

Abstract
Aims
The suppressors of cytokine signalling (SOCS) are identified inhibitors of cytokine and growth factor signalling that act via the Janus kinase (JAK) signal transducers and activators of transcription (STAT) pathways. Aberrant JAK/STAT signalling promotes progression from hypertrophy to heart failure. Little information is available concerning the role of SOCS in the transition from hypertrophy to heart failure. To this aim, we investigated the effects of SOCS1 overexpression obtained by in vivo adeno-associated gene transfer using an aortopulmonary cross-clamping technique in a chronic pressure-overload cardiac rat model.
Methods and results
Rats were randomized into four groups: sham-operated (n = 18), aortic banding (AB) (n = 18), AB + viral vector encoding for haemoagglutinin (AB + HA, n = 16), and AB + viral vector encoding for SOCS1 (AB + SOCS1, n = 18). Echocardiographic and haemodynamic measurements were performed 15 weeks after banding. While SOCS3 was upregulated during the hypertrophic phase, SOCS1 transcript levels increased significantly between 15 and 20 weeks. Remodelling was markedly worse in AB + SOCS1, showed larger left ventricular internal dimensions (+16%), higher end-diastolic pressures (+57%) and wall stress (+45%), and reduced fractional shortening (−32%) compared with AB + HA; apoptotic rate was increased threefold and the gp130 pathway was inhibited. Ex vivo experiments showed that mechanical stretch upregulated SOCS1 expression, which was in turn attenuated by tumour necrosis factor-α (TNF-α) inhibition.
Conclusion
Enhanced SOCS1 myocardial signalling is associated with accelerated transition from hypertrophy to failure in an established model of pressure overload. SOCS1 may represent an attractive target for the prevention of heart failure progression.
Keywords: SOCS, Gene therapy, Cardiac hypertrophy, Heart failure
1. Introduction
Suppressors of cytokine signalling (SOCS) are recently discovered cytoplasmic proteins that serve as negative regulators of cytokine signalling.1–3 In particular, SOCSs are implicated in the intricate cascade of myocardial growth and survival, since they control in negative feedback key intracellular pathways, including Janus kinase (JAK)/signal transducers and activators of transcription (STAT), Ras/MAPK and ERK, and PI3K/Akt.1–5 These pathways are hypertrophic and survival substrates of the common signal-transducing receptor chain gp130, which is activated by IL-6-type cytokines in response to pressure overload and ischaemic injury.6 While SOCS3 is mainly induced by IL-6-type cytokines, SOCS1 is highly susceptible to IFN-γ stimulation and modulates its effects in vivo, mainly through the gp130/JAK/STAT signalling pathway.7,8 SOCS1 was recently demonstrated to play a relevant role in virally induced heart disease because it negatively interferes with STAT3-dependent host-defence mechanisms.9 However, apart from this evidence mainly focused on its action in cardiac inflammation, the role of SOCS1 in response to increased haemodynamic load is currently unknown, despite its well-known effects on targets such as gp130/JAK signalling that traditionally plays a pivotal role during the transition to heart failure.6–10
To this aim, we overexpressed myocardial SOCS1 through adeno-associated viral gene transfer in a well-established model of experimental heart failure, the aortic banding (AB) model. Clarifying this issue would enhance the current understanding of the negative feedback loops of cytokine signalling and would provide further insights into the complex and delicate balance of stimulation and termination of growth and survival signals during the transition to cardiac failure.
2. Methods
2.1. Adeno-associated construct
The full-length cDNA of SOCS1 was subcloned and amplified with an N-terminal FLAG tag by using specific primer pairs. We used replication-defective adeno-associated serotype 1 viral vectors encoding for SOCS1 wild-type gene included in a frame haemoagglutinin (HA) epitope 30-tag (AAV.SOCS1) under the control of the cytomegalovirus promoter. As a control, we performed a similar adenoviral vector encoding for the HA epitope only (AAV.HA).
2.2. Animal model and cardiac gene transfer
Male Wistar rats (Charles River, Calco, Italy, body weight 60–80 g) were subjected to AB coupled to aortopulmonary cross-clamping and coronary retroperfusion. A method based on that reported by Hajjar et al.11 and subsequently modified in our laboratory12 was used to deliver 100 µL of the viral construct (5 × 1010 particle units) in the myocardium (see Supplementary material online). Rats were randomized into four groups: sham-operated (n = 18), rats subjected to AB (n = 18), rats subjected to AB and inoculated with either AAV.HA (AB + HA) (n = 16) or AAV.SOCS1 (AB + SOCS1) (n = 18). In vivo and in vitro analyses were performed 15 weeks after AB. Every adjunctive time point of analysis was performed with four rats for each group. In preliminary experiments, six sham-operated animals were inoculated with AAV.SOCS1 (Sham + SOCS1) and six with AAV.HA (Sham + HA) only to evaluate the effects of SOCS1 per se on cardiac architecture and function. All methods described conformed to Directive 2010/63/EU of the European Parliament and was approved by CESA of University ‘Federico II’ of Naples, and the anaesthesiological protocol [intraperitoneal injection of zolazepam hydrochloride (20 mg/kg), tiletamine hydrochloride (20 mg/kg) (Zoletil®, Virbac, Milan, Italy), and atropine (0.05 mg/kg) (Galenica Senese, Siena, Italy)] was approved and monitored by a veterinary physician of the Animal Vet Service of the University of Naples.
2.3. Echocardiography
Transthoracic echocardiograms were performed in all surviving animals, according to previously described methods13,14 with an SONOS 5500 electronic system equipped with a 7 or 12 MHz probe (Agilent Technologies, Wakefield, MA, USA) under light anaesthesia (zolezapam and tiletamine 10 mg/kg). Anterior and posterior end-diastolic wall thickness and left ventricular (LV) diastolic and systolic internal dimensions were measured as previously described13,14 according to standard procedures as well as calculations of LV per cent fractional shortening. All measurements, performed with an off-line analysis system by one observer who was blinded to prior results, were based on the average of three to six consecutive cardiac cycles.
2.4. Haemodynamic studies
Within 12 h of the final echocardiogram, rats were anaesthetized with zolezapam and tiletamine 20 mg/kg, and a calibrated 2F micromanometer-tipped catheter (Millar Instruments, Houston, TX, USA) was passed via the carotid artery into the left ventricle under constant pressure monitoring. The haemodynamic data were recorded and analysed as previously described in detail.13,14 Rats were euthanized by cervical dislocation under anaesthesia and the hearts were quickly excised and processed for the following analysis.
2.5. Isolated whole heart
A subgroup of rats (n = 6) was employed to evaluate SOCS1 activation under acute mechanical stretch, independent of the confounding effects of circulating substances that are upregulated under in vivo haemodynamic overload. The hearts were isolated and perfused according to previously described methods.15 The intraventricular pressure was raised up to reach an end-diastolic pressure of ∼5 mmHg (unstretched control myocardium), 15 mmHg (moderate stretch), or 35 mmHg (severe stretch) for 60 min (see Supplementary material online).
Four hearts were isolated and perfused as described above, and neutralizing polyclonal TNF-α antibody (50 ng/mL, Santa Cruz Biotechnology, Santa Cruz, CA, USA) was added to the perfusate while the hearts were stretched at 35 mmHg.
2.6. Northern blotting
Left ventricles were removed and homogenized in TRIzol reagent (Invitrogen Life technologies, Monza, Italy), and total RNA was isolated according to the manufacturer's instructions. Samples of total RNA were then separated, transferred to Biodyne membranes (Pall Corp., Glen Cove, NY, USA), and hybridized with 32P-labelled probes as previously described.14,16 28S rRNA was considered as standard, and autoradiograms were normalized with GADPH densitometric signal. Every probe was assayed in duplicate and three northern blotting were performed.
2.7. Western blotting
Left ventricles were homogenized in RIPA-SDS buffer and total protein concentration was measured by BCA assay.14–16 Proteins were resolved by PAGE and transferred to a nitrocellulose membrane, pre-incubated with PBS and 2% haemoglobin, to reduce non-specific binding. The membrane was incubated with the STAT3, phospho-STAT3, JAK-1, phosphor-JAK-1, VEGF-A, and angiopoietin-2 antibodies (Santa Cruz Biotechnology). Immunoblotting was detected by anti-rabbit horseradish peroxidase-conjugated secondary antibody (Santa Cruz Biotechnology) and visualized by using standard chemiluminescence.14–16 Three independent western blots were performed for each antibody and each sample was run in duplicate.
2.8. Histology, immunohistochemistry, and TUNEL assay
The hearts were weighed, formalin-fixed, paraffin-embedded, and cut into slices 6 µm thick.14–16 General morphology and collagen volume fraction were studied with classical histological stainings. SOCS1 cardiac gene transfer, SOCS-mediated gp130 inhibition signalling, capillary density (taken as the ratio of capillaries per cardiomyocyte), and inflammation markers were studied with immunohistochemistry, as previously described. Apoptotic cardiomyocytes were labelled with TUNEL reaction and anti-activated caspase-3 immunoreaction as previously described.17 All the observations and measurements were performed by two blinded researchers. Histology, immunohistochemistry, and TUNEL were described in detail in the Supplementary material online.
2.9. Plasma markers
Plasma electrolytes were measured with a Radiometer ABL 520 Blood Analyser (Brønshøj, Denmark). Activity of aspartate transaminase (AST), alanine transaminase (ALT), alkaline phosphatase, creatinine, and total bilirubin was determined using kits and controls supplied by Roche Diagnostic and COBAS analyser. Plasma concentration of malondialdehyde was determined by HPLC.
2.10. Statistics
Data are reported as mean ± SEM. Between-group comparisons (SHAM, AB, AB + HA, AB + SOCS1) of echocardiographic indexes were performed using a two-way ANOVA with repeated measure in one factor (time), followed by the Neumann–Keuls test. One-way ANOVA was employed for the other comparisons, also followed by the Neumann–Keuls test. A P-value <0.05 was considered significant.
3. Results
3.1. Cardiac gene transfer
Approximately 70% of cardiomyocytes were infected and marked with the HA tag included in the viral construct, consistent with previous evidence.11,12 Tissue histology analysis demonstrated a uniform distribution of the adenoviral infection with SOCS1 throughout the myocardium in AB + SOCS1 and Sham + SOCS1 groups (Figure 1A–L). Cardiomyocytes were abundantly infected as well as few leucocytes, while endothelial cells and fibroblasts displayed no signs of infection. Immunostaining was not detectable in the sham-operated group (Figure 1F) and in control sections obtained by omitting the primary antibody. We also found evidence of cardiac expression of SOCS1 in other organs. Specifically, following myocardial gene transfer, ectopic expression of SOCS1 in the kidney and liver was in the range of 3–5% of the myocardial expression. The proximal tract of the descending aorta, lung, and spleen showed few SOCS1-positive cells. Renal function was unaffected by the combination of gene transfer and AB, as shown by plasma values of creatinine, creatinine clearance, sodium, and potassium, which were not significantly different among the experimental groups (Table 1). Liver function was equally unaffected, serum transaminasemia, ALP, and bilirubin levels were not different among the study groups, while malondialdehyde, an index of oxidative stress, was increased in all rats subjected to AB (Table 1).
Figure 1.
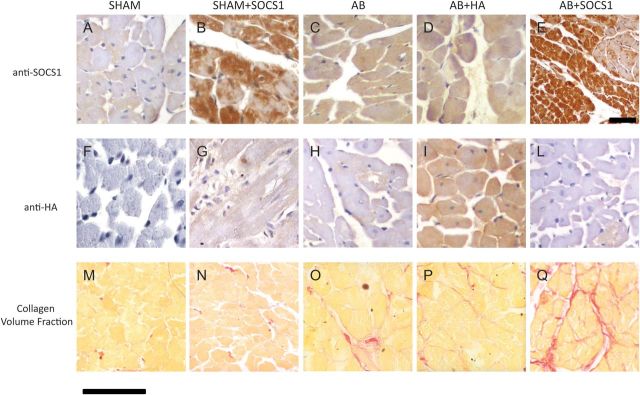
Cardiac expression of SOCS1 15 weeks after AB detected by immunohistochemistry. SOCS1 is slightly expressed in Sham animals (A); compensatory hypertrophy leads to a mild increase in SOCS1-positive cardiomyocytes in AB (C) and AB + HA groups (D). Overexpression of SOCS1 and its distribution is clearly depicted in (B) and (E) (at lower magnification). (F–L) Cardiac gene expression of AAV.HA enhanced with an anti-HA antibody. (I) It is shown that ≈70% of cardiomyocytes overexpressed the antigen. (M–Q) Myocardial collagen content in Sham, Sham + SOCS1, AB, AB + HA, and AB + SOCS1, respectively. The interstitial fibrosis following haemodynamic overload (O and P) is markedly augmented following SOCS1 infection (Q). SOCS1 overexpression in absence of AB did not alter interstitial fibrosis (N). Scale bar = 50 μm.
Table 1.
Plasma parameters of hepatic and renal function and oxidative stress after 15 weeks of treatment
| Anova |
Sham (n = 18) | AB (n = 18) | AB + HA (n = 16) | AB + SOCS1 (n = 18) | ||
|---|---|---|---|---|---|---|
| F | P | |||||
| Liver wet weight (mg) | 0.634 | 0.59 | 275 ± 12 | 280 ± 10 | 277 ± 10 | 278 ± 12 |
| Kidney wet weight (mg) | 0.535 | 0.660 | 52 ± 1.4 | 51.7 ± 1.9 | 52.3 ± 1.2 | 51.8 ± 1.3 |
| Plasma Na2+, (mM) | 2.292 | 0.086 | 139 ± 1.3 | 140 ± 1.9 | 139.4 ± 1.9 | 138.4 ± 2.3 |
| Plasma K+, (mM) | 5.161 | 0.003 | 5.0 ± 1.8 | 6.7 ± 1.4* | 6.5 ± 1.3* | 6.8 ± 1.7* |
| Plasma creatinine, (µM) | 0.219 | 0.883 | 0.25 ± 0.04 | 0.26 ± 0.05 | 0.26 ± 0.05 | 0.26 ± 0.04 |
| Plasma creatinine clearance, (µM/min) | 0.207 | 0.891 | 2.54 ± 0.67 | 2.37 ± 0.70 | 2.39 ± 0.78 | 2.43 ± 0.67 |
| Plasma ALT activity, (U/L) | 2.808 | 0.046 | 40.07 ± 0.8 | 41.3 ± 0.4 | 40.9 ± 0.7 | 41.2 ± 0.8 |
| Plasma AST activity, (U/L) | 1.071 | 0.367 | 107 ± 4.8 | 104 ± 3.7 | 107 ± 6.2 | 105 ± 8.5 |
| Plasma ALP activity, (U/L) | 0.08 | 0.967 | 175 ± 18 | 182 ± 21 | 179 ± 13 | 184 ± 15 |
| Bilirubin, (µM/l) | 0.392 | 0.759 | 1.58 ± 0.18 | 1.61 ± 0.19 | 1.57 ± 0.24 | 1.64 ± 0.23 |
| Malondialdehyde, (µM/l) | 30.41 | <0.001 | 25.9 ± 1.2 | 29.7 ± 1.2* | 29.5 ± 1.8* | 30.07 ± 1.7* |
Data are mean ± SEM; Sham, sham-operated AB group; AB + HA, rats subjected to AB plus inoculation of AAV.HA epitope only; AB + SOCS1, rats subjected to AB plus SOCS1 myocardial gene transfer.
*P < 0.05 vs. corresponding sham.
3.2. Effect of SOCS1 overexpression on cardiac structure and function
Sham-operated rats infected with the AAV.SOCS1 gene construct had a normal cardiac phenotype with morphological and echographic values similar to those reported in the sham-operated (Table 2) and sham-HA groups (see Supplementary material online, Figure S1), as revealed by preliminary experiments. No differences were found between sham-operated rats and sham + SOCS1 with regard to STAT3 and JAK-1 expression and phosphorylation, an expected finding since such pathways are not activated in normal hearts (see Supplementary material online, Figure S1).
Table 2.
Animal characteristics, morphometric histology, and LV in vivo dynamics after 15 weeks of treatment
| ANOVA |
Sham (n = 18) | AB (n = 18) | AB + HA (n = 16) | AB + SOCS1 (n = 18) | ||
|---|---|---|---|---|---|---|
| F | P | |||||
| LV wet/body weight (mg/g) | 5.1 | 0.003 | 1.6 ± 0.2 | 2.6 ± 0.1* | 2.5 ± 0.4* | 3.0 ± 0.3* |
| Collagen volume fraction (%) | 508 | <0.001 | 3.6 ± 0.1 | 10.4 ± 0.4* | 9.1 ± 0.3*‡ | 18.3 ± 0.2*†‡ |
| Capillary density (no./mm2) | 33.2 | <0.001 | 1550 ± 26 | 1255 ± 40* | 1195 ± 30 | 860 ± 24*†‡ |
| Capillary/myocyte ratio | 40 | <0.05 | 3.1 ± 0.9 | 2.9 ± 0.4* | 2.8 ± 0.6* | 1.2 ± 0.1*†‡ |
| LV diastolic diameter (mm) | 4 | 0.01 | 7.8 ± 0.7 | 8.8 ± 0.6 | 9.0 ± 0.5 | 10.5 ± 0.4* |
| LV fractional shortening (%) | 6 | 0.001 | 47 ± 2 | 44 ± 3 | 43 ± 4 | 29 ± 4*†‡ |
| LV end-diastolic pressure (mmHg) | 7.9 | <0.001 | 4 ± 1 | 14 ± 3 | 12 ± 4 | 22 ± 2*†‡ |
| LV meridional diastolic wall stress (kdyne/cm2) | 23.59 | <0.001 | 7 ± 2 | 35 ± 3* | 40 ± 6* | 58 ± 5*†‡ |
Data are mean ± SEM; Sham, sham-operated AB group; AB + HA, rats subjected to AB plus inoculation of AAV.HA epitope only; AB + SOCS1, rats subjected to AB plus SOCS1 myocardial gene transfer.
*P < 0.05 vs. corresponding sham.
‡P < 0.05 vs. corresponding AB.
†P < 0.05 vs. AB + HA group.
The in vivo consequences of myocardial SOCS1 overexpression on LV architecture and function were assessed 15 weeks after AB. This time point was chosen since it represents the transition from compensated hypertrophy, which typically occurs between 8 and 12 weeks, to overt heart failure, which develops in most banded rats 18–24 weeks following AB. At this time point, while the AB and AB + HA groups displayed compensatory hypertrophy, SOCS1 overexpression induced the rapid onset of the heart failure phenotype. Specifically, SOCS1-treated animals exhibited marked LV hypertrophy and dilation, as documented by higher LV mass/body weight and larger diastolic LV internal dimensions compared with the AB and the AB + HA group (Table 2). Moreover, both systolic and diastolic function were markedly impaired in the active treatment group, as demonstrated by lower values of endocardial fractional shortening and elevated end-diastolic pressures, which were all significantly different compared with the AB and AB + HA groups (Table 2). The AB + HA group exhibited the typical phenotype of compensated LV hypertrophy that has been described consistently by ourselves and other groups at that time point characterized by LV hypertrophy associated with preserved pump function.13,16 Myocardial architecture and haemodynamics of AB and AB + HA groups were similar, demonstrating no significant influence of viral load per se. The evolution from compensatory hypertrophy to heart failure is described in Supplementary material online, Figure S2.
3.3. Histology
We hypothesized that the inhibition of the gp130 pathway may lead to maladaptive remodelling, which was evaluated at the cellular level by analysing cardiomyocyte size and myocardial fibrosis. As expected, the AB group showed an increase in myocyte diameter (23.6 ± 1.8 µm) compared with shams (17.4 ± 2.5 and 18.1 ± 1.4). The myocyte diameter was more marked in the AB + SOCS1 group (27.3 ± 1.4 μm) than in the AB + HA group (24.2 ± 2.3 μm, P < 0.05) and was associated with a dramatic increase in myocardial fibrosis (Figure 1M,O,P, and Q) and reduced capillary density (see subsequently). Myocardial collagen content was not altered in the Sham + SOCS1 group (Figure 1N).
3.4. In vivo SOCS3 and SOCS1 induction following mechanical stress
The activation of SOCS3 but not of SOCS1 during in vivo pressure overload was previously documented in the literature in other models of aortic constriction.18 In that study, the second peak of SOCS3 expression was consistent with the onset of cardiac hypertrophy. To document the time course of SOCS1 expression in the rat model of pressure overload secondary to AB, we sacrificed rats 10, 15, and 20 weeks following AB. At 10 weeks, we confirm Yasukawa et al.'s18 findings in our animal model of pressure overload, with an increase in SOCS3 expression occurring in conjunction with myocardial hypertrophy, while SOCS1 mRNA induction remained at baseline levels. At 15 weeks, SOCS3 mRNA levels began to decrease compared with baseline while SOCS1 transcript levels displayed a mild increase, reaching a marked induction at 20 weeks, consistent with the onset of overt heart failure (Figure 2A).
Figure 2.

(A). SOCS1, SOCS3, and TNF-α mRNA are detected with northern blot and are differentially expressed in the myocardium after AB. SOCS1 mRNA show a delayed expression at the onset of heart failure, 20 weeks after AB, while SOCS3 is expressed in the hypertrophic phase. *P ≤ 0.05 vs. SOCS1; ¶P ≤ 0.05 vs. same mRNA of baseline time point; †P ≤ 0.05 vs. same mRNA of 10 weeks time point; ‡P ≤ 0.05 vs. same mRNA of 15 weeks time point. In (B) is shown a representative western blot for SOCS1 protein and other proteins involved in the JAK/STAT cascade at 15 weeks after AB. At 15 weeks, the group treated with AAV.HA displays a strong activation of the JAK/STAT pathway compared with the sham group. *P ≤ 0.05 vs. Sham; ¶P ≤ 0.05 vs. AAV.HA. The AAV.SOCS1 group exhibits a marked downregulation of all phosphorylated proteins. (C) Northern blot at 15 weeks after AB. AB + HA and AB + SOCS1 groups show similar transcript levels of STAT3, JAK-1, and gp130 (C). *P ≤ 0.05 vs. Sham; ¶P ≤ 0.05 vs. AAV.HA. Representative bands from the same gel are cropped and mounted together in the final image.
3.5. Long-term inhibition of the gp130 pathway
To confirm the negative feedback mechanism of the adeno-associated mediated SOCS1 overexpression on the gp130 pathway, we examined the effect of SOCS1 on STAT3, JAK-1, and gp130 expression and phosporylation. At 15 weeks, the inhibitory mechanism of SOCS1 was still active in the myocardium of AB + SOCS1 animals, which showed suppressed phosphorylation of STAT3, JAK-1, and gp130 compared with sham and AB + HA groups (Figure 2B), while mRNA transcripts levels were not significantly different among the study groups (Figure 2C).
3.6. SOCS1 overexpression induces massive cardiomyocyte apoptosis and inflammation
Since gp130-dependent pathways activate anti-apoptotic signal, we next assessed whether the rapid onset of cardiac dysfunction observed in the AB + SOCS1 group was associated with increased cardiomyocyte apoptosis death (Figure 3A). SOCS1 overexpression induced a marked apoptosis, as showed by the apoptotic rate that was significantly higher in AB + SOCS1 animals compared with AB + HA (6.8 ± 5.2% vs. 3.4 ± 1, AB + SOCS1 vs. AB + HA, P < 0.05) (Figure 3B).
Figure 3.

Cardiac apoptosis analysed by TUNEL and anti-activated caspase-3 antibody. (A) Apoptotic nuclei are marked by the TUNEL reaction and rhodamine fluorescent dye and digitally turned to yellowish colour, anti-caspase-3 antibody was revealed with fluorescin dye (green) as was anti-troponin antibody with Texas Red. DAPI (blue) was utilized as nuclear stain. Sham animals do not exhibit apoptotic cardiomyocytes (a), while apoptosis is present in animals subjected to AB. AB + HA animals exhibit an early apoptotic process, with few cardiomyocytes caspase-3-positive but TUNEL-negative (b). In SOCS1-treated animals, the apoptotic process is significantly increased with numerous cardiomyocytes that appear both TUNEL- and caspase-3-positive (c), leading to a marked elevation of the apoptotic rate (B). *P ≤ 0.05 vs. Sham; ¶P ≤ 0.05 vs. AAV.HA. Scale bar = 50 μm.
SOCS1 overexpression at 15 weeks resulted in a massive inflammation reaction, with the production of cytoplasmic TNF-α (Figure 4Ac) and a marked macrophage recruitment in the interstitium (Figure 4Af). On the contrary, AB + HA rats only displayed mild inflammation (Figure 4Ab–e)
Figure 4.

(A) Markers of inflammatory response 15 weeks after AB. AB induces an inflammatory response as demonstrated by TNF-α and macrophage localization in AB + HA animals (b and e) compared with shams (a and d). SOCS1 overexpression markedly enhances such inflammatory response with immunoreactions for TNF-α in a large number of cardiomyocytes (c) and evident proliferation of CD68-positive cells in the interstitium (f) as is visible in the corresponding bar graphs. In (B) is depicted a western blot for TNF-α protein, which confirms a sustained increase in TNF-α myocardial production in AAV-SOCS1-treated rats and in (C) densitometric values are showed. *P ≤ 0.05 vs. Sham; ¶P ≤ 0.05 vs. AB + HA. Scale bar = 50 μm.
3.7. Angiogenic markers
Since reduced myocardial production of VEGF-A and angiopoietin-2 has been postulated to play a pivotal role in the transition to heart failure, we evaluated the protein levels of these angiogenic markers. Compared with the AB group, SOCS1 infection was associated with an anticipated increase in angiogenic growth factor myocardial content at 10 weeks, which was followed by a rapid decrease at 15 weeks (Figure 5A and B). Capillary density was investigated by immunohistochemistry at 15 weeks after AB, and data showed a significant reduction of the capillary number in AB and AB + HA that further decreased in AB + SOCS1 (Figure 5C)
Figure 5.

(A) Representative western blots of the angiogenic proteins VEGF-A and angiopoietin-2, with the corresponding densitometric analysis (B). (C) A representative histological microphotograph of capillary density at 20 weeks in sham (a), AB + HA (b), and AB + SOCS1 (c) is depicted to the left. Capillaries/cardiomyocyte ratio at the various time points is depicted in the histogram to the right. AB induces an upregulation of angiopoietin-2 and VEGF-A followed by a marked decrease (A) that is paralleled by a reduction of capillary density (C). In the AAV.SOCS1 group, the onset of the heart failure phenotype was anticipated as well as the downregulation of angiopoietin-2 and VEGF-A. In (C) is depicted vascular rarefaction induced by AB (b) and worsened by SOCS1 gene transfer (c). Capillaries were stained with BSL-I biotin. *P ≤ 0.05 vs. Sham; ¶P ≤ 0.05 vs. AAV.HA. Scale bar = 50 μm.
3.8. Isolated whole-heart mechanical stretching
Isolated heart experiments were performed to gain further insight into potential triggers of SOCS1 upregulation. Compared with hearts perfused under control conditions (unstretched myocardium, 5 mmHg), no significant changes were detected during 60 min of stretch in diastolic and systolic function or in developed pressure, indicating absence of relevant ischaemia and mechanical tissue damage (data not shown).
Autoradiograms (Figure 6A) and corresponding densitometry (Figure 6B) of total myocardial RNA by northern blotting for TNF-α and SOCS1 showed that in unstretched hearts, TNF-α was undetectable while SOCS1 was mildly expressed; induction of moderate stretch associated with de novo TNF-α expression and a modest increase in SOCS1 gene expression. Under severe stretch, a progressive and significant increase in TNF-α and SOCS1expression was detectable. We next employed a neutralizing TNF-α antibody (Santa Cruz Biotechnology) in the perfusate to evaluate whether SOCS1 upregulation would be affected by blocking a well-known cytokine signalling pathway that displayed a robust overexpression in the in vivo studies. SOCS1 protein levels were reduced by ∼40% following TNF-α blockade. SOCS1 was not blocked by an aspecific antibody as anti-rabbit immunoglobulin.
Figure 6.

(A) Representative northern blots for TNF-α, SOCS1, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in control (unstretched myocardium: 5 mmHg), moderately (15 mmHg), and severely (35 mmHg) stretched myocardium. (B) Densitometric analysis of autoradiographic bands, expressed as means ± SEM of three independent experiments for each value of stretch. *P≤0.05 vs. 5 mmHg; ¶P≤0.05 vs. the corresponding 15 mmHg group. (C) The effects of a neutralizing TNF-α antibody applied in the perfusate on SOCS1 protein levels during stretch are displayed. In (D) is depicted the corresponding densitometric analysis, carried out on lanes from the same blot. Inhibition of TNF-α reduces SOCS1 myocardial production by ∼40% while the use of an aspecific antibody did not affect the SOCS1 protein level.
4. Discussion
The current study demonstrates for the first time that SOCS1 expression is upregulated in decompensated experimental pressure overload and that enhanced SOCS1 myocardial signalling accelerates the transition to heart failure. We also show that myocardial SOCS1 overexpression under normal circumstances does not influence the cardiac phenotype. Finally, we demonstrate for the first time that mechanical stretch alone is sufficient to trigger SOCS1 upregulation independent of circulating mediators that are increased following chronic haemodynamic overload, and that SOCS1 upregulation is partially dependent upon the TNF-α signalling pathway.
We provide the first evidence that SOCS1 is upregulated in the decompensated phase of pressure overload. Specifically, SOCS3 mRNA was upregulated mostly 10 weeks after AB and its activation decreased at 15 weeks, returning to baseline levels at 20 weeks, when the heart failure phenotype was prevalent. On the other hand, SOCS1 mRNA levels began to increase at 15 weeks, during the late phase of compensated hypertrophy, reaching the maximal induction at 20 weeks, when the heart failure phenotype prevailed. Interestingly, SOCS1 upregulation was paralleled by TNF-α-increased myocardial production, which in turn appears related to the haemodynamic overload and the inflammation state typical of chronic heart failure.19 Indeed, at 15 weeks, marked TNF-α production and macrophage infiltrates were evident in AB + SOCS1 hearts compared with AB + HA. Despite mRNAs being extracted from whole-heart samples (cardiac tissue), our immunohistochemical data confirmed that mRNA-related proteins SOCS1, SOCS3, and TNF- α were localized mostly in the cardiomyocytes.
The second novel finding is that overexpression of SOCS1 is associated with an overt heart failure phenotype: left ventricle was dilated with a marked reduction of systolic phase indexes, end-diastolic pressures were elevated, and wall stress dramatically increased compared with controls that exhibited the typical compensated hypertrophy, characterized by LV hypertrophy and preserved pump function. At the cellular level, dilated hearts with SOCS1 upregulation displayed massive interstitial fibrosis associated with increased cardiomyocyte apoptotic death and inflammation. At the molecular level, such phenotype associated with sustained inhibition of gp130 and gp130 signalling involved proteins STAT2 and JAK-1.
In this context, the current study provides further support to the concept that an intact gp130/STAT cascade is necessary for cardioprotection since its inhibition through SOCS1 overexpression accelerates transition to heart failure. The likely explanation for this finding is that the sustained and augmented inhibition of a critical survival (adaptive) pathway (i.e. gp130/Stat/Jak), particularly in the setting of increased haemodynamic load, induces maladaptive cardiac growth characterized by activation of the apoptotic cascade, markedly increased fibrosis, and vascular rarefaction with reduction of capillary density with consequent early transition to heart failure. In this regard, we show that the attenuation of angiopoietin- 2 and VEGF-A myocardial production, consistently found in pathological hypertrophy, is anticipated and more pronounced in SOCS1-infected rats vs. the AB group. This finding is in agreement with the notion that the lack of angiopoietin-2 and VEGF-A-induced angiogenesis is a key event leading to marked interstitial fibrosis and vascular rarefaction that disrupt the coordination between growth and angiogenesis, in turn promoting the transition from hypertrophy to failure.
The neutral effects of SOCS1 overexpression on LV architecture and function in normal hearts support the concept that SOCS1 inhibition induces significant phenotype changes only when the heart is challenged by stimuli such as pressure overload that activate the gp130 pathway.
In vitro acute experiments were designed in order to provide potential triggers of SOCS1 upregulation. The two magnitudes of acute myocardial stretch (15 and 35 mmHg) were chosen as representative loads that may occur in most clinical conditions, including systemic hypertension, aortic stenosis, and mild-to-moderate (15 mmHg) and severe myocardial infarction (35 mmHg). Chronic haemodynamic overload is indeed characterized by the synthesis of a portfolio of endogenous autocrine/paracrine cytokines and growth-promoting factors that initiate and modulate critical responses within the overloaded myocardium, including myocyte growth, apoptosis, reactive inflammation, and fibrosis.15 However, it is difficult to separate the effects of myocardial ischaemia and neurohormonal interference that play a relevant role in chronic overload from those of acute mechanical stretch. The results of the current study lend support to the hypothesis that SOCS1 is part of the complex cascade of events that take place once that mechanical stimulus is received by specific mechanosensors and then integrated into major intracellular cross-talking signal transduction pathways (MAPK, JAK/STAT, calcineurin), which ultimately modulate gene expression that determines the final phenotype. Not only is mechanical stretch sufficient to induce a robust and sustained SOCS1 upregulation, but its activation is in part dependent on TNF-α activation, since its blocking reduced SOCS1 protein levels by ∼40%. Therefore, SOCS1 does not appear as a mere marker of systemic inflammation that commonly occurs in chronic heart failure but as a molecule that upregulates within the myocardium in response to biomechanical stretch and that might play a role in the progression to heart failure by blocking the gp130 pathway.
In contrast to the thoroughly characterized mechanisms of positive regulation within cytokine signalling pathways, our knowledge of negative feedback loops is comparatively sparse. Very few reports are available regarding SOCS1 role in cardiovascular disease. It is known that SOCS1 is constitutively mildly expressed in the normal myocardium, and that its expression is strongly induced by IFN-γ, CT-1, LPS, and TNF-α, and that does not seem to affect the gp130-mediated ras/ERK and PI3k/Akt signalling, but the gp130/JAK/STAT pathway.1–8 Although SOCS1 suppresses gp130-mediated myocyte hypertrophy and survival in vitro, it is not induced by the IL-6-type cytokines and gp130, nor by pressure overload in vivo.18 Such observations have led to the hypothesis that SOCS1 may not be in a physiological negative feedback loop with gp130 signalling, but may play a relevant role in cardiomyopathies characterized by marked inflammatory reactions such as following viral infection or sepsis. In this regard, Tanimoto et al.19 recently reported that CT-1-mediated expression of SOCS1 in cardiomyocytes may confer cardioprotection in sepsis-induced myocardial depression.
The results of the current study shed new light on SOCS1 role in cardiac disease. Specifically, they suggest that SOCS1, by modifying the gp130–JAK–STAT signalling mechanism, exerts profound effects on the maintenance of cardiac function in response to haemodynamic overload. This is in agreement with a growing body of evidence showing that cytokines operating via gp130 pathways might play a critical role in the onset of cardiac failure, and that an intact gp130 cascade is necessary for cardioprotection.6,10 In this respect, our data are in agreement with those reported in an elegant study by Yasukawa et al.18 who showed that overexpression of SOCS3 and SOCS1 in isolated cardiomyocytes inhibited CT-1-induced hypertrophy. As mentioned earlier, the authors did not find SOCS1 induction in the mouse subjected to AB. However, we found SOCS1 induction during the later failing but not the early hypertrophic phase. Also, the experiments in the isolated perfused heart provided evidence of SOCS1 upregulation 60 min after acute mechanical stretch, when TNF-α is already maximally expressed. In this in vitro model, we reported that the initial response of the myocardium is to upregulate ‘adaptive’ molecules such as IGF-1 and IL-6 with low expression of TNF-α. With a further increase in the degree and the duration of mechanical stretch, maximal activation of TNF-α associates with progressive downregulation of IGF-1 and Il-6. The hypothesis can be put forward that with the development of the heart failure phenotype, the elevated mechanical stretch on the myocardium coincides with the known activation of the cytokine system including TNF-α and IFN that may promote SOCS1 expression. Supporting this concept, TNF-α overexpression paralleled the increase in SOCS1 transcript levels in chronic pressure overload, and, more importantly, SOCS1 myocardial production was attenuated by blocking the TNF-α pathway. Congruent with these findings, Wang et al.20 described the stimulation of the SOCS1 protein by TNF-α infusion in isolated perfused hearts. It is of note that upregulation of SOCS1 was recently reported by Sandek et al.21 in patients with end-stage chronic heart failure.
With regard to the neutral effects of SOCS1 overexpression in normal hearts, our data are in agreement with those reported by Yasukawa et al.22 who found no changes in cardiac structure and function in a mouse model overexpressing SOCS1, unless the mice were challenged with CBV3 infection.
The results of the current study appear relevant with the role of SOCS1 in the intricated molecular mechanisms of adaptive and maladaptive hypertrophy and failure in response to biomechanical stress. A prevailing concept predicts that an intricate balance between cell death-promoting and cell survival mechanisms determines heart failure progression. Among the molecules promoting maladaptive hypertrophy are second messengers and G-proteins including Gsα, Gαq, ras, kinases including PKA, PKCβ2, p38α, calcineurin, ERK5, MEKK1-JNK, transcription factors such as NF-AT3, and recently SOCS3. On the other hand, gp130, STAT3, Akt, PI3K, MEKK1-JNK, GATA4, and Gβγ are known to induce adaptive hypertrophy and to promote cell survival.23 Given this intricate scenario, SOCS1 may be listed as a maladaptive molecule, since its overexpression is detrimental in the settings of increased biochemical stress.
4.1. Clinical implications
Extrapolation of the current findings to the clinical arena appears extremely difficult in view of the complex signalling orchestration that involves SOCS pathways. On the one hand, overexpression of SOCS1 has been proposed as a cardioprotective agent in sepsis-induced myocardial depression;19 on the other hand, SOCS inhibition has been suggested as a potential novel strategy to treat heart failure since it ultimately promotes myocyte survival by stimulating gp130-dependent pathways.18 To reconcile these apparently divergent observations, the hypothesis has been put forward that in balanced systems, inhibition of SOCS in cardiomyocytes ensures the termination of the hypertrophic growth without interfering with survival, while in unbalanced situations, long-term overexpression induces insufficient hypertrophic response, disturbs sarcomeric organization, and increases apoptotic death. It is likely that this negative regulatory circuit serves to prevent hyperstimulation by gp130 cytokines, which may have independent pathological effects on cardiac function. In this context, the results of the acute experiments in the isolated heart suggest that SOCS1 upregulation may occur in all clinical conditions characterized by haemodynamic overload, including myocardial infarction, systemic hypertension, and valve diseases.
4.2. Future directions
The role of SOCS1 in cardiac disease should be investigated also with a loss-of-function approach, generating an inducible SOCS1 KO mouse or blocking SOCS1 in acute or chronic experiments of cardiac hypertrophy.
In conclusion, SOCS appear to play a relevant role as negative cytokine regulators in cardiovascular pathophysiology in order to eventually modulate their function to favour adaptive mechanisms while maintaining a well-balanced gp130 receptor system.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
This work was supported by the Italian Ministry of Health and the Italian Ministry of Education.
Conflict of interest: none declared.
Supplementary Material
References
- 1.Starr R, Wilson TA, Viney EM, Murray LJ, Rayner JR, Jenkins BJ, et al. A family of cytokine-inducible inhibitors of signaling. Nature. 1997;387:917–921. doi: 10.1038/43206. doi:10.1038/43206. [DOI] [PubMed] [Google Scholar]
- 2.Nicola NA, Greenhalgh CJ. The suppressor of cytokine signaling (SOCS) proteins: important feedback inhibitors of cytokine action. Exp Hemat. 2000;28:1105–1112. doi: 10.1016/s0301-472x(00)00525-7. doi:10.1016/S0301-472X(00)00525-7. [DOI] [PubMed] [Google Scholar]
- 3.Yasukawa H, Sasaki A, Yoshimura A. Negative regulation of cytokine signaling pathways. Annu Rev Immunol. 2000;18:143–164. doi: 10.1146/annurev.immunol.18.1.143. doi:10.1146/annurev.immunol.18.1.143. [DOI] [PubMed] [Google Scholar]
- 4.Hilfiker-Kleiner D, Landmesser U, Drexler H. Molecular mechanism in heart failure. Focus on cardiac hypertrophy, inflammation, angiogenesis, and apoptosis. J Am Coll Cardiol. 2006;48:A56–A66. doi:10.1016/j.jacc.2006.07.007. [Google Scholar]
- 5.Hamanaka I, Saito Y, Yasukawa H, Kishimoto I, Kuwahara K, Miyamoto Y, et al. Induction of JAB/SOCS-1/SSI-1 and CIS3/SOCS-3/SSI-3 is involved in gp130 resistance in cardiovascular system in rat treated with cardiotrophin-1 in vivo. Circ Res. 2001;88:727–732. doi: 10.1161/hh0701.088512. doi:10.1161/hh0701.088512. [DOI] [PubMed] [Google Scholar]
- 6.Fisher P, Hilfiker-Kleiner D. Role of gp130-mediated signalling pathways in the heart and its impact on potential therapeutic aspects. Br J Pharmacol. 2008;153:S414–S427. doi: 10.1038/bjp.2008.1. doi:10.1038/bjp.2008.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hanada T, Kobayashi T, Chinen T, Saeki K, Koga K, Minoda Y, et al. IFNgamma-dependent, spontaneous development of colorectal carcinomas in SOCS1-deficient mice. J Exp Med. 2006;203:1391–1397. doi: 10.1084/jem.20060436. doi:10.1084/jem.20060436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davey GM, Heath WR, Starr R. SOCS1: a potent and multifaceted regulator of cytokines and cell-mediated inflammation. Tissue Antigens. 2006;67:1–9. doi: 10.1111/j.1399-0039.2005.00532.x. doi:10.1111/j.1399-0039.2005.00532.x. [DOI] [PubMed] [Google Scholar]
- 9.Yakusawa H, Yasukawa H, Ohishi M, Mori H, Murakami M, Chinen T, et al. IL-6 induces an anti-inflammatory response in the absence of SOCS3 in macrophages. Nat Immunol. 2003;4:551–556. doi: 10.1038/ni938. doi:10.1038/ni938. [DOI] [PubMed] [Google Scholar]
- 10.Hirota H, Chen J, Betz UA, Rajewsky K, Gu Y, Ross J, et al. Loss of a gp130 cardiac muscle cell survival pathway is a critical event in the onset of heart failure during biomechanical stress. Cell. 1999;97:189–198. doi: 10.1016/s0092-8674(00)80729-1. doi:10.1016/S0092-8674(00)80729-1. [DOI] [PubMed] [Google Scholar]
- 11.Hajjar RJ, Schmidt U, Matsui T, Guerrero JL, Lee KH, Gwathmey JK, et al. Modulation of ventricular function through gene transfer in vivo. Proc Natl Acad Sci USA. 1998;95:5251–5256. doi: 10.1073/pnas.95.9.5251. doi:10.1073/pnas.95.9.5251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cittadini A, Monti MG, Iaccarino G, Di Rella F, Tsichlis PN, Di Gianni A, et al. Adenoviral gene transfer of Akt enhances myocardial contractility and intracellular calcium handling. Gene Ther. 2006;13:8–19. doi: 10.1038/sj.gt.3302589. doi:10.1038/sj.gt.3302589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cittadini A, Stromer H, Katz SE, Clark R, Moses AC, Morgan JP, et al. Differential cardiac effects of GH and IGF-1 in the rat: a combined in vivo and in vitro evaluation. Circulation. 1996;93:800–809. doi: 10.1161/01.cir.93.4.800. doi:10.1161/01.CIR.93.4.800. [DOI] [PubMed] [Google Scholar]
- 14.Cittadini A, Isgaard J, Monti MG, Casaburi C, Di Gianni A, Serpico R, et al. Growth hormone prolongs survival in experimental postinfarction heart failure. J Am Coll Cardiol. 2003;41:2154–2163. doi: 10.1016/s0735-1097(03)00483-2. doi:10.1016/S0735-1097(03)00483-2. [DOI] [PubMed] [Google Scholar]
- 15.Cittadini A, Monti MG, Isgaard J, Casaburi C, Strömer H, Di Gianni A, et al. Aldosterone receptor blockade improves left ventricular remodelling and increases ventricular fibrillation threshold in experimental heart failure. Cardiovasc Res. 2003;58:555–564. doi: 10.1016/s0008-6363(03)00251-7. doi:10.1016/S0008-6363(03)00251-7. [DOI] [PubMed] [Google Scholar]
- 16.Palmieri EA, Benincasa G, Di Rella F, Casaburi C, Monti MG, De Simone G, et al. Differential expression of TNF-α, IL-6, and IGF-1 by graded mechanical stress in normal rat myocardium. Am J Physiol. 2002;282:H926–H934. doi: 10.1152/ajpheart.00436.2001. [DOI] [PubMed] [Google Scholar]
- 17.Baldi A, Abbate A, Bussani R, Patti G, Melfi R, Angelini A, et al. Apoptosis and post-infarction left ventricular remodeling. J Mol Cell Cardiol. 2002;34:165–174. doi: 10.1006/jmcc.2001.1498. doi:10.1006/jmcc.2001.1498. [DOI] [PubMed] [Google Scholar]
- 18.Yasukawa H, Hoshijima M, Gu Y, Nakamura T, Pradervand S, Hanada T, et al. Suppressor of cytokine signaling-3 is a biochemical stress-inducible gene that suppresses gp130-mediated cardiac myocyte hypertrophy and survival pathways. J Clin Invest. 2001;108:1459–1467. doi: 10.1172/JCI13939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tanimoto K, Saito Y, Hamanaka I, Kuwahara K, Harada M, Takahashi N, et al. SOCS1/JAB likely mediates the protective effect of cardiotrophin-1 against lipopolysaccharide-induced left ventricular dysfunction in vivo. Circ J. 2005;69:1412–1417. doi: 10.1253/circj.69.1412. doi:10.1253/circj.69.1412. [DOI] [PubMed] [Google Scholar]
- 20.Wang M, Markel T, Crisostomo P, Herring C, Meldrm KK, Lillemoe KD, et al. Deficiency of TNFR1 protects myocardium through SOCS3 and IL-6 but not p38 MAPK or IL-1β. Am J Physiol. 2007;292:H1694–H1699. doi: 10.1152/ajpheart.01063.2006. [DOI] [PubMed] [Google Scholar]
- 21.Sandek A, Wuest F, von Haehling S, Szabo T, Misgeld BJ, Doehner W, et al. Upregulation of STAT-3, SOCS-1, pro-inflammatory and anti-inflammatory circulating mRNA gene expression in CHF patients with and without cachexia compared with healthy control subjects. Eur Heart J. 2008;29:423. doi: 10.1093/eurheartj/ehm551. (Abstract) doi:10.1093/eurheartj/ehm551. [DOI] [PubMed] [Google Scholar]
- 22.Yasukawa H, Yajima T, Duplain H, Iwatate M, Kido M, Hoshijima M, et al. The suppressor of cytokine signalling-1 (SOCS1) is a novel therapeutic target for enterovirus-induced cardiac injury. J Clin Invest. 2003;111:469–478. doi: 10.1172/JCI16491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morisco M, Sadoshima J, Trimarco B, Arora R, Vatner DE, Vatner SF. Is treating cardiac hypertrophy salutary or detrimental: the two faces of Janus. Am J Physiol. 2003;284:H1043–H1047. doi: 10.1152/ajpheart.00990.2002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.