

Abstract
Proteostasis, defined as the combined processes of protein folding/biogenesis, refolding/repair, and degradation, is a delicate cellular balance that must be maintained to avoid deleterious consequences 1. External or internal factors that disrupt this balance can lead to protein aggregation, toxicity and cell death. In humans this is a major contributing factor to the symptoms associated with neurodegenerative disorders such as Huntington's, Parkinson's, and Alzheimer's diseases 10. It is therefore essential that the proteins involved in maintenance of proteostasis be identified in order to develop treatments for these debilitating diseases. This article describes techniques for monitoring in vivo protein folding at near-real time resolution using the model protein firefly luciferase fused to green fluorescent protein (FFL-GFP). FFL-GFP is a unique model chimeric protein as the FFL moiety is extremely sensitive to stress-induced misfolding and aggregation, which inactivates the enzyme 12. Luciferase activity is monitored using an enzymatic assay, and the GFP moiety provides a method of visualizing soluble or aggregated FFL using automated microscopy. These coupled methods incorporate two parallel and technically independent approaches to analyze both refolding and functional reactivation of an enzyme after stress. Activity recovery can be directly correlated with kinetics of disaggregation and re-solubilization to better understand how protein quality control factors such as protein chaperones collaborate to perform these functions. In addition, gene deletions or mutations can be used to test contributions of specific proteins or protein subunits to this process. In this article we examine the contributions of the protein disaggregase Hsp104 13, known to partner with the Hsp40/70/nucleotide exchange factor (NEF) refolding system 5, to protein refolding to validate this approach.
Keywords: Genetics, Issue 77, Molecular Biology, Microbiology, Cellular Biology, Biochemistry, Bioengineering, Biomedical Engineering, Proteins, Saccharomyces cerevisiae, Protein Folding, yeast, protein, chaperone, firefly luciferase, GFP, yeast, plasmid, assay, microscopy
Introduction
In humans neurodegenerative disorders including Alzheimer's, Parkinson's, and Huntington's diseases have been linked to protein misfolding and aggregation 10. Cells employ molecular chaperones to prevent kinetic trapping of cellular proteins into misfolded inactive structures 6. Chaperones participate in intricate interaction networks within the cell, but it is not completely understood how the sum of these interactions contributes to organismal proteostasis. One of the main chaperones responsible for the majority of cytosolic protein folding is the 70 kD heat shock protein (Hsp70) family 19. It has been shown that in yeast loss of Hsp70 decreases the ability to fold nascent heterologously expressed FFL and to refold the endogenous protein, ornithine transcarbamoylase, in vivo 18, 7. The ability to analyze folding with near-real time resolution will facilitate understanding how additional cellular factors contribute to this Hsp70-dependent process. In addition, folding/refolding reactions may not be completely dependent on these contributing proteins, so assays must be sensitive enough to detect both large and small changes in kinetics and efficiency.
The yeast cell disaggregase, Hsp104, plays a vital role in repairing aggregated misfolded proteins. Although Hsp104 homologs have been identified in fungi and plants, this family appears to be absent in metazoans. It has been proposed that other chaperones such as those of the Hsp110 family perform some of the known Hsp104 activities in mammals 16. Hsp104 is an AAA+, hexameric protein complex that functions in yeast to remodel protein aggregates, contributing to refolding and repair 13. Hsp104, along with the yeast Hsp70, Ssa1, and the yeast Hsp40, Ydj1, is required for recovery of denatured FFL in yeast cells 5, 8. The small heat shock protein, Hsp26, has also been shown to be required for Hsp104-mediated disaggregation of FFL 2.
FFL is a two-domain protein that binds the substrate luciferin in the active site and following a conformational change that requires ATP and oxygen, decarboxylates the substrate releasing oxyluciferin, carbon dioxide (CO2), adenosine monophosphate (AMP), and light 11, 9, 3. The commercially available FFL substrate, D-luciferin, results in light emission between 550-570 nm that can be detected using a luminometer 15. FFL is exquisitely sensitive to denaturation from chemical or heat treatments and aggregates rapidly upon unfolding. When exposed to temperatures between 39-45 °C FFL is reversibly unfolded and inactivated 12. In contrast, GFP and its derivatives are highly resistant to protein unfolding stresses 14. Therefore fusion of these two proteins allows FFL to function as an experimentally labile moiety capable of targeting functional GFP to intracellular deposits that can be visualized using fluorescence microscopy at both the population and single cell levels. Application of the enzymatic assay in a semi-automated multimode plate reader coupled with automated microscopy allows unprecedented simultaneous assessment of kinetics and yield of refolding reactions. In addition, the facile molecular genetics of the model eukaryote Saccharomyces cerevisiae allows both precise manipulation of the protein quality control network and the opportunity for discovery-based approaches to identify novel players contributing to cellular stress response and proteostasis.
In this study, wild-type (WT) and HSP104 deletion strains expressing FFL-GFP are subject to protein denaturing heat shock. FFL-GFP refolding is monitored through both an enzymatic assay and microscopy as a proxy readout for repair of the expressed proteome over a recovery time course. When compared to WT cells, we show the Hsp104 deletion strain is ~60% less efficient at refolding FFL-GFP, supporting previous findings establishing a role for Hsp104 in reactivation of denatured FFL 2.
Protocol
1. Construction of Strains Containing FFL-GFP Plasmid
For this study, Saccharomyces cerevisiae strain BY4741 (MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0) was used along with an HSP104 deletion strain from the yeast knockout collection (Open Biosystems/Thermo Scientific). The deletion was confirmed using an Hsp104-specific antibody for Western blot analysis.
FFL-GFP was expressed from p426MET25-FFL-GFP, constructed from a LEU2-based source plasmid obtained from the Glover laboratory at the University of Toronto 17 and obtainable upon request to the authors. Expression of the FFL-GFP plasmid fusion protein is controlled by the MET25 methionine-repressible promoter. The plasmid was transformed into each strain using a protocol was adapted from Gietz et. al, 1995:
Centrifuge 25 ml of log phase cells in a 50 ml conical tube at 4,400 rpm for 2 min, wash with 500 μl of double distilled H20 (ddH2O), and discard supernate.
Resuspend cells in 250 μl of TE/LiAc (Tris-HCl 10 mM, pH=7.5, 1 mM EDTA, 0.1 M lithium acetate). Incubate at 30 °C without mixing for 20 min.
Transfer 50 μl of competent cells to a new tube along with 5 ml (50 μg) of carrier DNA and 5 μl (0.1-5 μg) of plasmid DNA. Add 300 μl of PEG/TE/LiAc (same as TE/LiAc plus 40% polyethylene glycol) to this mixture, and incubate cells at 30 °C for another 30 min.
Add 1/10 volume (v/v) DMSO (36 μl) to this mixture and heat shock at 42 °C for 6 min.
Centrifuge mixture at 6,000 rpm for 30 sec and remove supernate. Resuspend cells in 100 μl of sterile ddH20 and plate cells onto synthetic complete media lacking uracil (SC-URA).
2. Induction of FFL-GFP
FFL-GFP is induced once cells are in log phase so the majority of the protein prior to heat-shock is newly made to avoid proteins that have terminally aggregated over time due to aging-associated cellular inadequacies.
Day 1: Incubate cells in 5 ml of SC-URA at 30 °C rotating O/N.
Day 2: Measure OD600 and inoculate fresh cultures in 5 ml of SC-URA OD600~0.2.
Incubate cultures with rotation at 30 °C until they reach log phase OD600~0.8-1.0.
Centrifuge cells in 15 ml conical tubes at 4,400 rpm for 3 min, decant supernate and resuspend cell pellet in 500 μl of ddH20; transfer solution to a microcentrifuge tube.
Centrifuge at 6,000 rpm and discard supernate.
Resuspend cells in 5 ml of SC-URA-MET. Cells should be incubated rotating in this media for 1 hr at 30 °C.
Centrifuge cells and repeat wash in ddH20 and discard supernate.
Resuspend cells in 5 ml SC-URA media containing 100 μg/ml of cycloheximide to inhibit protein expression, and proceed immediately to Step 3.
3. Enzymatic FFL-GFP Recovery Assay
This assay is a quantitative approach to determine the levels of active enzyme in a population.
- Preparation for this assay:
- Measure OD600 for each sample.
- Prepare necessary amount of D-luciferin needed based on the number of samples and desired number of replicates plus approximately 1,200 μl for the tubing. Final concentration of D-luciferin is ~23 μg per 100 μl of cells. Initially it is dissolved in water at a stock solution concentration of 7.5 mg/ml and diluted to 455 μg/ml in SC prior to experiment. In Figure 2, three replicates for each sample were analyzed resulting in a total of 2.4 ml of D-luciferin being used.
- Program plate reader for flash luminescence assay (adapted from BioTek Gen5 "Luminescence Flash Assay with Injection"; for other instruments follow manufacturer's instructions)
- Set temperature to 30 °C
- Set plate reader to add D-luciferin, shake, and read each well individually.
- Read luminescence (endpoint reading, 5 sec integration time, 180 sensitivity level)
- For recovery assay between each reading shake for 5 min, delay 20 min, and shake an additional 5 min.
- Dispense 50 μl of D-luciferin from an injector.
- Preheat-shock luminescence is measured immediately after cells are added to media containing cycloheximide to halt protein synthesis.
- Transfer 100 μl of cells for each sample with desired number of replicates into a solid white 96 well plate.
- Controls should include a water blank and cells containing an empty vector.
- Start the read with the specifications listed above.
To denature the FFL moiety of FFL-GFP, incubate the 5 ml culture in a glass culture tube at 42 °C shaking for 25 min.
Recovery assay luminescence readings start immediately after cells are removed from the incubator, which is time point zero. Measure luminescence same as described above for the first time point and every 30 min for 90 min.
Normalize samples to preheat-shock luminescence values that have been adjusted based on the OD600 (divide the preheat-shock values by OD600).
4. Fluorescence Microscopy
This assay is a semi-quantitative method to determine solubilization of aggregates over time in a population of cells.
Induction is the same as described in Section 2, Steps 1-8 of protocols.
The same time points are taken as described above (Section 3, Step 4), but for microscopy collect 1 ml of cells at preheat-shock and each recovery time point, and incubate samples rotating at 30 °C in culture tube.
- Visualization:
- Centrifuge 1 ml of cells at 6,000 rpm for 30 sec and remove supernate.
- Resuspend cell pellet in 2 μl of ddH2O.
- Mix 2 μl of cells plus 2 μl of 2% low melt agarose on the slide and add a cover slip. In order to obtain a single plane of cells lightly press finger down in the center of the cover slip and rotate until the cover slip is difficult to move.
- Cells are visualized using 100X oil objective on fluorescent microscope; use DIC to visualize cells and the FITC filter to visualize GFP fluorescence.
- Take multiple pictures for each strain at each time point for quantitation. Fields for pictures should contain ~15-30 cells for quantitative post-experimental analysis.
- To calculate percentage of cells containing aggregates, count 50-100 cells total and divide the number of cells containing aggregates.
5. Single Cell Microscopy
This method is used to follow the solubilization of individual aggregates in a single cell over time.
Induce FFL-GFP expression as described in Section 2, Steps 1-8.
After heat-shock, centrifuge cells in 15 ml conical tube at 4,400 rpm for 2 min.
Wash cells with 500 μl of water and resuspend in 20 μl of ddH2O.
For each strain, cut out a 5x5 mm section of an SC-URA agar plate and using the surface that was in contact with Petri dish, pipette 4 μl of cells and spread around surface of agar with pipette tip. Let stand for ~1 min.
Use tweezers to place agar cube section face-down on a ~55 mm glass bottom dish No. 0 and press down lightly.
- Visualization:
- See i-iii in Section 4.
- Set coordinates for 2-4 focal points for each strain depending on density of culture.
- Set camera to take pictures with a Z-stack with appropriate range (-/+ 15 μm with a 0.5-1.0 μm slice thickness) every 5 min for a 90 min period.
Representative Results
Yeast is dependent on the disaggregase, Hsp104 to efficiently refold heat-denatured proteins. The activity of FFL-GFP was monitored after a 25 min heat shock, using a luminescence flash assay Figure 1. The results of this automated assay shown in Figure 2 revealed a stepwise increase in activity over 90 min that ultimately led to a >80% recovery in WT cells. The hsp104Δ strain recovered 19% of the original activity over the same time frame. Moreover, the extent of initial inactivation was much higher in the chaperone mutant strain (26% activity in WT and 11% in hsp104Δ) suggesting that Hsp104 may be serving a protective role pre-stress, or rapidly associates with denaturing FFL-GFP during the thermal inactivation step to reduce loss of enzyme activity.
Population microscopy corroborated the enzymatic activity assay. Nearly all cells in the hsp104D mutant strain maintained at least one FFL-GFP aggregate compared to the number of aggregates in the WT strain, which decreased by 62% within 90 min after heat-shock (Figure 3). While a substantial number of aggregates formed immediately after heat-shock in both strains, the number of WT cells containing aggregates decreased rapidly while hsp104Δ cells failed to clear the observable puncta. These data corroborate the activity assay, which showed a 52% recovery of activity in WT versus a minimal 8% recovery in hsp104Δ strain.
Single cell automated microscopy revealed that in WT cells versus the hsp104Δ strain, FFL-GFP was solubilized at a higher rate (representative still images are shown in Figure 4; see supplemental movies for time-lapse series of FFL-GFP re-solubilization). In addition, the dynamics of the protein aggregates were very different; in the WT cells aggregates tended to fuse before being solubilized, and this was not observed in the hsp104Δ strain. This assay not only supports the results from the other two methods, but also reveals insight into a possible mechanism for how the aggregated protein is solubilized and refolded.
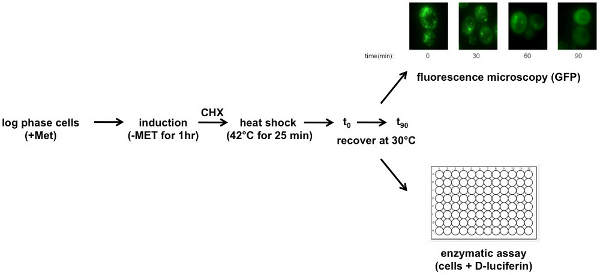 Figure 1. Schematic of FFL-GFP heat-shock recovery assay. FFL-GFP expression is induced in log phase in cells that have been incubated in SC-URA. To induce cells incubate in SC-URA-MET for 1 hr. Centrifuge cells and resuspend in SC-URA containing 100 μg/ml cycloheximide (CHX). For heat-shock, incubate cells at 42 °C for 25 min. Allow cells to recover by incubating at 30 °C. Collect samples for a 90 min time course. For enzymatic assays D-luciferin is added before each reading. Click here to view larger figure.
Figure 1. Schematic of FFL-GFP heat-shock recovery assay. FFL-GFP expression is induced in log phase in cells that have been incubated in SC-URA. To induce cells incubate in SC-URA-MET for 1 hr. Centrifuge cells and resuspend in SC-URA containing 100 μg/ml cycloheximide (CHX). For heat-shock, incubate cells at 42 °C for 25 min. Allow cells to recover by incubating at 30 °C. Collect samples for a 90 min time course. For enzymatic assays D-luciferin is added before each reading. Click here to view larger figure.
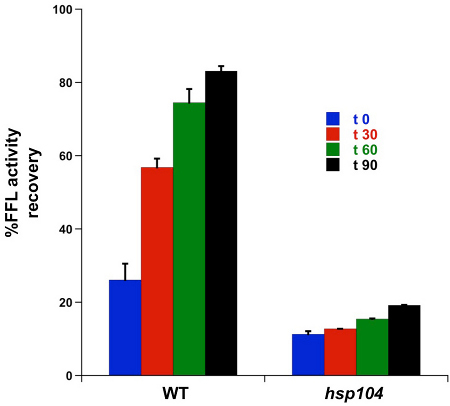 Figure 2. FFL-GFP enzymatic refolding/recovery assay. Samples of WT and hsp104Δ cells (100 μl) were collected prior to heat shock and immediately after heat-shock for each time point (0, 30, 60, and 90 min). Three replicates of each sample were aliquoted into wells of a 96-well white plate for each time point. For readings, the plate reader was set to measure luminescence every 30 min for 90 min at 30 °C.
Figure 2. FFL-GFP enzymatic refolding/recovery assay. Samples of WT and hsp104Δ cells (100 μl) were collected prior to heat shock and immediately after heat-shock for each time point (0, 30, 60, and 90 min). Three replicates of each sample were aliquoted into wells of a 96-well white plate for each time point. For readings, the plate reader was set to measure luminescence every 30 min for 90 min at 30 °C.
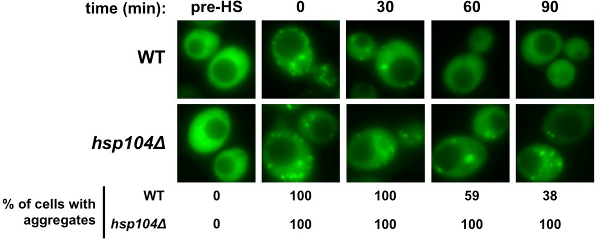 Figure 3. FFL-GFP refolding microscopy. 1 ml of cells at each time point (pre-HS, 0, 30, 60, and 90 min) were collected and centrifuged. Supernate was aspirated and cells were resuspended in 2 μl of water. Cells were prepared by mixing with 2 μl of low melt agarose. Cells were visualized using the 100X oil objective. Representative images of each time point are shown. Quantitation was done by counting cells in 4-5 fields and calculating the percent of cells containing aggregates (n~100).
Figure 3. FFL-GFP refolding microscopy. 1 ml of cells at each time point (pre-HS, 0, 30, 60, and 90 min) were collected and centrifuged. Supernate was aspirated and cells were resuspended in 2 μl of water. Cells were prepared by mixing with 2 μl of low melt agarose. Cells were visualized using the 100X oil objective. Representative images of each time point are shown. Quantitation was done by counting cells in 4-5 fields and calculating the percent of cells containing aggregates (n~100).
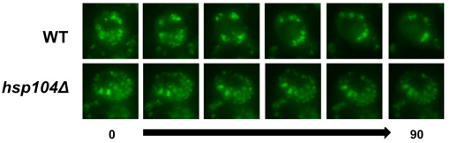 Figure 4. Single cell refolding microscopy time course of FFL-GFP. Post heat-shock 5 ml of cells were collected, centrifuged, and resuspended in 20 μl of ddH2O. 4 μl of cells were placed on a 5 mm2 section of SC-URA agar, and visualized using an automated fluorescent microscope over a 90 min period. The images are projections from a Z-stack taken during the time course.
Figure 4. Single cell refolding microscopy time course of FFL-GFP. Post heat-shock 5 ml of cells were collected, centrifuged, and resuspended in 20 μl of ddH2O. 4 μl of cells were placed on a 5 mm2 section of SC-URA agar, and visualized using an automated fluorescent microscope over a 90 min period. The images are projections from a Z-stack taken during the time course.
Discussion
In this article the model protein FFL-GFP was used to show that the yeast disaggregase, Hsp104 contributes to protein re-solubilization and repair. The enzymatic assays and microscopy differentially interrogated the status of the same substrate protein to determine refolding efficiency and yield. Results of the enzymatic recovery assays suggest that not only is the maximal recovery in the hsp104D mutant strain inefficient, but the initial magnitude of the unfolding stress was greater in the mutant strain (Figure 2). The analysis also shows that while hsp104D cells appeared to be attempting repair, they were unable to refold the protein as quickly as WT cells. This method allowed sensitive and quantitative analysis of FFL-GFP activity revealing the essential role of Hsp104 in repair of this protein.
Microscopy results suggest that the reason for the inefficient repair of denatured FFL enzyme is due to protein trapping within aggregates. The population analysis showed that in cells lacking Hsp104, no cells could completely clear all the aggregates in 90 min (Figure 3). In addition, the single cell analyses revealed that aggregates fuse over time, suggesting that consolidation of smaller aggregates into larger structures may aid the repair and refolding process (Figure 4). While this result is not obvious from the still images, time-lapse movies allow one to follow the dynamic behavior of independent aggregates. Observation of aggregates over the time course uncovered decreased aggregate fusion in the hsp104Δ strain, indicating that these cells do not form the larger structures as efficiently and suggesting that Hsp104 is required for this facet of protein quality control. A further speculation is that cells may solubilize protein more quickly from these larger aggregates, which may additionally contain the disaggregation and repair machinery. Single-cell microscopy can also be used to determine if other chaperones and co-chaperones are present in the larger versus smaller aggregates, and if this residency pattern varies over time.
Together these methods allow both biochemical and cell biological analysis of protein refolding and repair in living cells. Integration of the results from the three methods described affords multi-dimensional insight into the kinetics and efficiency of cellular recovery after proteotoxic heat-shock. Automation of some or all of the steps in the protocol also allows for greater sample sizes and biological replicates in a given experiment, increasing the robustness and ultimate confidence in the outcome. In addition, these methods theoretically can be extended to use in human cells and not only for genetic analyses, but also to investigate chemicals that alter proteostasis.
Disclosures
No conflicts of interest declared.
Acknowledgments
This work was supported by a grant from the National Institutes of Health (NIGMS-074696) to K.A.M. and an American Society of Microbiology Robert D. Watkins Graduate Student Research Fellowship to J.L.A. We also thank John Glover at the University of Toronto for providing the FFL-GFP source plasmid.
References
- Balch WE, Morimoto RI, Dillin A, Kelly JW. Adapting proteostasis for disease intervention. Science. 2008;319:916–919. doi: 10.1126/science.1141448. [DOI] [PubMed] [Google Scholar]
- Cashikar AG, Duennwald M, Lindquist SL. A chaperone pathway in protein disaggregation. Hsp26 alters the nature of protein aggregates to facilitate reactivation by Hsp104. J. Biol. Chem. 2005;280:23869–23875. doi: 10.1074/jbc.M502854200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti E, Lloyd LF, Akins J, Franks NP, Brick P. Crystallization and preliminary diffraction studies of firefly luciferase from Photinus pyralis. Acta Crystallogr. D. Biol. Crystallogr. 1996;52:876–878. doi: 10.1107/S0907444996002405. [DOI] [PubMed] [Google Scholar]
- Gietz RD, Schiestl RH, Willems AR, Woods RA. Studies on the transformation of intact yeast cells by the LiAc/SS-DNA/PEG procedure. Yeast. 1995;11:355–360. doi: 10.1002/yea.320110408. [DOI] [PubMed] [Google Scholar]
- Glover JR, Lindquist S. Hsp104, Hsp70, and Hsp40: a novel chaperone system that rescues previously aggregated proteins. Cell. 1998;94:73–82. doi: 10.1016/s0092-8674(00)81223-4. [DOI] [PubMed] [Google Scholar]
- Grantcharova V, Alm EJ, Baker D, Horwich AL. Mechanisms of protein folding. Curr. Opin. Struct. Biol. 2001;11:70–82. doi: 10.1016/s0959-440x(00)00176-7. [DOI] [PubMed] [Google Scholar]
- Kim S, Schilke B, Craig EA, Horwich AL. Folding in vivo of a newly translated yeast cytosolic enzyme is mediated by the SSA class of cytosolic yeast Hsp70 proteins. Proc. Natl. Acad. Sci. U.S.A. 1998;95:12860–12865. doi: 10.1073/pnas.95.22.12860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lum R, Niggemann M, Glover JR. Peptide and protein binding in the axial channel of Hsp104. Insights into the mechanism of protein unfolding. J. Biol. Chem. 2008;283:30139–30150. doi: 10.1074/jbc.M804849200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques SM, Esteves da Silva JC. Firefly bioluminescence: a mechanistic approach of luciferase catalyzed reactions. IUBMB Life. 2009;61:6–17. doi: 10.1002/iub.134. [DOI] [PubMed] [Google Scholar]
- Morimoto RI. The heat shock response: systems biology of proteotoxic stress in aging and disease. Cold Spring Harb. Symp. Quant. Biol. 2011;76:91–99. doi: 10.1101/sqb.2012.76.010637. [DOI] [PubMed] [Google Scholar]
- Nakatsu T, et al. Structural basis for the spectral difference in luciferase bioluminescence. Nature. 2006;440:372–376. doi: 10.1038/nature04542. [DOI] [PubMed] [Google Scholar]
- Nathan DF, Vos MH, Lindquist S. In vivo functions of the Saccharomyces cerevisiae Hsp90 chaperone. Proc. Natl. Acad. Sci. U.S.A. 1997;94:12949–12956. doi: 10.1073/pnas.94.24.12949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsell DA, Kowal AS, Singer MA, Lindquist S. Protein disaggregation mediated by heat-shock protein Hsp104. Nature. 1994;372:475–478. doi: 10.1038/372475a0. [DOI] [PubMed] [Google Scholar]
- Penna TC, Ishii M, Junior AP, Cholewa O. Thermal stability of recombinant green fluorescent protein (GFPuv) at various pH values. Appl. Biochem. Biotechnol. 2004. pp. 113–116. [DOI] [PubMed]
- Seliger HH, Buck JB, Fastie WG, McElroy WD. The Spectral Distribution of Firefly Light. J. Gen. Physiol. 1964;48:95–104. doi: 10.1085/jgp.48.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shorter J. The mammalian disaggregase machinery: Hsp110 synergizes with Hsp70 and Hsp40 to catalyze protein disaggregation and reactivation in a cell-free system. PLoS One. 2011;6:e26319. doi: 10.1371/journal.pone.0026319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tkach JM, Glover JR. Nucleocytoplasmic trafficking of the molecular chaperone Hsp104 in unstressed and heat-shocked cells. Traffic. 2008;9:39–56. doi: 10.1111/j.1600-0854.2007.00666.x. [DOI] [PubMed] [Google Scholar]
- Unno K, Kishido T, Hosaka M, Okada S. Role of Hsp70 subfamily, Ssa, in protein folding in yeast cells, seen in luciferase-transformed ssa mutants. Biol. Pharm. Bull. 1997;20:1240–1244. doi: 10.1248/bpb.20.1240. [DOI] [PubMed] [Google Scholar]
- Verghese J, Abrams J, Wang Y, Morano KA. Biology of the heat shock response and protein chaperones: budding yeast (Saccharomyces cerevisiae) as a model system. Microbiol. Mol. Biol. Rev. 2012;76:115–158. doi: 10.1128/MMBR.05018-11. [DOI] [PMC free article] [PubMed] [Google Scholar]