex as well as of cells in the striatum and the olfactory bulb (Marin et al. 2000; Wichterle et al. 2001; Stenman et al. 2003; Wonders and Anderson 2006; Chédotal and Rijli 2009; Huang 2009; Nóbrega-Pereira and Marín 2009; Rakic 2009). There are conflicting reports about whether RA is necessary for DARPP-32 expression in striatal cells (Waclaw et al. 2004; Molotkova et al. 2007), a protein required for dopaminergic transmission, but RA is believed to be instrumental for inducing a network of dopaminergic signal transduction in the GE (Wang and Liu 2005). In particular, the dopamine D2-receptor is under overriding control by RA (Farooqui 1994; Samad et al. 1997; Krezel et al. 1998; Valdenaire et al. 1998), and deficiency of RALDH3 expression in the GE leads to a reduction of this receptor in the basal ganglia (Molotkova et al. 2007).
Dopamine receptors are expressed in the embryonic GE before their canonical role in neurotransmission is apparent, where they modulate several developmental processes including cell proliferation and differentiation (Popolo et al. 2004). We have previously described a function of the D2 receptor in cell migration: activation of the D2 receptor inhibits the tangential migration of GABAergic neurons from the GE into the cortex (Crandall et al. 2007). Because transcription of the D2 receptor is regulated by RA, and because the GE is enriched in RA, we asked whether RA can influence cell migration into the cortex. In a slice culture system of embryonic mouse forebrains we found that exogenous RA potently inhibits cell migration into the cortex via a mechanism that is completely reversed by a D2 dopamine receptor antagonist. Abolishing endogenous RA signalling by addition of a pan-RA receptor antagonist to the cultures, however, similarly reduced cell migration into the cortex, indicating that a minimum level of endogenous RA is necessary for cortical migration. To ask which of the two opposite RA effects observed in the slice cultures is likely the one that patho-physiologically prevails, we counted GABAergic interneurons in the cortices of adult mice with three different types of vitamin A deficiency syndromes: in all three mouse models we detected significant reductions of some GABAergic populations.
Materials and methods
Animals
For the slice cultures, timed-pregnant CD-1 mice were purchased from Charles River Laboratories (Wilmington, MA, USA). The RA-reporter strain (Rossant et al. 1991), which is also on a CD-1 background, was bred in the animal facility of the E.K. Shriver Center. The Raldh knockout mice (Molotkov et al. 2006) maintained on a C57BL background, were raised and perfused in G. Duester’s laboratory (Sanford-Burnham Medical Research, La Jolla, CA, USA), and brain tissue was transferred to Boston for further analyses. The retinol-binding protein (RBP) null mutants on a C57BL background, which represent an animal model for tuneable nutritional vitamin A deficiency (Quadro et al. 2005), were generously provided by Dr William Blaner and raised on vitamin A deficient food (VitA Def PD 0007184, Test Diet, Richmond, IN, USA) from embryonic day 10 to postnatal day 21. The day of vaginal plug was designated embryonic day 0 (E0). All of the experimental procedures were in full compliance with institutional guidelines and the NIH Guide for the Care and Use of Laboratory Animals.
RALDH3 and tyrosine hydroxylase immunohistochemistry
The embryos were removed from dams deeply anesthetized by an intraperitoneal injection of a mixture of Ketamine (50 mg/kg body weight) and Xylazine (10 mg/kg body weight), fixed by immersion in 4% paraformaldehyde and sectioned in the coronal plane on a cryostat. RALDH3 immunohistochemistry was performed on slidemounted sections. We used a RALDH3 polyclonal antiserum initially developed and characterized by Luo et al. (2004). To improve its specificity, cross-reactivity of the RALDH3 antiserum with intermediate-filament like structures was reduced by preabsorbing with a Triton-insoluble extract of formalin-fixed brain (7% Triton X-100 and 0.1 M ethylenediaminetetraacetic acid); a volume of 50 μL of brain homogenate was used for absorption of 0.2 mL of antibody. For double labelling of RALDH1 and tyrosine hydroxylase, we used an antiserum generated by Lindahl et al. (1983), and the tyrosine hydroxylase was visualized with a mouse monoclonal antibody from Boehringer Mannheim Biochemicals (Indianapolis, IN, USA). Brains were sectioned at 15 μm on a cryostat; the sections were melted onto slides, and the preparations were incubated with a mixture of primary antibodies overnight at 4°C, followed by incubations with secondary antibodies for 2 h at 22°C. The secondary antibodies were labelled with fluorescein isothiocyanate or tetramethyl rhodamine iso-thiocyanate.
Brain slice preparation
Neuronal migration from the striatum/GE to the cerebral wall was assayed in slice preparations of the embryonic telencephalon as described previously (Tobet et al. 1994; Anderson et al. 1997; Crandall et al. 2004). Briefly, E15 embryos were removed one at a time from deeply anesthetized dams (see above). Age of the embryos was confirmed by crown-rump length (13–15 mm) and other external features (Theiler 1972). The embryos were decapitated and the heads were embedded in 8% agarose (Type VII, Sigma, St Louis, MO, USA). Coronal sections of the embryonic head were cut at a thickness of 250 μm on a Leitz Vibratome. The sections were cultured individually on polycarbonate membrane filters of 4.5 cm2 area and 8 μm pore size (Costar, Corning, NY, USA) and placed in a 6-well cell culture plate. Neurobasal medium (GIBCO-BRL) containing 2% B27 supplement, penicillin, streptomycin and glutamine were added beneath the filter into each well. The slices were maintained in culture for 2 days. The medium and additives were replaced at the end of the first 24 h.
Application of drugs
An all-trans RA stock solution in dimethylsulfoxide (DMSO) was prepared under low yellow light and kept under nitrogen. Aliquots of this stock were diluted in tissue culture medium to give a final concentration of 1 nM or 100 nM RA and 0.1% DMSO, and added to the brain slices 2–3 h after plating; the control cultures contained 0.1% DMSO. In other experiments, the D2-receptor antagonist eticlopride (20 μM; Sigma) was added to the culture medium 2–3 h after plating the slices, and 2 h later RA (1 or 100 nM) was added in the presence of eticlopride. The eticlopride concentration used here selectively blocks the D2-receptor (Ohtani et al. 2003; Popolo et al. 2004). Trichostatin (Sigma) was used at a final concentration of 50 nM and valproate at 0.5 mM, concentrations at which these drugs are known to inhibit histone deacetylase activity (Stockhausen et al. 2005; Marinova et al. 2009). As in the previously described experiments, the drugs were added to the tissue culture medium 2–3 h after the slices were plated. In experiments in which drug treatments were combined with RA, the drugs were added 2 h after the RA. The pan-retinoic acid receptor (RAR) antagonist LG100815 and the pan-retinoid X receptors (RXR) antagonist LG100849, gracious gifts from Ligand Pharmaceuticals Inc. (La Jolla, CA, USA), were employed at a concentration of 5 μM, the same concentration used to block RA signalling pathways controlling cell growth (Chen et al. 2006), and were added 2–3 h after the slices were plated.
In vitro culture of forebrain flat mounts from RA reporter mice
E15 embryos of RA reporter mice (Rossant et al. 1991) were removed from a pregnant dam, and the brains were rapidly dissected in ice-cold tissue culture medium. The meninges were removed and the telencephala were cut off at their connection with the diencephalon. The peripheral parts of the dorsal telencephalon were trimmed away, and the remaining telencephalic vesicle was flattened onto the membrane filters of polycarbonate transwells with the ventricular surface facing up and the meninegeal surface adhering to the filter. The same conditions and tissue culture medium were used as for the cell-migration assay in slice cultures. Explants were cultured for 2 days in the presence of drugs or vehicle. After this time the tissue was fixed in 1% glutaraldehyde with 2 mM MgCl2 for 15 min, washed in phosphate-buffered saline and incubated with the X-gal reaction buffer (33 mM potassium ferrocyanide, 33 mM potassium ferricyanide, 1 mM MgCl2 and 2 mg/mL X-gal [5-bromo-4-choloro-3-indoyl-β-d-galactoside in phosphate-buffered saline) for 1 h in the dark at 37°C to allow for blue color to develop. Because of variability in the lacZ reporter expression between individual mice, the eyes of each embryo from several litters were pre-tested for β-galactosidase activity. Only those with a strong signal after 1 h incubation at 37°C were used.
DiI-labelling of cells in the cultured slices
1,1′-Dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI) crystals (Molecular Probes, Eugene, OR, USA) were reconstituted after dissolving in 95% ethanol in glass Petri dishes and drying at 60°C. The crystals were inserted into the GE, at approximately 200–400 μm ventromedial to the pallial–striatal angle, with the aid of sharpened needles under a stereomicroscope, immediately after plating each slice. The DiI placements were such that they labelled cells originating from both the lateral and medial ganglionic eminences (Crandall et al. 2004). Following 2 days in culture, the slices were fixed with 4% paraformaldehyde in 0.1 M phosphate buffer, pH 7.2 and viewed as whole-mounts under a fluorescence microscope fitted with tetramethyl rhodamine isothiocyanate filters.
Analysis of cell migration
The position of DiI-labelled neurons was recorded with respect to the boundaries of the slice, using an image analysis system (IPLab, v3.6, Scanalytics Inc., Rockville, MD, USA) interfaced to a Zeiss Axioplan widefield microscope (Crandall et al. 2004). A single investigator blinded to the identity of the experimental group performed the analyses. A laser printer plot for each case showed the location of each DiI-labelled cell in the entire slice. An overlay grid of rectangles (each 640 μm × 480 μm) – ‘grid boxes’ – was placed on the image of the slice to measure the overall extent of neuronal migration. The average number of grid boxes containing DiI-labelled cells was calculated. As GE cells migrating to the cortex take non-linear pathways, and because the cells migrate in different directions upon their arrival in the cortex (Gupta et al. 2003; Nadarajah et al. 2003), the grid box analysis rather than point-to-point linear measurement of migration is meaningful.
We also calculated the percentage of DiI-labelled cells that entered the cerebral wall. For this measurement, any DiI-labelled cell found dorsal and lateral to the caudate-pallial angle was considered to have reached the cortex. The number of such cells in a slice was expressed as a percentage of the total number of DiI-labelled cells in that slice, to take into account the variability in numbers of DiI-labelled cells in the slices. The mean values for each measure were statistically compared between each group using Student’s t-test. The average total number of DiI-labelled cells per hemisphere was not statistically different between experimental groups (mean ± SEM: control = 284 ± 126.7; 10−7M RA = 240 ± 49.5; 10−9M RA = 237 ± 38.8) suggesting that the size of the DiI deposit and the DiI labelling were comparable across the different experimental conditions.
Measurement of RA release
This assay was performed on the supernatants from the forebrain slices used for the analysis of cell migration. The slices were incubated for 24 h in polycarbonate transwells in Neurobasal medium, as described above. Supernatant was removed, and 100 μL was assessed for RA concentration with the Sil-15 RA reporter cell line grown on a 96-well plate (Wagner et al. 1992). β-Galactosidase synthesis in response to RA was measured by X-gal reaction and quantified on an ELISA plate reader at 630 nm. RA concentrations were determined by comparison with a RA standard curve. We analyzed RA release in three slices each from the rostral and caudal regions of the forebrain. The forebrain slices containing the lateral ganglionic eminence (LGE) (three rostral-most slices) were considered the rostral samples and the slices containing the caudal ganglionic eminence (CGE) (three caudal-most) were included in the caudal samples (Crandall et al. 2007). The intervening slices (on average, 2–3) were not included in the analysis. RA reporter animals were from Dr Janet Rossant (University of Toronto, Canada) and are transgenic for an hsp-lacZ transgene (Rossant et al. 1991).
Immunohistochemistry analysis of parvalbumin, calretinin and calbindin in the adult cortex
Adult (2 months old) mice were perfused with 4% paraformaldehyde in 0.1 M phosphate buffer, pH 7.2, and the brains were fixed overnight at 4°C in the same fixative, cryoprotected in 30% sucrose, and frozen-sectioned at 40 μm thickness in the coronal plane on a sliding microtome. The sections were processed for immunohistochemistry using antibodies to parvalbumin (mouse monoclonal, cat. no. P3088; Sigma), calretinin (mouse monoclonal, cat. no. 18–0291; Invitrogen, Carlsbad, CA, USA), and calbindin (rabbit, cat. no. C2724; Sigma), biotinylated secondary antibody (Jackson Immuno Research, West Grove, PA, USA), avidin– biotin kit (Vector Laboratories, Burlingame, CA, USA), and nickel ammonium sulfate (0.02%; Sigma) for enhancement of the diaminobenzidine reaction product as described previously (Crandall et al. 2004). Immunopositive cells were counted by a single investigator blinded to experimental treatment groups in the 40-μm-thick sections through the width of the primary somatosensory cortex, using a 40× objective lens. The cell counts were expressed as number of cells per 300 μm2.
Results
RA-mediated inhibition of cell migration from the GE to the cerebral wall can be reversed by a dopamine D2 receptor antagonist
Prior to its function as a neurotransmitter, dopamine is known to regulate neuronal development in the embryo. The region of the ganglionic eminence (GE) is particularly rich in dopamine, and the dopaminergic ligand-receptor system controls cell proliferation, differentiation and migration of neurons from the GE to the cerebral cortex (Ohtani et al. 2003; Popolo et al. 2004; Crandall et al. 2007). The D1 and D2 receptors often perform complementary actions; the D1 receptor promotes migration and the D2 receptor is a strong inhibitor. Transcription of the D2 receptor is potently stimulated by RA (Samad et al. 1997; Valdenaire et al. 1998). To address the question of whether RA also influences neuronal migration from the GE, we employed an in vitro slice culture system from mouse E15 brains for quantifying migration of DiI-labelled cells from the GE to cortex (Crandall et al. 2007). Addition of 1 or 100 nM RA (with 0.1% DMSO) to the culture wells, significantly decreased cell migration from the GE into the cerebral wall by 57% (Fig. 1a and b) indicating that cell migration from the GE to the cortex is highly sensitive to exogenous RA.
Fig. 1.
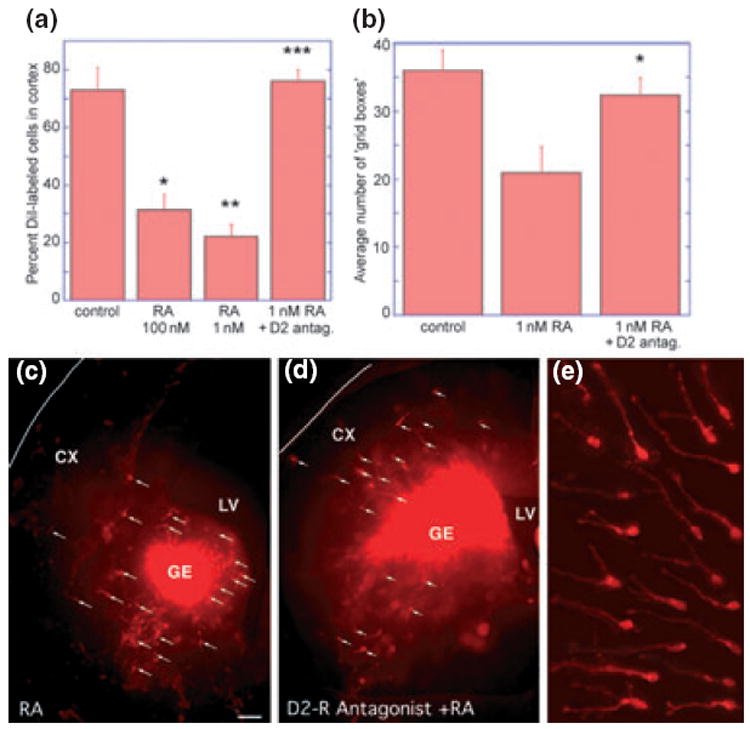
Retinoic acid reduces the number of cells migrating from the ganglionic eminence (GE) to the cortex in slice cultures, and this reduction can be reversed by addition of a D2 receptor antagonist. (a) Addition of all-trans RA (100 or 1 nM) significantly reduced the percentage of DiI-labelled cells migrating to the cortex. This reduction could be reversed, if the D2-dopamine receptor was blocked with a specific antagonist (20 μM eticlopride). (*100 nM RA vs. control t = 4.43, p = 0.003, **1 nM RA vs. control t = 6.40, p = 0.0001, ***1 nM RA vs. 1 nM RA + eticlopride t = 7.80, p < 0.0001. (b) Similarly, migration indicated by the average number of grid boxes that contained DiI-labelled cells was reduced, following 1 nM RA treatment, but approached control levels by the addition of the D2-dopamine receptor antagonist eticlopride (*1 nM RA vs. 1 nM RA + eticlopride t = 2.38, p = 0.03. (c–e) Micrographs of experimental brain slices photographed at one focal plane. E15 forebrain slices were labelled with identical amounts of DiI crystals placed in the ganglionic eminence (GE); Panels (c) and (d) are overviews, and panel (e) shows assorted migrating precursors with axonal processes and growth cones heading to the left and up. Far fewer DiI-labelled cells with processes (white arrows) can be seen in the cerebral wall (CX) after 2 days of all-trans RA exposure (c), as compared with the slice exposed to RA and the dopamine D2-receptor antagonist eticlopride (d). LV, lateral ventricle.
To test whether the RA effect on migration was primarily mediated by the D2 receptor, the D2 antagonist eticlopride (20 μM) was added to the culture medium prior to the addition of RA. The D2 receptor antagonist completely reversed the effect of RA on migration (Fig. 1a). The estimated migration distance, that is, the average number of grid boxes containing at least one DiI-labelled cell, was also significantly reduced by 1nM RA and this effect was reversed by the D2 receptor antagonist (Fig. 1b). A visual impression of these experiments is given in Figure 1c–e: in the overviews of representative injection sites (Fig. 1c and d) the locations of migrating precursors are indicated by white arrows, and Fig. 1e shows enlarged views of assorted cells arranged as migrating towards the left and up. RA treatment resulted in reductions of DiI-labelled cell migration into the cortex (Fig. 1c), and addition of the D2 receptor antagonist (Fig. 1d) reversed the RA effect, allowing many more DiI-labeled cells to move into the cortex. These results indicate that the D2-receptor signalling pathway represents a target through which RA reduces GE-to-cortex cell migration.
Endogenous synthesis of RA by forebrain slices
The effective total RA concentration required to inhibit neuronal migration from the GE to the cortex is unknown, because the RA added to the slice preparations augments endogenous RA synthesized in the ventral telencephalon. To obtain an estimate of the rate and gross topography of RA synthesis in the slice preparations used, we employed a sensitive RA reporter cell line (Wagner et al. 1992) for assessing the RA released from the slices into the medium after 1 day of incubation. Significant quantities of RA were detected (Fig. 2a), and the amounts released from rostral slices were about three times higher than those from caudal slices. This difference most likely reflects the patterns of endogenous RALDHs in the slices.
Fig. 2.
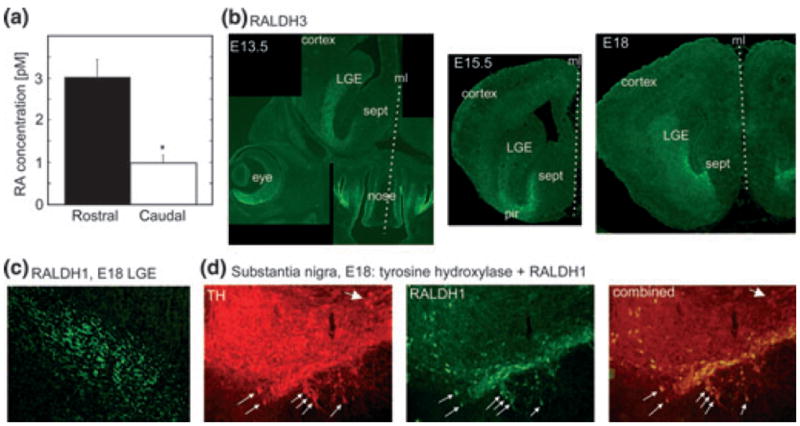
Release of RA from rostral and caudal forebrain slices and expression of RALDHs. (a) Measurements of RA released from rostral versus caudal regions of the forebrain with a RA reporter cell line (Wagner et al. 1992). Supernatants were collected from E15 forebrain slices cultured overnight. The rostral forebrain released three-fold more RA than the caudal forebrain (*t2 = 4.45, p = 0.011 vs. control). (b) Immunohistochemistry for RALDH3 on coronal head and forebrain sections from three ages, embryonic days E13.5, E15.5 and E18. RALDH3 is strongly expressed in the rostrally located LGE and in adjoining regions of the ventral telencephalon, the piriform cortex (pir) and the septum (sept). The section through the head of an E13.5 embryo illustrates in addition the strong RALDH3 expression in the face: the ventral retina (eye) and the olfactory epithelium (nose). RALDH3 is not expressed in more caudal regions of the forebrain (not shown). The midlines (ml) of the head/brains are marked by the dotted lines. (c) RALDH1-expressing fibres within the LGE. (d) The tyrosine hydroxylase (TH)-positive neurons of the substantia nigra include a subset of RALDH1 positive neurons that are the source of the RALDH1 positive fibres in the LGE. Thick arrow: cells containing TH only; thin arrows: double-labeled cells containing both TH and RALDH1.
The ventral telencephalon contains two RALDHs, whose anatomical distributions and quantities undergo continuous changes throughout embryonic and postnatal development. The RALDH3 enzyme becomes detectable at the outer wall of the ventral telencephalon at E12; during subsequent embryonic development its expression first increases and then diminishes, and RALDH3-positive cells shift gradually centripetally across the LGE and adjoining structures to the ventricular zone (Smith et al. 2001). Immunohistochemistry for RALDH3 on fixed coronal sections from E13.5 to E18 showed strong expression in the rostral-most forebrain in the LGE, the adjacent rostral piriform cortex and the septum (Fig. 2b). The medial GE (MGE) contained in the illustrated sections was entirely devoid of RALDH3 expression and similarly, from the CGE RALDH3 labelling was absent (not shown).
The RALDH1 protein is transported anterogradely from Raldh1-mRNA containing cell bodies in the ventral midbrain, which are a subpopulation of the dopaminergic projection neurons in the substantia nigra and adjoining ventral tegmentum (McCaffery and Dräger 1994). The presence of RALDH1 in the LGE within the axons of the dopaminergic projection neurons is shown in Fig. 2c and the corresponding cell bodies of the RALDH1-positive fibres are double labelled for tyrosine hydroxylase in the substantia nigra (small arrows, Fig. 2d). The tyrosine-hydroxylase and RALDH1 expressing axonal processes thus provide dopamine and RA to the same cellular target regions. The axons enter the forebrain by E13/14, and gradually increase extensively by profuse branching in the postnatal basal ganglia. At early embryonic stages RALDH3 is thus the only RA synthesizing enzyme in the GE, but from embryonic day 14 onwards, axon terminals from the RALDH1-containing meso-telencephalic projection neurons make a growing contribution, eventually replacing RALDH3 as the major local RA source (McCaffery and Dräger, 1994). The greater RA release from rostral versus caudal slices is presumably the additive result of RA synthesis by both RALDH3 and RALDH1, whereas caudal RA may derive only from RALDH1.
Effects of enhanced RA signalling on neuronal migration
The results described above demonstrate that RA is synthesized endogenously within the embryonic basal forebrain, and that addition of exogenous RA suppresses cell migration from the GE into the cortex via the D2 dopamine receptor, which is also endogenously expressed in the GE (Ohtani et al. 2003; Araki et al. 2007). A possible interpretation of these data is that the endogenous RA concentration must be very low at the location where the decision is made for cells to migrate into the cortex. To investigate this hypothesis further, we used the histone-deacetylase inhibitors trichostatin A and valproate, which can potentiate endogenous RA actions (Menegola et al. 2006; Epping et al. 2007). Inhibition of histone deacetylation leads to hyperacetylation of histones and promotes activation of genes, including those regulated by nuclear receptors (Jones and Shi 2003); the RA signalling pathway can predominate, and this pathway is the rate-limiting target of the histone deacetylase inhibitors in their action to inhibit cell proliferation (Epping et al. 2007). Trichostatin A and valproate were applied in the in vitro GE slice culture cell migration assay at concentrations known to inhibit histone deacetylases (Stockhausen et al. 2005; Marinova et al. 2009), and both drugs significantly reduced migration of DiI-labeled cells to the cortex in the slice cultures by 39 and 48% respectively (Fig. 3a). The observation that the inhibition was not further increased by addition of exogenous RA (Fig. 3a) supported a predominant action of these drugs through enhancement of endogenous RA.
Fig. 3.
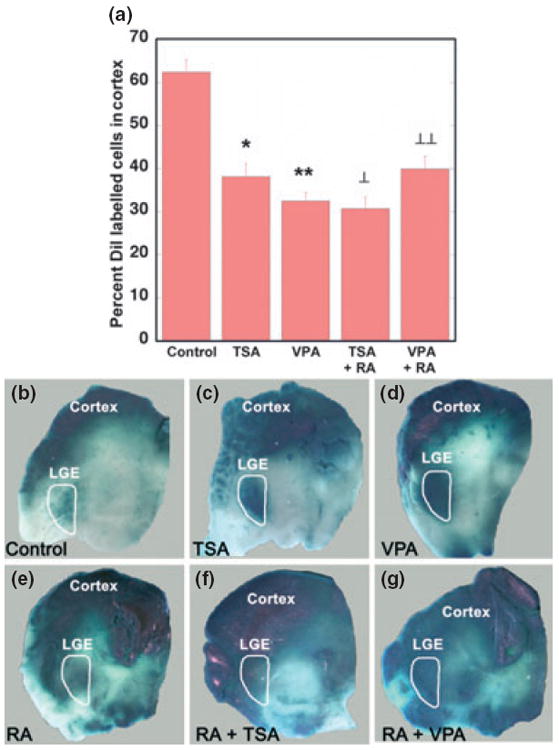
The histone deacetylase inhibitors trichostatin A (TSA) and valproate (VPA) stimulate RA signalling in the brain. (a) When the drugs were investigated for their influence on cell migration in slice cultures from E15 forebrain, both TSA and VPA significantly reduced cell migration from GE to cortex, and combination of the drugs with RA did not result in a further decline in migration (*control vs. VPA t = 8.73, p = 2.87 × 10−7, **control vs. TSA t = 5.19, p = 6.14 × 10−5, ⊥control vs. RA+VPA t = 5.35, p = 0.00013, ⊥⊥control vs. RA+TSA t = 7.99, p = 2.27 × 10−6). (b–g) RA signalling visualized as reporter expression in explant cultures of E15 forebrain from RARE-lacZ reporter mice (Rossant et al. 1991). LacZ expression is relatively weak in the LGE (outlined in white), which is the region of endogenous RA synthesis by RALDH3 (b). This endogenous signal is enhanced by 2- day culture with either TSA (c) or VPA (d). Addition of 100 nM RA increased the spread of RA reporter response in the forebrain, but without a greater response in the LGE, compared with the influence of TSA or VPA alone (e). Similarly, the combination of RA + TSA (f) or RA + VPA (g), did not visibly enhance the strength of RA reporter response in the LGE, although it expanded the distribution of the signal in the forebrain.
Because RA cannot be directly visualized in the tissue, nor are RA’s molecular actions immediately detectable by histological means, two indirect indicators can be used to identify the locations of RA signalling in vivo: the RALDH expression sites and expression of histological markers in transgenic RA reporter animals (Ross et al. 2000, Clagett-Dame and DeLuca 2002). The RA reporter mice contain multiple copies of the beta-galactosidase (lacZ) gene driven by the sensitive RA response element (RARE) of RARβ (Rossant et al. 1991). At many sites in the developing embryo, the lacZ expression in RARE-lacZ reporter mice colocalizes with RALDH expression sites. In the forebrain, however, the two signs tend to diverge, which is particularly obvious in the telencephalon: the ventral telencephalon with its prominent RA synthesis shows only low RA reporter expression (Luo et al. 2004). No global explanations for the discrepancies between local RA levels and RA-reporter expression exist (Liao et al. 2005; Sakai and Dräger 2010). To assay for components involved in the unexplained suppression of RA signalling in the ventral telencephalon, we applied the histone deacetylase inhibitors to in vitro preparations of the RARE-lacZ strain (Rossant et al. 1991). Figure 3b shows that in the LGE, which is the region outlined in white where RALDH3 is present, RA-reporter expression is low. Addition of trichostatin or valproate to the RARE-lacZ cultures resulted in selective reporter-expression increase in the LGE (Fig. 3c and d). Addition of exogenous RA intensified the RARE-lacZ signal throughout most the forebrain preparation (Fig. 3e). Combinations of RA with trichostatin or valproate increased RARE-lacZ signalling throughout almost all of the forebrain, but the intensity of the signal in the LGE was not greater than with trichostatin or valproate alone (Fig. 3f and g).
A RAR antagonist inhibits GE-to-cortex migration
The observations above demonstrate that exogenous RA inhibits migration of cells from the GE to the cortex. What, however, might be the result of an abnormal loss of endogenous RA signal in the GE? To address this question, we used pan-RA receptor antagonists in the slice cultures, because multiple RA receptor transcripts are expressed in the GE and adjoining cortex around E15, the developmental stage examined in this study (Allen Brain Atlas http:// www.brain-map.org/, Eurexpress http://www.eurexpress.org/ee/: Lein et al., 2007; Diez-Roux et al., 2011). To inhibit any endogenous RA receptor, the pan-RAR antagonist LG100815 was employed at a concentration of 5 μM, the concentration used in a previous in vitro culture system to block RA signalling pathways regulating cell growth (Chen et al. 2006). The RAR antagonist significantly decreased by 56% the average percentage of DiI labelled cells reaching the cortex (Fig. 4). We also tested the pan-RXR antagonist LG100815, which blocks signalling via the second group of RA receptors, the RXRs. When the RXR antagonist was applied at the same concentration as the pan-RAR antagonist, the average percentage of DiI labelled cells reaching the cortex was not significantly different from controls. These results demonstrate that loss of endogenous RA signalling is similarly disruptive for GE-to-cortex cell migration as excess RA, indicating that a minimal amount of RA signalling via RAR is necessary for GE-to-cortex neuronal migration.
Fig. 4.
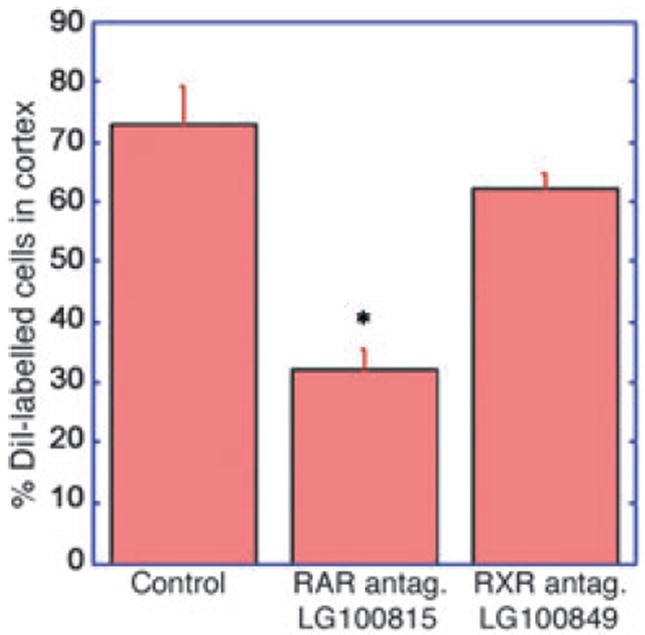
RA receptor antagonists inhibit migration of precursor cells from of the ganglionic eminence to the cortex. In the slice cultures, migration of cells from the GE to cortex could be significantly inhibited by treatment with a pan-RAR antagonist (LG100814), but was not blocked by a pan-RXR antagonist (LG100849), both at a concentration of 5 μM (*control vs. RAR-antagonist t = 6.16, p = 0.0016).
Influence of reduced RA synthesis on numbers of GABAergic interneurons in the adult cortex
The slice culture experiments show that retinoid perturbations at E15 affect the migration of cells from the GE into the cortex: both a global excess as well as global loss in RA signalling in the in vitro preparations reduced the migration. In normal development, the majority of dorsally migrating cells will differentiate into neurons and practically all GABAergic interneurons in the murine cerebral cortex originate from the ventral telencephalon (Chédotal and Rijli 2009; Huang 2009; Nóbrega-Pereira and Marín 2009; Rakic 2009). To extend the in vitro observations at E15 to the postnatal cortex, we counted GABAergic interneurons in the barrel field of the primary somatosensory cortex of adult mice with developmental retinoid perturbations. We used mice with three kinds of reductions in RA signalling: firstly, homozygous Raldh1 null-mutants, which are viable and fertile (Fan et al. 2003); secondly, heterozygous Raldh3 null-mutants, since the homozygous mutation is lethal in newborns (Dupe et al. 2003; Molotkova et al. 2007); and thirdly, mutants lacking serum RBP, whose mothers were fed a vitamin A-deficient diet from gestational day 10 to postnatal day 21, in order to potentiate the vitamin A deficiency caused by lack of the Rbp4 gene function alone (Quadro et al. 1999, 2005). In all three types of mice, the overall RA supply for the developing cortical GABAergic neurons is reduced, but the degrees, locations and temporal characteristics of developmental deficiencies are different in each mutant strain.
Figure 5 illustrates reductions in GABAergic interneurons in each of the three mutant strains. Among the Raldh defective mice, calretinin-positive cells were significantly reduced both in Raldh1 and Raldh3 null-mutants, and the number of calbindin cells was mainly impaired in Raldh1 null-mutants (Fig. 5a–c). In the developmentally vitamin A-deficient RBP mutants (Rbp4−/−, vitamin A deficiency), calbindin-positive interneurons were significantly reduced by 48% to almost half of normal counts, whereas the parvalbumin neurons showed only a marginal decrease (Fig. 5d and e). Thus, just as inhibition of RA signalling in the in vitro slice preparations can lead to a 50% reduction in cortical migration, developmental impairments of RA actions in vivo can reduce the number of GABAergic interneurons overall to comparable extents, and a proportion of these in vivo changes may be the result of a reduction in precursor migration.
Fig. 5.
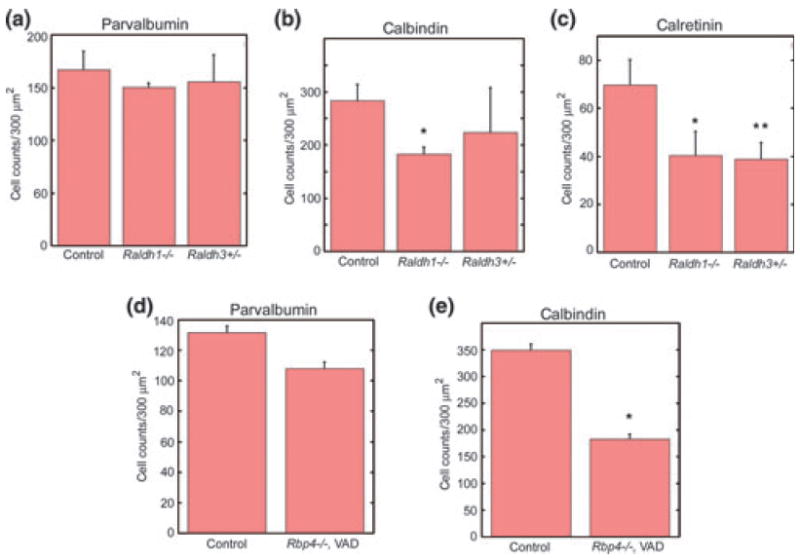
Mutations of the RA synthesizing Raldh1 and Raldh3 genes, as well as global reductions in vitamin A supply, decrease numbers of GABAergic interneurons in the cortex. (a) The density of parvalbuminpositive cells was not influenced by the Raldh mutations, but (b) calbindin-expressing cells were diminished in the homozygous Raldh1 null mutants, and (c) density of calretinin-positive cells were reduced in both the Raldh1 null mutants and Raldh3-heterozygous null mutants (density = number of cells per 300 mm2, *control vs. Raldh1−/− t = 3.01, p < 0.02, **control vs. Raldh3−/− t = 2.47, p < 0.03). Panels (d) and (e) show data from adult retinol-binding protein (RBP) mutants, which were exposed in addition to a vitamin A-deficient (VAD) diet during the entire time of cortex development. In these mice (Rbp4−/−, VAD) (d) the parvalbumin-positive cells were only marginally affected, but (e) calbindin-labelled cells were reduced to almost half their normal counts (density = number of cells per 300 mm2, *control vs. Rbp4−/−, VAD t = 11.12, p < 0.0001).
Discussion
Retinoic acid patterns the developing brain by influencing expression of factors for neuronal migration and progenitor zones
This study demonstrates an effective influence of RA on tangential neuronal migration from the GE to the cerebral cortex; both excess and deficiency interfere with this migration, and the action of excessive RA levels is likely to be mediated, at least in part, via the dopamine D2 receptor. Although RA has never been implicated in the tangential migration of neurons, RA generated by RALDH2 in the meninges was proposed to play a role in regulating cell division in the cerebral cortex by diffusing to the adjacent endfeet of the radial glia (Siegenthaler et al. 2009); however, studies of Raldh2−/− embryos did not support this view (Chatzi et al. 2011). Studies on Raldh3−/− embryos provided evidence that RA synthesized by RALDH3 influences LGE-derived GABAergic interneuron differentiation (Chatzi et al. 2011). A proportion of the change in GABAergic subpopulations seen on genetic or dietary depletion of RA signalling might be accounted for by such an influence on differentiation, but future work will be required to determine what proportion is due to changes in migration driven by the dopamine receptors versus effects on differentiation.
There is plenty of evidence for a RA influence on neuronal migration in other systems, (for example, Pratt et al. 1987; Maden and Holder 1992; Voigt and Zintl 2003; Yamamoto et al. 2003; Zhang et al. 2003; Joshi et al. 2006). Moreover, the same classes of molecules demonstrated to act in migration to the cortex (Chédotal and Rijli 2009; Huang 2009; Nóbrega-Pereira and Marín 2009; Rakic 2009) are known to be involved in RA-regulated cell migration at other sites (Rossino et al. 1991; Gross et al. 1992; Gao et al. 2001; Singh et al. 2003; Day et al. 2006; Wu et al. 2006, 2007; Bogoch and Linial 2008). The most important predictor for the locations and times of RA signalling are known to be elevated RA concentrations at the particular tissue sites (McCaffery and Dräger 1994; Duester 2008). Because RA cannot be directly visualized histologically, and because the effective RA concentration at a cellular site represents a sum of RA diffusing from locally expressed RALDHs and systemic supplies, the precise details of the RA patterns in the developing GE remain elusive. From the temporally, spatially and quantitatively changing RALDH3 and RALDH1 expression patterns in the developing striatum, however, it is obvious that local RA patterns undergo continuous changes throughout the entire period of GE-to-cortex migration.
The GE stands out as a region in which most of the components of the RA signalling pathway have been reported (McCaffery and Dräger 1994; Samad et al. 1997; Valdenaire et al. 1998; Wolf 1998; Toresson et al. 1999; Mollard et al. 2000; Smith et al. 2001; Waclaw et al. 2004; Liao et al. 2005; Wang and Liu 2005; Molotkova et al. 2007), and this signal may provide local instruction to GE subregions. All neuronal precursors destined for the cerebral cortex in rodents are known to originate from the medial and caudal GE portions, the MGE and CGE, which are devoid of the RALDH3 enzyme, the most potent local RA source; the RALDH3-expressing, RA-rich LGE gives rise exclusively to precursors for ventral telencephalic sites and the olfactory bulb (Marin et al. 2000; Wichterle et al. 2001; Stenman et al. 2003; Wonders and Anderson 2006). The observations that experimental RA excess inhibits GE-to-cortex migration in the slice cultures make it likely that endogenous RA is a significant factor in the normal determination of the gross ventro-dorsal dichotomy of precursors from the RA-rich LGE and RA-poor MGE: the excess RA probably confers an LGE-like milieu onto emerging MGE precursors, which might be redirected from cortical to subcortical targets, pathways not testable in the slice preparations used here.
The observations that experimental deficiency in RA signalling also reduces GE-to-cortex migration in the slice cultures, and that in the adult cortex it results in a net deficit in interneuron content, indicate that RA contributes to the diversity of cortical interneurons. Elucidation of the cell-biological and molecular mechanisms of neuronal migrations for the cerebral cortex has been a central focus in neuroscience for over half a century, and in recent years detailed clues to the determination of the migratory path and future biochemical identity of the different cortical GABAergic interneuron types are being identified (Chédotal and Rijli 2009; Huang 2009; Nóbrega-Pereira and Marín 2009; Rakic 2009). The fate of interneurons is dependent on the times and locations of their birth in the GE; it is determined in regionally and temporally changing progenitor zones. A combinatorial expression of multiple transcription factors delineates distinct domains in the subpallium (Flames et al. 2007), and within the MGE, comparisons of dorsal and ventral parts reveal significant heterogeneity in gene expression and distinct origins of future GABAergic neurons (Wonders et al. 2008). For the developing hindbrain, which contains, at least initially, a geometrically simple caudorostral RA gradient, RA is well known to play multiple spatio-temporally changing roles in axial patterning, determinations of neuronal fates and target innervations (Marshall et al. 1992; Dupe and Lumsden 2001); directly or indirectly RA contributes to differential expressions of hundreds of genes in the hindbrain (Chambers et al. 2009). By comparison to the hindbrain, the changing RA patterns in the GE, as they are bound to be created by the local in-growth of RALDH1-containing axons and shifting diffusion conditions in the enlarging brain, are geometrically much more complex. Similar to the distinct roles of RA in the developing hindbrain, RA is likely involved in the combinatorial expressions of transcription-factor domains, in the regionalization of gene expression in the MGE, and hence in the fate of GABAergic precursors.
Retinoid perturbations and mechanisms of retinoic acid actions in the brain
As a first experimental procedure, we added RA to the slice cultures, because the rapid creation of RA excess is by far the most effective and commonly used method to probe for a RA role in a particular process. The reason for the high effectiveness of RA excess is that it practically never occurs naturally, and no defence mechanisms have evolved to protect from it. Moreover, no physiological event has ever been identified which causes a sudden local RA increase; all naturally occurring changes in RA levels, as deduced from changes in RALDH expression, take place over hours to days (e.g. Li et al. 2000; Smith et al. 2001). The opposite perturbation, RA deficit or vitamin A deficiency, however, occurs relatively frequently, and it stimulated the evolution of an elaborate bulwark of physiological defences against it. It is extremely difficult to achieve vitamin A deficiency in a normal animal solely by nutritional restrictions, which was the reason here for using three gene-knockout models. Although each of the three null-mutants differs in the presumed details of the RA-pattern alterations in the GE, in the adult cortex they all revealed significant reductions in some GABAergic cell counts. While the occurrence of vitamin A deficiency based on poor nutrition is largely restricted to developing countries, transient states of insufficient vitamin A supply are relatively common everywhere, because activation of an immune response shuts off acutely the systemic circulation of retinol bound to its carrier protein RBP, by switching off the transcription of the Rbp4 gene in the liver (Rosales and Ross 1998). In addition, mutations in most retinoid genes, as well as mutation in RA-regulated genes, result in similar phenotypes as nutritional vitamin A deficiency (reviewed in Luo et al. 2009).
Relevance to complex human brain diseases
The observations of this study are likely to be patho-physiogically relevant for human brain diseases, beyond RA’s reputation as one of the most potent embryonic teratogens (Lammer et al. 1985). The 13-cis isomer of RA isotretinoin, which is used orally to treat acne, is known to lead to severe embryopathy, with the CNS being particularly sensitive to its actions. Depending on the time of isotretinoin exposures, the RA teratology ought to include a decline in the number of cells migrating from the GE, and thus a reduction in the cortical GABAergic interneuron population. As already mentioned above, accidental or experimental creation of RA excess is so effective because practically all endogenous mechanisms in retinoid homeostasis protect against vitamin A deficiency. The experimental value of RA excess is that it causes similar abnormalities as RA deficit, although the cellular events leading to the final abnormalities may differ in details. This phenotypic overlap in excess and deficiency symptoms derives as a general insight from over 50 years of retinoid research, and it also sums up the present results: both too much and too little RA signalling cause interneuron reductions for the cerebral cortex. The three types of knockout mice used here for the adult cell counts appear normal in behaviour and gross morphology, but their reductions in GABAergic interneurons diminish the inhibitory latitude in the excitatory–inhibitory balance of their cortical circuitry. These mice ought to be more prone to epileptic discharges, when subjected to additional stressors of cortical homeostasis.
In addition to epilepsy, our observations on perturbed RA signalling may be relevant to autism and complex neuropsychiatric diseases in general, which are now regarded as disturbances in cortical homeostasis (Hill 2004; Ozonoff et al. 2004; Ramocki and Zoghbi 2008). Electrical activity in the normal cortex is maintained within a narrow functional range by continuous adjustments of the balance between excitatory glutamatergic and inhibitory GABAergic synaptic activities (Maffei and Fontanini 2009). Quantitative changes in GABA signalling have been associated with autism (Fatemi et al. 2002; Casanova et al. 2003; McCauley et al. 2004). A third of the population with autism spectrum disorder show signs of epilepsy, and cortical hyperexcitability caused by an imbalance of the normal excitatory–inhibitory equilibrium is part of the autistic core pathology (Hussman 2001; Rubenstein and Merzenich 2003; Belmonte et al. 2004; Polleux and Lauder 2004).
As an alternative method for generating an excess in RA signaling we applied the histone-deacetylase (HDAC) inhibitors trichostatin and valproate, which resulted in the same effect as adding RA excess, a result that is consistent with the action of HDAC inhibitors via potentiation of RA signalling (Menegola et al. 2006; Epping et al. 2007). Well before valproic acid was identified as an HDAC inhibitor (Göttlicher et al. 2001; Phiel et al. 2001), the drug has been widely used clinically as an anti-epileptic medication for many years, and much was learned about its teratogenic side effects, when taken during pregnancy. Valproate teratogenicity in human includes autism as a clinical phenotype (Christianson et al. 1994; Williams and Hersh 1997; Moore et al. 2000; Williams et al. 2001; Rasalam et al. 2005), and embryonic exposures of rodents have identified teratologies and symptoms equivalent to an autistic syndrome (Ingram et al. 2000; Schneider and Przewlocki 2005; Wagner et al. 2006; Rinaldi et al. 2007, 2008; Dufour-Rainfray et al. 2009). The human valproate syndrome includes specific malformations in the hindbrain and brainstem similar to those found in patients with idiopathic autism (Ingram et al. 2000; Miyazaki et al. 2005). Valproate was proposed to affect hoxa1 gene expression in the hindbrain through augmentation of RA action (Stodgell et al. 2006). The current study suggests that the cortex may be another brain area in which development is disrupted by valproate in a way that contributes to autism. Indicative of the disruptive effects of valproate on neuronal migration, cortical as well as hippocampal dysplasias have been described in offspring of rats exposed to valproate during the last 5 days of pregnancy (Manent et al. 2007). Although the neuronal dysplasia was interpreted to result from the capacity of valproate to increase GABA signalling, results of this study indicate that valproate’s action as a HDAC inhibitor ought to contribute significantly to disruption of neuronal migration.
From extensive genomic research over recent years it has become apparent that the characteristic symptomatology of autism spectrum disorders can be caused by disturbances in many different genes. Analyses of copy number variations that cause autism show that both increases and decreases in gene dosage can result in similar, overlapping clinical phenotypes. These observations on the relevance of proper gene dosage is behind the notion that failure of neuronal homeostasis is a common denominator of the different genetic causes of autism (Ramocki and Zoghbi 2008). Historically, the vitamin A system is among the longest known contributors to the regulation of body homeostasis, the maintenance of a constant internal environment. The experimental observations of similar symptoms in vitamin A excess and deficiency underscore the function of this system in homeostasis, a feature also shared with neuropsychiatric disease genes (Ramocki and Zoghbi 2008). Another commonality in genetic causes of autism spectrum disorders is a convergence onto the ERK and PI3K signalling cascades (Levitt and Campbell 2009), which are the same pathways that are also prevalently involved in non-genomic RA actions (Canon et al. 2004; Masia et al. 2007). A common denominator of genes implicated in autism and neuropsychiatric diseases thus might be differential regulation at locations of RA signalling in the developing brain.
Acknowledgments
We thank Tuanlian Luo and Elisabeth Wagner for preparations of the data on adult vitamin A-deprived cortex, Bill Blaner for the RBP mutant mice, Janet Rossant for the RARE-lacZ mice, Michael Wagner for the RARE-lacZ cell line, Ron Lindahl for the RALDH1 antiserum, and Ligand Pharmaceuticals for the RA receptor antagonists. This work was supported by United States Public Health Service grants NS 43426, DA02076, HD 05515, EY01938, EY13272, EY013969; Autism Speaks; the Royal Society and the Edinburgh Royal Society.
Abbreviations used
- CGE
caudal ganglionic eminence
- DiI
1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate
- DMSO
dimethylsulfoxide
- GE
ganglionic eminence
- HDAC
histone deacetylase
- LGE
lateral ganglionic eminence
- MGE
medial ganglionic eminence
- RA
retinoic acid
- RALDH
retinaldehyde dehydrogenase
- RAR
retinoic acid receptors
- RARE
RA response element
- RBP
retinolbinding protein
- RXR
retinoid X receptors
- VAD
vitamin A deficiency
Footnotes
The authors have no conflicts of interests to declare.
References
- Anderson SA, Eisenstat DD, Shi L, Rubenstein JL. Interneuron migration from basal forebrain to neocortex: dependence on Dlx genes. Science. 1997;278:474–476. doi: 10.1126/science.278.5337.474. [DOI] [PubMed] [Google Scholar]
- Araki KY, Sims JR, Bhide PG. Dopamine receptor mRNA and protein expression in the mouse corpus striatum and cerebral cortex during pre- and postnatal development. Brain Res. 2007;1156:31–45. doi: 10.1016/j.brainres.2007.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belmonte MK, Cook EH, Jr, Anderson GM, et al. Autism as a disorder of neural information processing: directions for research and targets for therapy. Mol Psychiatry. 2004;9:646–663. doi: 10.1038/sj.mp.4001499. [DOI] [PubMed] [Google Scholar]
- Bogoch Y, Linial M. Coordinated expression of cytoskeleton regulating genes in the accelerated neurite outgrowth of P19 embryonic carcinoma cells. Exp Cell Res. 2008;314:677–690. doi: 10.1016/j.yexcr.2007.12.003. [DOI] [PubMed] [Google Scholar]
- Canon E, Cosgaya JM, Scsucova S, Aranda A. Rapid effects of retinoic acid on CREB and ERK phosphorylation in neuronal cells. Mol Biol Cell. 2004;15:5583–5592. doi: 10.1091/mbc.E04-05-0439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casanova MF, Buxhoeveden D, Gomez J. Disruption in the inhibitory architecture of the cell minicolumn: implications for autisim. Neuroscientist. 2003;9:496–507. doi: 10.1177/1073858403253552. [DOI] [PubMed] [Google Scholar]
- Cawley S, Bekiranov S, Ng HH, et al. Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of noncoding RNAs. Cell. 2004;116:499–509. doi: 10.1016/s0092-8674(04)00127-8. [DOI] [PubMed] [Google Scholar]
- Chambers D, Wilson LJ, Alfonsi F, et al. Rhombomere-specific analysis reveals the repertoire of genetic cues expressed across the developing hindbrain. Neural Dev. 2009;4:6. doi: 10.1186/1749-8104-4-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatzi C, Brade T, Duester G. Retinoic Acid functions as a key GABAergic differentiation signal in the Basal Ganglia. PLoS Biol. 2011;9:e1000609. doi: 10.1371/journal.pbio.1000609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chédotal A, Rijli FM. Transcriptional regulation of tangential neuronal migration in the developing forebrain. Curr Opin Neurobiol. 2009;19:139–145. doi: 10.1016/j.conb.2009.04.005. [DOI] [PubMed] [Google Scholar]
- Chen Y, Dokmanovic M, Stein WD, Ardecky RJ, Roninson IB. Agonist and antagonist of retinoic acid receptors cause similar changes in gene expression and induce senescence-like growth arrest in MCF-7 breast carcinoma cells. Cancer Res. 2006;66:8749–8761. doi: 10.1158/0008-5472.CAN-06-0581. [DOI] [PubMed] [Google Scholar]
- Christianson AL, Chesler N, Kromberg JG. Fetal valproate syndrome: clinical and neuro-developmental features in two sibling pairs. Dev Med Child Neurol. 1994;36:361–369. doi: 10.1111/j.1469-8749.1994.tb11858.x. [DOI] [PubMed] [Google Scholar]
- Clagett-Dame M, DeLuca HF. The role of vitamin A in mammalian reproduction and embryonic development. Annu Rev Nutr. 2002;22:347–381. doi: 10.1146/annurev.nutr.22.010402.102745E. [DOI] [PubMed] [Google Scholar]
- Crandall JE, Hackett HE, Tobet SA, Kosofsky BE, Bhide PG. Cocaine exposure decreases GABA neuron migration from the ganglionic eminence to the cerebral cortex in embryonic mice. Cereb Cortex. 2004;14:665–675. doi: 10.1093/cercor/bhh027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crandall JE, McCarthy DM, Araki KY, Sims JR, Ren JQ, Bhide PG. Dopamine receptor activation modulates GABA neuron migration from the basal forebrain to the cerebral cortex. J Neurosci. 2007;27:3813–3822. doi: 10.1523/JNEUROSCI.5124-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day RM, Lee YH, Park AM, Suzuki YJ. Retinoic acid inhibits airway smooth muscle cell migration. Am J Respir Cell Mol Biol. 2006;34:695–703. doi: 10.1165/rcmb.2005-0306OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diez-Roux G, Banfi S, Sultan M, et al. A high-resolution anatomical atlas of the transcriptome in the mouse embryo. PLoS Biol. 2011;9:e1000582. doi: 10.1371/journal.pbio.1000582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duester G. Retinoic acid synthesis and signaling during early organogenesis. Cell. 2008;134:921–931. doi: 10.1016/j.cell.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufour-Rainfray D, Vourc’h P, Le Guisquet AM, Garreau L, Ternant D, Bodard S, et al. Behavior and serotonergic disorders in rats exposed prenatally to valproate: a model for autism. Neurosci Lett. 2009;470:55–59. doi: 10.1016/j.neulet.2009.12.054. [DOI] [PubMed] [Google Scholar]
- Dupe V, Lumsden A. Hindbrain patterning involves graded responses to retinoic acid signalling. Development. 2001;128:2199–2208. doi: 10.1242/dev.128.12.2199. [DOI] [PubMed] [Google Scholar]
- Dupe V, Matt N, Garnier JM, Chambon P, Mark M, Ghyselinck NB. A newborn lethal defect due to inactivation of retinaldehyde dehydrogenase type 3 is prevented by maternal retinoic acid treatment. Proc Natl Acad Sci USA. 2003;100:14036–14041. doi: 10.1073/pnas.2336223100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epping MT, Wang L, Plumb JA, Lieb M, Gronemeyer H, Brown R, Bernards R. A functional genetic screen identifies retinoic acid signaling as a target of histone deacetylase inhibitors. Proc Natl Acad Sci USA. 2007;104:17777–17782. doi: 10.1073/pnas.0702518104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan X, Molotkov A, Manabe S, Donmoyer CM, Deltour L, Foglio MH, Cuenca AE, Blaner WS, Lipton SA, Duester G. Targeted disruption of Aldh1a1 (Raldh1) provides evidence for a complex mechanism of retinoic acid synthesis in the developing retina. Mol Cell Biol. 2003;23:4637–4648. doi: 10.1128/MCB.23.13.4637-4648.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farooqui SM. Induction of adenylate cyclase sensitive dopamine D2-receptors in retinoic acid induced differentiated human neuroblastoma SHSY-5Y cells. Life Sci. 1994;55:1887–1893. doi: 10.1016/0024-3205(94)00520-6. [DOI] [PubMed] [Google Scholar]
- Fatemi SH, Halt AR, Stary JM, Kanodia R, Schulz SC, Realmuto GR. Glutamic acid decarboxylase 65 and 67 kDa proteins are reduced in autistic parietal and cerebellar cortices. Biol Psychiatry. 2002;52:805–810. doi: 10.1016/s0006-3223(02)01430-0. [DOI] [PubMed] [Google Scholar]
- Flames N, Pla R, Gelman DM, Rubenstein JL, Puelles L, Marin O. Delineation of multiple subpallial progenitor domains by the combinatorial expression of transcriptional codes. J Neurosci. 2007;27:9682–9695. doi: 10.1523/JNEUROSCI.2750-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Bian W, Yang J, Tang K, Kitani H, Atsumi T, Jing N. A role of N-cadherin in neuronal differentiation of embryonic carcinoma P19 cells. Biochem Biophys Res Commun. 2001;284:1098–1103. doi: 10.1006/bbrc.2001.5089. [DOI] [PubMed] [Google Scholar]
- Göttlicher M, Minucci S, Zhu P, et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 2001;20:6969–6978. doi: 10.1093/emboj/20.24.6969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross N, Favre S, Beck D, Meyer M. Differentiation-related expression of adhesion molecules and receptors on human neuroblastoma tissues, cell lines and variants. Int J Cancer. 1992;52:85–91. doi: 10.1002/ijc.2910520116. [DOI] [PubMed] [Google Scholar]
- Gupta A, Sanada K, Miyamoto DT, Rovelstad S, Nadarajah B, Pearlman AL, Brunstrom J, Tsai LH. Layering defect in p35 deficiency is linked to improper neuronal-glial interaction in radial migration. Nat Neurosci. 2003;6:1284–1291. doi: 10.1038/nn1151. [DOI] [PubMed] [Google Scholar]
- Hill EL. Executive dysfunction in autism. Trends Cogn Sci. 2004;8:26–32. doi: 10.1016/j.tics.2003.11.003. [DOI] [PubMed] [Google Scholar]
- Huang Z. Molecular regulation of neuronal migration during neocortical development. Mol Cell Neurosci. 2009;42:11–22. doi: 10.1016/j.mcn.2009.06.003. [DOI] [PubMed] [Google Scholar]
- Hussman JP. Suppressed GABAergic inhibition as a common factor in suspected etiologies of autism. J Autism Dev Disord. 2001;31:247–248. doi: 10.1023/a:1010715619091. [DOI] [PubMed] [Google Scholar]
- Ingram JL, Peckham SM, Tisdale B, Rodier PM. Prenatal exposure of rats to valproic acid reproduces the cerebellar anomalies associated with autism. Neurotoxicol Teratol. 2000;22:319–324. doi: 10.1016/s0892-0362(99)00083-5. [DOI] [PubMed] [Google Scholar]
- Jones PL, Shi YB. N-CoR-HDAC corepressor complexes: roles in transcriptional regulation by nuclear hormone receptors. Curr Top Microbiol Immunol. 2003;274:237–268. doi: 10.1007/978-3-642-55747-7_9. [DOI] [PubMed] [Google Scholar]
- Joshi S, Guleria R, Pan J, Dipette D, Singh US. Retinoic acid receptors and tissue-transglutaminase mediate short-term effect of retinoic acid on migration and invasion of neuroblastoma SH-SY5Y cells. Oncogene. 2006;25:240–247. doi: 10.1038/sj.onc.1209027. [DOI] [PubMed] [Google Scholar]
- Krezel W, Ghyselinck N, Samad TA, Dupe V, Kastner P, Borrelli E, Chambon P. Impaired locomotion and dopamine signaling in retinoid receptor mutant mice. Science. 1998;279:863–867. doi: 10.1126/science.279.5352.863. [DOI] [PubMed] [Google Scholar]
- Kurlandsky SB, Gamble MV, Ramakrishnan R, Blaner WS. Plasma delivery of retinoic acid to tissues in the rat. J Biol Chem. 1995;270:17850–17857. doi: 10.1074/jbc.270.30.17850. [DOI] [PubMed] [Google Scholar]
- Lammer EJ, Chen DT, Hoar RM, et al. Retinoic acid embryopathy. N Engl J Med. 1985;313:837–841. doi: 10.1056/NEJM198510033131401. [DOI] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–176. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- Levitt P, Campbell DB. The genetic and neurobiologic compass points toward common signaling dysfunctions in autism spectrum disorders. J Clin Invest. 2009;119:747–754. doi: 10.1172/JCI37934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Wagner E, McCaffery P, Smith D, Andreadis A, Drager UC. A retinoic acid synthesizing enzyme in ventral retina and telencephalon of the embryonic mouse. Mech Dev. 2000;95:283–289. doi: 10.1016/s0925-4773(00)00352-x. [DOI] [PubMed] [Google Scholar]
- Liao WL, Tsai HC, Wu CY, Liu FC. Differential expression of RARbeta isoforms in the mouse striatum during development: a gradient of RARbeta2 expression along the rostrocaudal axis. Dev Dyn. 2005;233:584–594. doi: 10.1002/dvdy.20344. [DOI] [PubMed] [Google Scholar]
- Lindahl R, Clark R, Evces S. Histochemical localization of aldehyde dehydrogenase during rat hepatocarinogenesis. Cancer Res. 1983;43:5972–5977. [PubMed] [Google Scholar]
- Luo T, Wagner E, Grun F, Drager UC. Retinoic acid signaling in the brain marks formation of optic projections, maturation of the dorsal telencephalon, and function of limbic sites. J Comp Neurol. 2004;470:297–316. doi: 10.1002/cne.20013. [DOI] [PubMed] [Google Scholar]
- Luo T, Wagner E, Drager UC. Integrating retinoic acid signaling with brain function. Dev Psychol. 2009;45:139–150. doi: 10.1037/0012-1649.45.1.139. [DOI] [PubMed] [Google Scholar]
- Maden M, Holder N. Retinoic acid and development of the central nervous system. Bioessays. 1992;14:431–438. doi: 10.1002/bies.950140702. [DOI] [PubMed] [Google Scholar]
- Maffei A, Fontanini A. Network homeostasis: a matter of coordination. Curr Opin Neurobiol. 2009;19:168–173. doi: 10.1016/j.conb.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manent JB, Jorquera I, Mazzucchelli I, Depaulis A, Perucca E, Ben-Ari Y, Represa A. Fetal exposure to GABA-acting antiepileptic drugs generates hippocampal and cortical dysplasias. Epilepsia. 2007;48:684–693. doi: 10.1111/j.1528-1167.2007.01056.x. [DOI] [PubMed] [Google Scholar]
- Marin O, Anderson SA, Rubenstein JL. Origin and molecular specification of striatal interneurons. J Neurosci. 2000;20:6063–6076. doi: 10.1523/JNEUROSCI.20-16-06063.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinova Z, Ren M, Wendland JR, Leng Y, Liang MH, Yasuda S, Leeds P, Chuang DM. Valproic acid induces functional heat-shock protein 70 via Class I histone deacetylase inhibition in cortical neurons: a potential role of Sp1 acetylation. J Neurochem. 2009;111:976–987. doi: 10.1111/j.1471-4159.2009.06385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall H, Nonchev S, Sham MH, Muchamore I, Lumsden A, Krumlauf R. Retinoic acid alters hindbrain Hox code and induces transformation of rhombomeres 2/3 into a 4/5 identity. Nature. 1992;360:737–741. doi: 10.1038/360737a0. [DOI] [PubMed] [Google Scholar]
- Masia S, Alvarez S, de Lera AR, Barettino D. Rapid, nongenomic actions of retinoic acid on phosphatidylinositol-3-kinase signaling pathway mediated by the retinoic acid receptor. Mol Endocrinol. 2007;21:2391–2402. doi: 10.1210/me.2007-0062. [DOI] [PubMed] [Google Scholar]
- McCaffery P, Dräger UC. High levels of a retinoic-acid generating dehydrogenase in the meso-telencephalic dopamine system. Proc Natl Acad Sci USA. 1994a;91:7772–7776. doi: 10.1073/pnas.91.16.7772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCaffery P, Dräger UC. Hotspots of retinoic acid synthesis in the developing spinal cord. Proc Natl Acad Sci USA. 1994b;91:7194–7197. doi: 10.1073/pnas.91.15.7194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCaffery P, Zhang J, Crandall JE. Retinoic acid signaling and function in the adult hippocampus. J Neurobiol. 2006;66:780–791. doi: 10.1002/neu.20237. [DOI] [PubMed] [Google Scholar]
- McCauley JL, Olson LM, Delahanty R, Amin T, Nurmi EL, Organ EL, Jacobs MM, Folstein SE, Haines JL, Sutcliffe JS. A linkage disequilibrium map of the 1-Mb 15q12 GABA(A) receptor subunit cluster and association to autism. Am J Med Genet B Neuropsychiatr Genet. 2004;131:51–59. doi: 10.1002/ajmg.b.30038. [DOI] [PubMed] [Google Scholar]
- Menegola E, Di Renzo F, Broccia ML, Giavini E. Inhibition of histone deacetylase as a new mechanism of teratogenesis. Birth Defects Res C Embryo Today. 2006;78:345–353. doi: 10.1002/bdrc.20082. [DOI] [PubMed] [Google Scholar]
- Miyazaki K, Narita N, Narita M. Maternal administration of thalidomide or valproic acid causes abnormal serotonergic neurons in the offspring: implication for pathogenesis of autism. Int J Dev Neurosci. 2005;23:287–297. doi: 10.1016/j.ijdevneu.2004.05.004. [DOI] [PubMed] [Google Scholar]
- Mollard R, Viville S, Ward SJ, Decimo D, Chambon P, Dolle P. Tissue-specific expression of retinoic acid receptor isoform transcripts in the mouse embryo. Mech Dev. 2000;94:223–232. doi: 10.1016/s0925-4773(00)00303-8. [DOI] [PubMed] [Google Scholar]
- Molotkov A, Molotkova N, Duester G. Retinoic acid guides eye morphogenetic movements via paracrine signaling but is unnecessary for retinal dorsoventral patterning. Development. 2006;133:1901–1910. doi: 10.1242/dev.02328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molotkova N, Molotkov A, Duester G. Role of retinoic acid during forebrain development begins late when Raldh3 generates retinoic acid in the ventral subventricular zone. Dev Biol. 2007;303:601–610. doi: 10.1016/j.ydbio.2006.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore SJ, Turnpenny P, Quinn A, Glover S, Lloyd DJ, Montgomery T, Dean JC. A clinical study of 57 children with fetal anticonvulsant syndromes. J Med Genet. 2000;37:489–497. doi: 10.1136/jmg.37.7.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadarajah B, Alifragis P, Wong RO, Parnavelas JG. Neuronal migration in the developing cerebral cortex: observations based on real-time imaging. Cereb Cortex. 2003;13:607–611. doi: 10.1093/cercor/13.6.607. [DOI] [PubMed] [Google Scholar]
- Nóbrega-Pereira S, Marín O. Transcriptional control of neuronal migration in the developing mouse brain. Cereb Cortex. 2009;19:107–113. doi: 10.1093/cercor/bhp044. [DOI] [PubMed] [Google Scholar]
- Ohtani N, Goto T, Waeber C, Bhide PG. Dopamine modulates cell cycle in the lateral ganglionic eminence. J Neurosci. 2003;23:2840–2850. doi: 10.1523/JNEUROSCI.23-07-02840.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozonoff S, Cook I, Coon H, et al. Performance on Cambridge Neuropsychological Test Automated Battery subtests sensitive to frontal lobe function in people with autistic disorder: evidence from the Collaborative Programs of Excellence in Autism network. J Autism Dev Disord. 2004;34:139–150. doi: 10.1023/b:jadd.0000022605.81989.cc. [DOI] [PubMed] [Google Scholar]
- Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J Biol Chem. 2001;276:36734–36741. doi: 10.1074/jbc.M101287200. [DOI] [PubMed] [Google Scholar]
- Polleux F, Lauder JM. Toward a developmental neurobiology of autism. Ment Retard Dev Disabil Res Rev. 2004;10:303–317. doi: 10.1002/mrdd.20044. [DOI] [PubMed] [Google Scholar]
- Popolo M, McCarthy DM, Bhide PG. Influence of dopamine on precursor cell proliferation and differentiation in the embryonic mouse telencephalon. Dev Neurosci. 2004;26:229–244. doi: 10.1159/000082140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt RM, Goulding EH, Abbott BD. Retinoic acid inhibits migration of cranial neural crest cells in the cultured mouse embryo. J Cranio Genet Dev Biol. 1987;7:205–217. [PubMed] [Google Scholar]
- Quadro L, Blaner WS, Salchow DJ, Vogel S, Piantedosi R, Gouras P, et al. Impaired retinal function and vitamin A availability in mice lacking retinol-binding protein. EMBO J. 1999;18:4633–4644. doi: 10.1093/emboj/18.17.4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quadro L, Hamberger L, Gottesman ME, Wang F, Colantuoni V, Blaner WS, Mendelsohn CL. Pathways of vitamin A delivery to the embryo: insights from a new tunable model of embryonic vitamin A deficiency. Endocrinology. 2005;146:4479–4490. doi: 10.1210/en.2005-0158. [DOI] [PubMed] [Google Scholar]
- Rakic P. Evolution of the neocortex: a perspective from developmental biology. Nat Rev Neurosci. 2009;10:724–735. doi: 10.1038/nrn2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramocki MB, Zoghbi HY. Failure of neuronal homeostasis results in common neuropsychiatric phenotypes. Nature. 2008;455:912–918. doi: 10.1038/nature07457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasalam AD, Hailey H, Williams JH, Moore SJ, Turnpenny PD, Lloyd DJ, Dean JC. Characteristics of fetal anticonvulsant syndrome associated autistic disorder. Dev Med Child Neurol. 2005;47:551–555. doi: 10.1017/s0012162205001076. [DOI] [PubMed] [Google Scholar]
- Rinaldi T, Kulangara K, Antoniello K, Markram H. Elevated NMDA receptor levels and enhanced postsynaptic long-term potentiation induced by prenatal exposure to valproic acid. Proc Natl Acad Sci USA. 2007;104:13501–13506. doi: 10.1073/pnas.0704391104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinaldi T, Silberberg G, Markram H. Hyperconnectivity of local neocortical microcircuitry induced by prenatal exposure to valproic acid. Cereb Cortex. 2008;18:763–770. doi: 10.1093/cercor/bhm117. [DOI] [PubMed] [Google Scholar]
- Rosales FJ, Ross AC. A low molar ratio of retinol binding protein to transthyretin indicates vitamin A deficiency during inflammation: studies in rats and a posterior analysis of vitamin Asupplemented children with measles. J Nutr. 1998;128:1681–1687. doi: 10.1093/jn/128.10.1681. [DOI] [PubMed] [Google Scholar]
- Ross SA, McCaffery PJ, Drager UC, De Luca LM. Retinoids in embryonal development. Physiol Rev. 2000;80:1021–1054. doi: 10.1152/physrev.2000.80.3.1021. [DOI] [PubMed] [Google Scholar]
- Rossant J, Zirngibl R, Cado D, Shago M, Giguère V. Expression of a retinoic acid response element-hsplacZ transgene defines specific domains of transcriptional activity during mouse embryogenesis. Genes Dev. 1991;5:1333–1344. doi: 10.1101/gad.5.8.1333. [DOI] [PubMed] [Google Scholar]
- Rossino P, Defilippi P, Silengo L, Tarone G. Up-regulation of the integrin alpha1/beta1 in human neuroblastoma cells differentiated by retinoic acid: correlation with increased neurite outgrowth response to laminin. Cell Reg. 1991;2:1021–1033. doi: 10.1091/mbc.2.12.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenstein JL, Merzenich MM. Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2003;2:255–267. doi: 10.1034/j.1601-183x.2003.00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai Y, Dräger UC. Detection of retinoic acid catabolism with reporter systems and by in-situ hybridization for CYP26 enzymes. Methods Mol Biol. 2010;652:277–294. doi: 10.1007/978-1-60327-325-1_16. [DOI] [PubMed] [Google Scholar]
- Samad TA, Krezel W, Chambon P, Borrelli E. Regulation of dopaminergic pathways by retinoids: activation of the D2 receptor promoter by members of the retinoic acid receptor-retinoid X receptor family. Proc Natl Acad Sci USA. 1997;94:14349–14354. doi: 10.1073/pnas.94.26.14349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider T, Przewlocki R. Behavioral alterations in rats prenatally exposed to valproic acid: animal model of autism. Neuropsychopharmacology. 2005;30:80–89. doi: 10.1038/sj.npp.1300518. [DOI] [PubMed] [Google Scholar]
- Siegenthaler JA, Ashique AM, Zarbalis K, et al. Retinoic acid from the meninges regulates cortical neuron generation. Cell. 2009;139:597–609. doi: 10.1016/j.cell.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh US, Pan J, Kao YL, Joshi S, Young KL, Baker KM. Tissue transglutaminase mediates activation of RhoA and MAP kinase pathways during retinoic acid-induced neuronal differentiation of SH-SY5Y cells. J Biol Chem. 2003;278:391–399. doi: 10.1074/jbc.M206361200. [DOI] [PubMed] [Google Scholar]
- Smith D, Wagner E, Koul O, McCaffery P, Drager UC. Retinoic acid synthesis for the developing telencephalon. Cereb Cortex. 2001;11:894–905. doi: 10.1093/cercor/11.10.894. [DOI] [PubMed] [Google Scholar]
- Stenman J, Toresson H, Campbell K. Identification of two distinct progenitor populations in the lateral ganglionic eminence: implications for striatal and olfactory bulb neurogenesis. J Neurosci. 2003;23:167–174. doi: 10.1523/JNEUROSCI.23-01-00167.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockhausen MT, Sjolund J, Manetopoulos C, Axelson H. Effects of the histone deacetylase inhibitor valproic acid on Notch signalling in human neuroblastoma cells. Br J Cancer. 2005;92:751–759. doi: 10.1038/sj.bjc.6602309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stodgell CJ, Ingram JL, O’Bara M, Tisdale BK, Nau H, Rodier PM. Induction of the homeotic gene Hoxa1 through valproic acid’s teratogenic mechanism of action. Neurotoxicol Teratol. 2006;28:617–624. doi: 10.1016/j.ntt.2006.08.004. [DOI] [PubMed] [Google Scholar]
- Theiler K. The House Mouse. Development and Normal Stages from Fertilization to 4 Weeks of Age. Springer-Verlag; Berlin, Heidelberg, New York: 1972. [Google Scholar]
- Tobet SA, Chickering TW, Hanna I, Crandall JE, Schwarting GA. Can gonadal steroids influence cell position in the developing brain? Horm Behav. 1994;28:320–327. doi: 10.1006/hbeh.1994.1028. [DOI] [PubMed] [Google Scholar]
- Toresson H, Mata de Urquiza A, Fagerstrom C, Perlmann T, Campbell K. Retinoids are produced by glia in the lateral ganglionic eminence and regulate striatal neuron differentiation. Development. 1999;126:1317–1326. doi: 10.1242/dev.126.6.1317. [DOI] [PubMed] [Google Scholar]
- Valdenaire O, Maus-Moatti M, Vincent JD, Mallet J, Vernier P. Retinoic acid regulates the developmental expression of dopamine D2 receptor in rat striatal primary cultures. J Neurochem. 1998;71:929–936. doi: 10.1046/j.1471-4159.1998.71030929.x. [DOI] [PubMed] [Google Scholar]
- Voigt A, Zintl F. Effects of retinoic acid on proliferation, apoptosis, cytotoxicity, migration, and invasion of neuroblastoma cells. Med Pediatr Oncol. 2003;40:205–213. doi: 10.1002/mpo.10250. [DOI] [PubMed] [Google Scholar]
- Waclaw RR, Wang B, Campbell K. The homeobox gene Gsh2 is required for retinoid production in the embryonic mouse telencephalon. Development. 2004;131:4013–4020. doi: 10.1242/dev.01272. [DOI] [PubMed] [Google Scholar]
- Wagner M, Han B, Jessell TM. Regional differences in retinoid release from embryonic neural tissue detected by an in vitro reporter assay. Development. 1992;116:55–66. doi: 10.1242/dev.116.1.55. [DOI] [PubMed] [Google Scholar]
- Wagner GC, Reuhl KR, Cheh M, McRae P, Halladay AK. A new neurobehavioral model of autism in mice: pre- and postnatal exposure to sodium valproate. J Autism Dev Disord. 2006;36:779–793. doi: 10.1007/s10803-006-0117-y. [DOI] [PubMed] [Google Scholar]
- Wang HF, Liu FC. Regulation of multiple dopamine signal transduction molecules by retinoids in the developing striatum. Neuroscience. 2005;134:97–105. doi: 10.1016/j.neuroscience.2005.04.008. [DOI] [PubMed] [Google Scholar]
- Wichterle H, Turnbull DH, Nery S, Fishell G, Alvarez-Buylla A. In utero fate mapping reveals distinct migratory pathways and fates of neurons born in the mammalian basal forebrain. Development. 2001;128:3759–3771. doi: 10.1242/dev.128.19.3759. [DOI] [PubMed] [Google Scholar]
- Williams PG, Hersh JH. A male with fetal valproate syndrome and autism. Dev Med Child Neurol. 1997;39:632–634. doi: 10.1111/j.1469-8749.1997.tb07500.x. [DOI] [PubMed] [Google Scholar]
- Williams G, King J, Cunningham M, Stephan M, Kerr B, Hersh JH. Fetal valproate syndrome and autism: additional evidence of an association. Dev Med Child Neurol. 2001;43:202–206. [PubMed] [Google Scholar]
- Wolf G. Vitamin A functions in the regulation of the dopaminergic system in the brain and pituitary gland. Nutr Rev. 1998;56:354–355. doi: 10.1111/j.1753-4887.1998.tb01678.x. [DOI] [PubMed] [Google Scholar]
- Wonders CP, Anderson SA. The origin and specification of cortical interneurons. Nat Rev Neurosci. 2006;7:687–696. doi: 10.1038/nrn1954. [DOI] [PubMed] [Google Scholar]
- Wonders CP, Taylor L, Welagen J, Mbata IC, Xiang JZ, Anderson SA. A spatial bias for the origins of interneuron subgroups within the medial ganglionic eminence. Dev Biol. 2008;314:127–136. doi: 10.1016/j.ydbio.2007.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu N, Zhang W, Yang Y, Liang YL, Wang LY, Jin JW, Cai XM, Zha XL. Profilin 1 obtained by proteomic analysis in all-trans retinoic acid-treated hepatocarcinoma cell lines is involved in inhibition of cell proliferation and migration. Proteomics. 2006;6:6095–6106. doi: 10.1002/pmic.200500321. [DOI] [PubMed] [Google Scholar]
- Wu JJ, Cantor A, Moscinski LC. beta2 Integrins are characteristically absent in acute promyelocytic leukemia and rapidly upregulated in vivo upon differentiation with all-trans retinoic acid. Leuk Res. 2007;31:49–57. doi: 10.1016/j.leukres.2006.04.012. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Zhang J, Smith D, Hayakawa Y, McCaffery P. A critical period for retinoic acid teratogenesis and loss of neurophilic migration of pontine nuclei neurons. Mech Dev. 2003;120:701–709. doi: 10.1016/s0925-4773(03)00047-9. [DOI] [PubMed] [Google Scholar]
- Zhang J, Smith D, Yamamoto M, Ma L, McCaffery P. The meninges is a source of retinoic acid for the late-developing hindbrain. J Neurosci. 2003;23:7610–7620. doi: 10.1523/JNEUROSCI.23-20-07610.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]