

Abstract
Retroviral gene transfer has proven therapeutic potential in clinical gene therapy trials but may also cause abnormal cell growth via perturbation of gene expression in the locus surrounding the insertion site. By establishing clonal marks, retroviral insertions are also used to describe the regenerative potential of individual cells. Deep sequencing approaches have become the method of choice to study insertion profiles in preclinical models and clinical trials. We used a protocol combining ligation-mediated polymerase chain reaction (LM-PCR) and pyrosequencing for insertion profiling and quantification in cells of various tissues transduced with various retroviral vectors. The presented method allows simultaneous analysis of a multitude of DNA-barcoded samples per pyrosequencing run, thereby allowing cost-effective insertion screening in studies with multiple samples. In addition, we investigated whether the number of pyrosequencing reads can be used to quantify clonal abundance. By comparing pyrosequencing reads against site-specific quantitative PCR and by performing spike-in experiments, we show that considerable variation exists in the quantification of insertion sites even when present in the same clone. Our results suggest that the protocol used here and similar approaches might misinterpret abundance clones defined by insertion sites, unless careful calibration measures are taken. The crucial variables causing this variation need to be defined and methodological improvements are required to establish pyrosequencing reads as a quantification measure in polyclonal situations.
Brugman and colleagues use a novel technique combining ligation-mediated polymerase chain reaction (LM-PCR) and 454 pyrosequencing for insertion profiling and quantification in cells of different tissues transduced with different retroviral vectors. Using this approach, they show that considerable variation exists in the quantification of insertion sites, even when present in the same clone.
Introduction
Retroviral marking has long been used as a tool to mark different clones in the hematopoietic system with the aim to study their fate in vivo (Schmidt et al., 2001; Barese et al., 2011). In addition, studies in freshly transduced cells revealed distinct integration patterns of various retroviral genera and derived gene vectors based on lentivirus (Schroder et al., 2002; Marshall et al., 2007), foamy virus (Beard et al., 2007), gammaretrovirus (Mitchell et al., 2004; Kustikova et al., 2007), and alpharetrovirus (Mitchell et al., 2004). Retroviral integration events may cause gene deregulation and disruption, and have thus been used to identify genes involved in leukemia and lymphoma development (Li et al., 2002; Lund et al., 2002). The clinical relevance of methods to detect retroviral marking methods became apparent, when they allowed the leukemic and preleukemic clones that were caused by insertional mutagenesis in gene therapy of X-linked severe combined immunodeficiency (SCID-X1) and X-linked chronic granulomatous disease (X-CGD) to be identified and monitored (Hacein-Bey-Abina et al., 2003; Ott et al., 2006; Howe et al., 2008; Stein et al., 2010; Wang et al., 2010).
The methods most often used for vector mark analysis are ligation-mediated polymerase chain reaction (LM-PCR) (Kustikova et al., 2009a) or the more sensitive linear amplification-mediated PCR (LAM-PCR) (Schmidt et al., 2007). In both methods, a resulting PCR product is generated starting from the known viral sequence. Restriction fragment length polymorphisms are introduced by digestion of the intermediate PCR products (LAM-PCR) or the flanking genomic DNA (LM-PCR) with various restriction endonucleases. In a next step, adapters are ligated to the resulting fragments and subsequently nested PCR with primers binding to linker and vector sequences is performed to generate amplicons of various lengths, which are then visualized on an agarose gel. The original methodology was shown to be limited by the availability of restriction sites in the DNA surrounding the retroviral integration site, which can be problematic when the aim is to identify the entire repertoire of insertions. However, this limitation could largely be overcome by the use of multiple restriction enzymes, because mixing of different enzymes minimizes the number of insertion sites that have no appropriate restriction site nearby (Harkey et al., 2007; Gabriel et al., 2009). Because the LAM-PCR and LM-PCR methods rely on nested exponential PCR, they are likely to be sensitive to differences in GC content and amplicon length. Quantification of clones therefore needs careful efficiency-corrected insertion-specific quantitative PCR (qPCR) measurements (Bozorgmehr et al., 2007; Kustikova et al., 2009b) to determine the contribution of a given clone to the entire repertoire of clones contributing to, for example, hematopoiesis. The development of pyrosequencing protocols for clonality studies (Schmidt et al., 2007; Wang et al., 2008; Uren et al., 2009) also allows different LM-PCR or LAM-PCR samples to be combined into one pyrosequencing library by the addition of DNA barcodes (Hamady et al., 2008; refined in Bystrykh, 2012) that mark individual PCRs. This allows the simultaneous analysis of a multitude of samples. Given the large number of sequences that can be analyzed at once, this technique facilitates clonal tracking in experiments with large numbers of samples. We employed an LM-PCR and 454 pyrosequencing protocol that allowed us to analyze a multitude of samples in a single 454 pyrosequencing run, facilitating the monitoring of multiple clones in cohorts of transplanted mice (Kustikova et al., 2009b; Maetzig et al., 2011; Suerth et al., 2012). Others have demonstrated the usefulness of pyrosequencing data as a surveillance tool in clinical gene therapy, for example, in clinical trials targeting adrenoleukodystrophy (Cartier et al., 2009), SCID-X1 (Hacein-Bey-Abina et al., 2010; Wang et al., 2010), severe combined immunodeficiency related to adenosine deaminase deficiency (SCID-ADA) (Biasco et al., 2011), β-thalassemia (Cavazzana-Calvo et al., 2010), X-CGD (Stein et al., 2010), and Wiskott–Aldrich syndrome (WAS) (Boztug et al., 2010). The authors of the aforementioned papers employed either LAM-PCR or LM-PCR coupled to pyrosequencing (Schmidt et al., 2007; Wang et al., 2008), using either barcoded primers or manufacturer-provided sample multiplexing procedures, and then calculated the fraction of read counts over total reads in a sample as a measure for abundance for specific clones. Alternatively, the more classical approach of quantification based on gel band density was performed (Stein et al., 2010). To provide reliable clonal surveillance data, it is important that the number of reads detected by pyrosequencing of LM-PCR or LAM-PCR samples have a tight correlation with the abundance of the clone in the sample. In addition to the field of gene therapy, the ability to track individual clones has important applications in stem cell biology, cancer biology, and aging research (Cavazzana-Calvo et al., 2010; Barese et al., 2011).
In the present study, we investigated the relation between pyrosequencing reads count and the actual clonal composition of samples, analyzing 20 clones that were tagged with gammaretroviral or lentiviral vectors. qPCR measurements were compared with reads obtained using our newly established LM-PCR and pyrosequencing protocols. In addition, we performed studies with a polyclonal lentivirally transduced liver sample and two sets of spike-in experiments, one with a gammaretrovirally tagged clone and one with an alpharetrovirally tagged clone, in which we analyzed the contribution of pyrosequencing reads from the clones with a known number of insertions compared with a polyclonal population of transduced cells.
The design of these spike-in experiments, with increasing presence of a dominant clone in a polyclonal background, is similar the clonal dynamics seen in the SCID-X1, X-CGD, and β-thalassemia gene therapy trials (Ott et al., 2006; Hacein-Bey-Abina et al., 2008; Howe et al., 2008; Cavazzana-Calvo et al., 2010), and should therefore give a good estimation of the performance of this method in gene therapy safety surveillance.
Materials and Methods
Retroviral transduction and transplantation of mouse bone marrow cells
Pyrosequencing of LM-PCR products was performed on peripheral blood and bone marrow obtained from mice that had been transplanted with LSK (lineageneg/loIL7RnegSca1highc-Kithigh) or short-term repopulating hematopoietic stem cells (ST-HSCs: LSK CD34+Flt3−) transduced with pRSF91.EGFP.wPRE gammaretroviral vector or pRRL.PPT.SF.EGFP.wPRE lentiviral vector as described previously (Schambach et al., 2006b; Kustikova et al., 2009b). The sorting procedure, transduction, transplantation, and sample collection were described previously (Kustikova et al., 2009b). In brief, sorted cells were immediately transduced with the pRRL.PPT.SF.EGFP.wPRE lentiviral vector or were cultured in StemSpan (Stem Cell Technologies, Grenoble, France) with murine stem cell factor (SCF, 50 ng/ml), human FLT3 ligand (FLT3L, 50 ng/ml), murine interleukin (IL)-3 (20 ng/ml), and human IL-11 (50 ng/ml) (all from Peprotech, Hamburg, Germany) for 2 days, after which the cells were transduced with the pRSF91.EGFP.wPRE gammaretroviral vector on two consecutive days (multiplicities of infection of 5 and 10 in the first and second transduction, respectively) on plates coated with RetroNectin CH296 (48 μg/cm2) (Takara Bio, Otsu, Japan). Peripheral blood samples were obtained 5, 11, 16, 23, and 27 or 8, 14, 20, 27, and 32 weeks posttransplantation. Genomic DNA isolation was performed with a DNeasy blood and tissue kit according to the instructions of the manufacturer (Qiagen, Hilden, Germany).
Lentiviral hepatocyte transduction
Hepatocytes were isolated from C57BL/6J mice by a modified two-step collagenase perfusion (Seglen, 1979) and plated on 6-well Primaria cell culture dishes (BD Biosciences, Heidelberg, Germany) at 1.1×106 viable cells per well in hepatocyte culture medium (HCM; Lonza, Basel, Switzerland) supplemented with 10% fetal bovine serum (FBS; PAA, Pasching, Austria). Cells were incubated at 37°C in a humidified environment at 5% CO2. After 2 hr the medium was changed to remove nonattached or dead cells in basal medium without FBS for 4 hr before overnight transduction with a self-inactivating lentiviral vector, pRRL.PPT.SF.eGFP.pre* (Schambach et al., 2006b). Cells were washed with phosphate-buffered saline (PBS; Invitrogen, Darmstadt, Germany) and harvested by incubation with 10×trypsin–EDTA solution (Biochrom, Berlin, Germany) at 37°C until hepatocytes detached from the culture plate. One milliliter of basal medium containing FBS was added to stop the reaction before the hepatocytes were centrifuged and used for DNA isolation with a DNeasy blood and tissue kit according to the instructions of the manufacturer (Qiagen). DNA (300 ng) from primary hepatocytes were digested with 5 U of the enzymes Tsp509I, TaqαI, and HaeIII (all from New England BioLabs, Ipswich, MA) according to the manufacturer's instructions for 5 hr and used for further integration site analysis.
Integration site determination and sequencing
Integration sites were identified by LM-PCR, using established methods (Kustikova et al., 2009a) with the following modifications: after the first exponential PCR, the internal control amplicon was digested with HindIII or SacI (New England BioLabs) at 37°C for 2 hr, after which the endonuclease was inactivated at 65°C for 20 min. The digested DNA was purified with a QIAquick PCR purification kit (Qiagen) and a secondary nested PCR was performed with barcoded primers carrying the primer A sequence and DNA barcode on the long terminal repeat (LTR) side and the primer B sequence on the linker side (according to Kustikova et al., 2009b), primers from Eurogentec (Seraing, Belgium) (primer sequences and multiplex primer design in Supplementary Tables S1 and S2; supplementary data are available online at http://www.liebertpub.com/hgtb). The secondary PCR products were then quantified with a NanoDrop spectrophotometer (Thermo Scientific, Wilmington, DE) and equal amounts of amplicons (1.7 ng/sample) were added for a total DNA amount of 500 ng. This amplicon library was then sequenced according to the manufacturer's standard protocols (AMPure bead amplification; Beckman Coulter, Bernried, Germany): median enrichment, 13.7% (5–21.1%) in the emulsion PCR; estimated product size of 300 bp, derived from Agilent DNA 1000 LabChip (Agilent, Böblingen, Germany) measurements of the library content. The library was then sequenced on a 454 genome sequencer FLX (Roche, Mannheim, Germany) according to the manufacturer's protocols. The resulting sequences belonging to the various samples were sorted according to their DNA barcode, using a custom BioPerl script.
Sequence clustering
With the sequencing parameters set up as described previously, we aimed for 20- to 50-fold oversampling, which was considered a good trade-off between the amount of samples present on the sequencing plate and the number of reads retrieved for every sequence. Because of this oversampling, every amplicon is represented by several sequences. To avoid the unnecessary alignment of identical sequences, we employed sequence-based clustering (CD-HIT [cluster database at high identity with tolerance; Li et al., 2006] settings: word size, 8; percentage identity, 95%). This resulted in the removal of identical sequences from the data set, while keeping track of the number of removed duplicated sequences. In addition, the contamination between samples present in the same pyrosequencing experiment was analyzed and collision sequences (identical sequences present in unique samples from independent transductions) and potential PCR artifacts were removed.
Sequence alignment and annotation
LTR and linker sequences were then removed from the sequences, using cross_match (P. Green, University of Washington, Seattle, WA; www.phrap.org). The resulting sequences were aligned to the mouse genome v37 database, obtained from the National Center for Biotechnology Information (NCBI, Bethesda, MD), using an in-house application running BLAST (Altschul et al., 1990) and using NCBI genome Map Viewer annotations. The alignment data set was further processed with R-2.10 (http://cran.r-project.org/). Identical BLAST alignments that occurred in multiple samples were considered artifacts and removed from the data. Of the aligned sequences, the alignment with the single lowest E value was considered to describe the vector insertion site. When multiple equal E values were observed, the alignment was considered ambiguous and removed from further analysis. Alignments that occurred within 20 bp in the same sample were considered to originate from the same vector insertion and the read numbers from these sequences were combined. For every insertion, the number of clustered sequences belonging to this sequence was annotated for quantification purposes.
Quantitative PCR on specific vector insertions: Site-specific qPCR
For selected insertions obtained from the mouse experiment, primers were designed to anneal to the LTR sequence and a sequence specific for the identified insertion (Kustikova et al., 2009b) (Supplementary Table S1). This allowed us to track the abundance of this specific sequence by SYBR green PCR (Qiagen), using the amount of the Flk1 genomic sequence as a reference for DNA amount. Abundance of specific clones was determined at five different time points after transplantation in peripheral blood samples and in the bone marrow (BM) obtained at the end of the experiment. The abundance of a clone was determined by the efficiency-corrected ΔCt method (Pfaffl, 2001). Similar site-specific qPCR measurements were performed for the lentivirally transduced hepatocyte samples (Supplementary Table S1).
Quantification of LM-PCR amplicons in polyacrylamide gels
To determine to what extent the LM-PCR procedure introduced variations in the abundance of specific insertion amplicons, we performed LM-PCR as described (Kustikova et al., 2009b), replacing the LTR primer in the nested PCR step of the protocol with a primer of the same sequence, labeled with IRDye 700 (gammaretroviral) or IRDye 800 (lentiviral) (both from Eurogentec). Because the fluorescent label present in the primer is present at only one position in the amplicon, this allowed us to more accurately determine the amount of amplicon obtained after the LM-PCR procedure. The amount of product was determined by fluorescence measurement in 10% polyacrylamide (Bio-Rad, Munich, Germany) DNA gels, using an Odyssey scanner (Li-Cor, Bad Homburg, Germany). Amplicon quantities are presented as median fluorescence per area for the amplicon of interest divided by the median fluorescence for the internal control amplicon.
Spike-in experiments
To determine the sensitivity of the presented method, two different spike-in experiment were performed. In one experiment we added known amounts of cells of a green fluorescent protein (GFP) clone (Modlich et al., 2006) to a polyclonal population of DsRed-transduced cells. In this experiment, the polyclonal pool was obtained from SC1 cells, cultured in Dulbecco's modified Eagle's medium (DMEM)–10% FCS, which were transduced once with a gammaretrovirus carrying pSRS11.SF.DsRed.PRE* (Schambach et al., 2006a). Afterward, DsRed-positive cells were sorted with a FACSAria cell sorter (BD Biosciences, San Jose, CA) and mixed with the GFP clone, mixing in 0.1, 0.3, 1, 3, 10, and 30%. Mixing ratios were confirmed by flow cytometry. From these cell mixtures, DNA was isolated with DNA extraction columns (Qiagen) and 100 ng of DNA was used for LM-PCR and pyrosequencing in triplicates, using Tsp509I restrictase. In the second experiment, an SC1 cell clone tagged with an alpharetroviral vector expressing enhanced green fluorescent protein (EGFP) from the spleen focus-forming virus promoter (Suerth et al., 2012) was obtained by limiting dilution. This clone was then mixed with bulk-transduced SC1 cells with the same avian sarcoma-leukosis viral (ASLV) vector and LM-PCR and pyrosequencing were performed. Restriction digest was performed with Tsp509I (New England BioLabs). LAM-PCR was performed as described (Schmidt et al., 2007) to compare against LM-PCR, and the internal control product was removed by digested with NotI (New England BioLabs) before the pyrosequencing library was generated.
Statistics
Pearson's product moment correlation coefficient and their associated p values between the qPCR, pyrosequencing, and gel density measurements were calculated and data analysis was performed in R-2.14 (R Foundation for Statistical Computing, 2012) and Perl. Insertion density data were collected with Odyssey software and image quantification was performed with ImageJ (Rasband, 1997–2012).
Results
Correlation of 454 reads and site-specific qPCR
In the current study we used a modified protocol of LM-PCR (Kustikova et al., 2009a) in combination with 454 pyrosequencing to investigate whether clones tagged by retrovirus insertion can be quantified using the number of reads that are obtained from 454 pyrosequencing (Fig. 1). To do so, we investigated the abundance of 20 different clones in genomic DNA samples obtained over time in a previously published experiment (Kustikova et al., 2009b), using efficiency-corrected integration site-specific qPCR (Supplementary Table S1). We compared the relative abundance retrieved this way against the number of 454 pyrosequencing reads obtained from the same samples after LM-PCR and pyrosequencing (Supplementary Table S3). To determine the exact amount of each respective amplicon after the LM-PCR procedure, we labeled the primer binding in the virus LTR region with IRDye and performed LM-PCR, followed by separation of the amplicons on a 10% polyacrylamide gel. This allowed us to measure the amplicon quantity without the need for product size correction. The gel fragments were then associated to the respective insertions by the size of the insert, which was previously sequenced and allowed us to identify the insertion site (Fig. 2A). The fluorescent signal for each of the amplicons was then quantified and normalized to the signal of the internal control. Because every amplicon has only one fluorescent tag, provided by the LTR primer, the measured fluorescence should be directly related to the amount of amplicon present. The signal is not related to the length of the amplicon, as opposed to the use of DNA dyes such as ethidium bromide. When comparing the data retrieved from qPCR with the normalized fluorescent signal from the polyacrylamide gels and the normalized pyrosequencing reads (Fig. 2B and C), considerable differences were found between the different quantification methods.
FIG. 1.
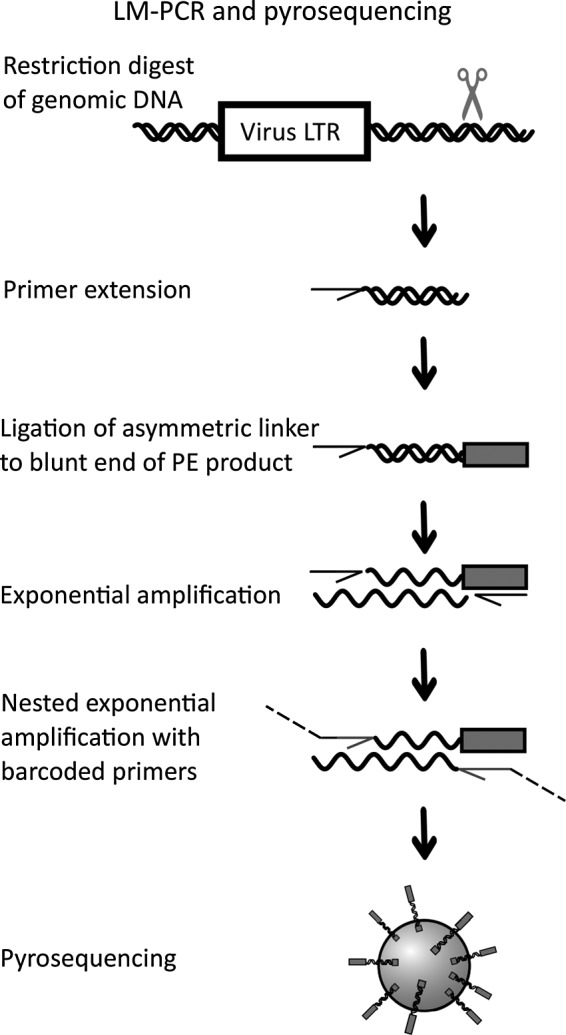
Experimental design. Ligation-mediated polymerase chain reaction (LM-PCR) is performed as previously described. The genomic DNA with integrated viral vectors is enzymatically digested. Starting from the long terminal repeat (LTR) of the integrated virus, linear amplification is performed with a biotinylated primer, resulting in a blunt product. Subsequently, an asymmetric linker cassette is ligated to the blunt end of the product. Exponential amplification is performed followed by a nested exponential PCR using primers with a DNA barcode and the pyrosequencing adapters. The resulting products of individual LM-PCRs were then mixed to constitute a library for pyrosequencing. PE, primer extension.
FIG. 2.

Different techniques for quantification of virally marked clones. Twenty vector insertion sites were analyzed by measuring the fluorescence of an IRDye 700-labeled primer in acrylamide gels, by measuring DNA content by site-specific qPCR and by counting pyrosequencing reads for a specific sequence. For the qPCR measurements, the insertion measurements were corrected for input DNA and subsequently normalized to the first time point measured. (A) Fluorescence measurements of polyacrylamide gels for samples of retrovirally transduced cells taken 5, 11, 16, 23, and 27 or 6, 12, 19, 26, and 31 weeks after transplantation. IC, internal LTR control. (B) Quantification of the fluorescent signal for the vector insertions identified on the gels in (A). (C) Amount of pyrosequencing reads for each vector insertion. The pyrosequencing reads were normalized to the highest number of sequences measured. (D) Comparison of normalized site-specific qPCR measurements and normalized pyrosequencing reads for the data shown in (A–C). For appropriate comparison, each time series was scaled to the highest value obtained in the time series. (E) Correlation comparison between fluorescence measurements on amplicons in acrylamide gels and normalized site-specific qPCR measurements. (F) Correlation comparison between fluorescence measurements of amplicons in the acrylamide gels and normalized pyrosequencing reads. The gray density in (D–F) shows a gridded bivariate interpolation of the R2 values, calculated for the correlation between the normalized number of pyrosequencing reads and the normalized clone abundance measured by qPCR, for each individual insertion. Higher R2 values are depicted with lighter shades.
Taking the qPCR data as the “gold standard” for quantification, similar to RT-qPCR being used for validation of gene expression microarrays, we normalized both the relative abundance of each clone measured by qPCR and the number of reads retrieved for this insertion by 454 pyrosequencing to the maximum quantity measured over time. Normalization was necessary, because both the maximum number of reads and the qPCR abundance vary and we intended to calculate Pearson's product moment correlation coefficient r for the data set consisting of the entire time series for all animals. To assess how well the 454 reads described the qPCR data, we also calculated r for each individual insertion. In 11 of 20 insertions studied, r was positive (meaning that an increase in the qPCR measurement results correlated with an increase in the pyrosequencing reads) and the Pearson correlation was p<0.05. In another 8 of 20 cases, the Pearson correlation was p>0.05 and we saw the sign of r change from positive to negative. This is counterintuitive, because it implies that an increase in pyrosequencing reads is in anticorrelation with the abundance of the insertion as measured by qPCR. In one case, LOC666619, r is negative and p<0.05, which indicates the existence of such an anticorrelation (Supplementary Tables S3 and S4).
We further investigated whether the relation between qPCR and pyrosequencing reads could be described best by a linear exponential log-linear model. Focusing on the residual sum of squares demonstrated that in each of the models, the parameters could be optimized to provide a good fit, which is clear because the sum-of-squares did not vary to a large extent (median standard deviation of the sum-of-squares divided by average sum-of-squares for all three models, 0.088; range, 0.003–1.068; Supplementary Fig. S1 and boldface in Supplementary Table S3).
Comparisons of the 454 reads with the qPCR data for the amplicons or the gel density also failed to show a stringent correlation (Fig. 2E). The overall r value calculated for nine clones for which accurate gel density measurements could be taken over all time points was 0.055, with p=0.592. The comparison of pyrosequencing reads and qPCR data shows a moderate correlation with r=0.515 and p=4.8×10−8. To investigate whether the absence of correlation already occurs at the level of the LM-PCR, we compared gel density measurements (Supplementary Table S5) with insertion-specific qPCR data (Fig. 2E). For nine clones for which accurate gel density measurements could be obtained over all time points, we obtained r=0.054 and p=0.592, showing that inaccuracy already exists at the LM-PCR level. A similarly poor correlation (r=0.1199, p=0.236) was also observed when gel density data were compared against pyrosequencing reads (Fig. 2F). Moreover, the individual R2 values of the site-specific qPCR to pyrosequencing read comparison, depicted by the shading in Fig. 2D–F, did not show a density of high R2 values along the diagonal. This indicates that the data points with better correlation between site-specific qPCR and pyrosequencing reads did not have high R2 values in the individual time series. These results show that pyrosequencing reads in the presented time course did not accurately represent the abundance of individual clones in a sample.
LM-PCR: 454 pyrosequencing sensitivity compared with site-specific qPCR in polyclonal samples
To address the question of sensitivity, we used a sample of hepatocytes, transduced with lentiviral vectors in which the integration sites were analyzed by LM-PCR–454 pyrosequencing. Clearly, the amount of reads that can be retrieved from one sample in a multiplexed setup is limited by the amount of genomic DNA used as input. In this experiment, 125 ng of genomic DNA containing amplicons was analyzed, resulting in 96,583 reads. Because 2078 different integrations were retrieved, this would yield 46 reads per integration. Because the lentivirally transduced hepatocytes investigated here were harvested shortly after transduction, large differences in abundances between the various transduced cells were not expected. Yet, the distribution of read numbers obtained from individual clones (Fig. 3A) showed that the majority of insertions had only one or two reads, whereas the maximum retrieved number of reads was 3574 (mean, 6.3; median, 2).
FIG. 3.

Quantification of lentiviral insertions from a lentiviral bulk transduction. From a bulk transduced sample of hepatocytes, virus insertions were determined by LM-PCR and pyrosequencing. (A) Distribution of reads associated with unique insertions. (B) Insertions with different amounts of reads associated with them were selected and the abundance of these cells carrying these insertions was measured by insertion site-specific qPCR. Gray circles indicate insertions associated with high numbers of pyrosequencing reads, but showing low abundance in qPCR.
Because of this unexpected distribution of read numbers, we investigated a set of insertions by insertion site-specific qPCR. A set of 15 different integrations was chosen (Table 1) in such a way that the read count ranged from low (1 read) to high (803 reads). In addition, the selected integrations had suitably long vector–genome boundaries so that site-specific qPCR primers could be designed (Supplementary Table S1). With these primers, site-specific qPCR was then performed on the original material also used for LM-PCR–454 pyrosequencing. The efficiency-corrected ΔCt method was used to measure the abundance of each of the 15 selected probes. A comparison of pyrosequencing reads and site-specific qPCR data showed, similar to the results obtained from the previously investigated gammaretroviral and lentiviral data set (Supplementary Table S3), that of the 15 insertions investigated, 3 had high read numbers (indicated in gray in Fig. 3B) but were actually present at low levels; the 10 other samples showed a low correlation (r=0.138, p=0.6687) with qPCR data. Furthermore, this experiment showed that in a set of insertions in which we expected no or little clonal expansion because these hepatocytes failed to divide in vitro (Rothe et al., 2012), we did find clones with associated high numbers of reads (Fig. 3A), which would have led us to the erroneous conclusion that expansion of several individual clones did occur. When verifying a sample of the lentiviral insertions in this experiment by qPCR, the discrepancy between reads and qPCR data was clear (Fig. 3B) and showed that the clone we selected for verification was indeed present in much lower amounts than the pyrosequencing reads suggested. Taken together these data again show, now using a different tissue and a different vector, that considerable differences exist between site-specific qPCR quantification and pyrosequencing reads and that a relatively large bias is contained in the data gathered by this approach.
Table 1.
Pyrosequencing Reads and Quantitative PCR Abundance for 15 Lentiviral Insertions
| Sequence no. | Abundance (efficiency-corrected qPCR) | Number of pyrosequencing reads |
|---|---|---|
| 1 | 0.425 | 803 |
| 2 | 20.627 | 211 |
| 3 | 0.114 | 123 |
| 4 | 0.154 | 105 |
| 5 | 0.369 | 91 |
| 6 | 0.295 | 27 |
| 7 | 1.000 | 26 |
| 8 | 0.266 | 20 |
| 9 | 0.402 | 17 |
| 10 | 0.021 | 12 |
| 11 | 0.000 | 5 |
| 12 | 0.002 | 4 |
| 13 | 0.108 | 1 |
| 14 | 0.259 | 1 |
| 15 | 0.295 | 1 |
Spike-in experiments to model clonal complexity
To establish the relation between LM-PCR–454 pyrosequencing and the number of clones inserted, we set up a spike-in experiment that should yield similar sample complexity as that observed in mice repopulated with a limited repertoire of stem cells (Kustikova et al., 2007; Maetzig et al., 2011). To this end, we took a well-established hematopoietic clone carrying eight known retrovirus insertions (Supplementary Fig. S2), which expresses GFP from a gammaretroviral vector (Modlich et al., 2006), as spike-in material and transduced freshly isolated lineage− cells with a gammaretroviral vector expressing DsRed (pRSF91.DsRed.wpre*) as the polyclonal background. The transduced cells were then sorted for DsRed and mixed with the GFP clone at ratios ranging from 30 to 0.1% in triplicates (Fig. 4A). Genomic DNA was prepared to perform LM-PCR and pyrosequencing. Clustered sequences were mapped using an in-house–developed pipeline for BLAST alignment and annotation using NCBI Map View data. To obtain an accurate measurement, the number of retrieved reads for a specific clone was divided by the total number of reads obtained for that sample. All of the insertions depicted in Fig. 4B should have been retrieved at the same rate, because they are present in one clone. Interestingly, LM-PCR and pyrosequencing methods generated a biased recovery that favored reads for some insertions. This resulted in an 11.7-fold difference in read retrieval rate (range, 1.59×10−3 to 2.04×10−2) with an average standard deviation in the triplicates of 33% of the mean of each sample for the example shown in Fig. 4B. Six of the insertions that were retrieved in the 30% spike-in sample could also be identified in the 10% spike-in sample (Fig. 4C). The expected dose response was found in four of these six insertions, where the average corrected reads increased approximately 3-fold, as expected, but showed considerable variation between the samples. In the 30% spike-in sample, 7.8±0.7% (Table 2, mean±standard deviation) of the reads were retrieved from the clone, whereas in the 10% spike-in sample 1.8±0.5% (mean±standard deviation) of the reads could be attributed to the clone.
FIG. 4.

Quantification differences in a spike-in experiment. (A) The spike-in experiments were performed by mixing a clone with known vector insertions with a bulk transduced population, and the resulting mixture was analyzed by LM-PCR–pyrosequencing. (B) Percentage of total reads for the 30% spike-in sample, in triplicate, for the insertions in the clone, identified by the closest gene to the insertion. Bars indicate sample means. (C) Comparison of the percentage of total reads in the 10 and 30% spike-in samples. The insertions shown as solid circles demonstrate a clear increase in percentage of total reads, whereas the insertions shown as gray circles did not show a comparable increase.
Table 2.
Contribution of Pyrosequencing Reads for a 30% Spike-In of a Clone with Eight Known Insertions
| Insertion | Reads for this insertion | % Spike-in | Total reads per sample | Reads (corrected for total reads per sample) | % of total | Average % |
|---|---|---|---|---|---|---|
| Akap6 | 55 | 30 | 2373 | 0.023 | 2.318 | 2.050 |
| 32 | 30 | 2431 | 0.013 | 1.316 | ||
| 73 | 30 | 2901 | 0.025 | 2.516 | ||
| Gm885 | 29 | 30 | 2373 | 0.012 | 1.222 | 1.563 |
| 29 | 30 | 2431 | 0.012 | 1.193 | ||
| 66 | 30 | 2901 | 0.023 | 2.275 | ||
| LOC100042073 | 30 | 30 | 2373 | 0.013 | 1.264 | 1.232 |
| 39 | 30 | 2431 | 0.016 | 1.604 | ||
| 24 | 30 | 2901 | 0.008 | 0.827 | ||
| Dnm2 | 21 | 30 | 2373 | 0.009 | 0.885 | 1.083 |
| 34 | 30 | 2431 | 0.014 | 1.399 | ||
| 28 | 30 | 2901 | 0.010 | 0.965 | ||
| Supt16h | 22 | 30 | 2373 | 0.009 | 0.927 | 1.080 |
| 21 | 30 | 2431 | 0.009 | 0.864 | ||
| 42 | 30 | 2901 | 0.014 | 1.448 | ||
| AI480653 | 11 | 30 | 2373 | 0.005 | 0.464 | 0.335 |
| 9 | 30 | 2431 | 0.004 | 0.370 | ||
| 5 | 30 | 2901 | 0.002 | 0.172 | ||
| Wdr45 | 9 | 30 | 2373 | 0.004 | 0.379 | 0.278 |
| 6 | 30 | 2431 | 0.002 | 0.247 | ||
| 6 | 30 | 2901 | 0.002 | 0.207 | ||
| Lig3 | 5 | 30 | 2373 | 0.002 | 0.211 | 0.160 |
| 4 | 30 | 2431 | 0.002 | 0.165 | ||
| 3 | 30 | 2901 | 0.001 | 0.103 |
Spike-in experiment using an alpharetroviral vector
To rule out specific effects related to the vector or clone used in this setting, a similar experiment was performed with a clone tagged with an alpharetroviral vector (Suerth et al., 2012). DNA from a previously established clone was diluted in bulk transduced cells (30% EGFP-positive cells) in concentrations ranging from 100 to 0.1%. The clone contained an insertion near the Irf8 gene and one near LOC666779. LM-PCR and LAM-PCR were performed as described and the read numbers specific for these two insertions were identified. Figure 5A and B shows that both insertions in the alpharetroviral clone were amplified by LM-PCR in a similar fashion, but differences in pyrosequencing read counts are obvious. The difference between the reads obtained for the Irf8 and LOC666779 insertions, which are both present only once in the clone, ranged from 3.2 to 18% (Table 3) with consistently higher reads for the LOC666779 insertion.
FIG. 5.

Alpharetroviral spike-in experiment. A clone with two vector insertions was spiked in at 0.1, 1, 10, 50, and 100% in a population of bulk transduced cells. (A) Percentage of spike-in compared with the fraction of total pyrosequencing reads, which were obtained by LM-PCR and pyrosequencing using Tsp509I for the restriction digest. Squares indicate the LOC666779 insertion and triangles indicate the Irf8 insertion. (B) An agarose electrophoresis gel fluorescence image, showing the increasing contribution of the two insertions with increased spike-in percentages of the clone. In (C) and (D), instead of LM-PCR, LAM-PCR was performed. (C) Percentage of spike-in, compared with the fraction of total reads; (D) agarose electrophoresis gel fluorescence image.
Table 3.
Contribution of Specific Pyrosequencing Reads in Avian Sarcoma-Leukosis Virus Spike-In Experiment
| % Spike-in | Irf8 (% of total reads) | LOC666779 (% of total reads) | % difference between clones | % of total reads used occupied by clone | |
|---|---|---|---|---|---|
| LM PCR | 100 | 33.6 | 52.3 | 18.6 | 85.9 |
| 50 | 20.3 | 26.9 | 6.6 | 47.2 | |
| 10 | 5.4 | 12.8 | 7.4 | 18.1 | |
| 1 | 0.0 | 3.2 | 3.2 | 3.2 | |
| 0.1 | 0.0 | 0.0 | NA | 0.0 | |
| LAM PCR | 100 | 14.0 | 82.4 | 68.5 | 96.4 |
| 50 | 7.3 | 18.3 | 10.9 | 25.6 | |
| 10 | 1.1 | 2.9 | 1.7 | 4.0 |
PCR, polymerase chain reaction; LAM-PCR, linear amplification-mediated PCR; LM-PCR, ligation-mediated PCR.
To investigate whether LAM-PCR would reduce variations between samples, we replaced the LM-PCR protocol by LAM-PCR. The LAM-PCR protocol resulted in similar results as LM-PCR, showing consistent higher reads for the LOC666779 insertion (Fig. 5C and D), but a higher variation (1.7–68%) between the two insertions (Table 3).
In the LM-PCR experiments, we observed that the total number of reads associated with the clonal insertions was disproportionately higher than the percentage spike-in contribution when less than 10% of the sample consisted of the clone, whereas LAM-PCR gave consistent underestimation of the clone (Table 3). Differences between amplification methods might therefore result in differences in the estimated clonal contribution.
Typical PCR parameters do not explain the inconsistency of the quantification method
To find parameters that would allow us to correct the quantification differences between the site-specific qPCR quantification and the amount of pyrosequencing reads, we analyzed a series of parameters relevant to the qPCR, such as qPCR efficiency, GC percentage, and amplicon length, together with parameters relevant to the pyrosequencing method, such as GC percentage and amplicon length in the LM-PCR product or pyrosequencing product (Fig. 6). These parameters were then compared with the Pearson correlation coefficient (R2) for the time series shown in Fig. 2, revealing no evidence that they contribute to the correlation. In addition, multiple linear regression analysis of the data in Fig. 6 did not lead us to a model that explained the differences in correlation between the pyrosequencing reads and the abundance of a specific clone (data not shown) even if we focused on the 11 insertions with good correlation between pyrosequencing reads and qPCR data (Supplementary Fig. S3). Other sources of variation, probably inherent to the highly competitive nature of the multiple PCRs, may thus contribute to the observed inaccuracy of read numbers in polyclonal samples.
FIG. 6.

Correlation plots for various amplicons, showing amplicon length, GC percentage in amplicon, qPCR efficiency for each specific amplicon, the GC percentage in the LM-PCR product, the length of the LM-PCR product, the length of the resulting pyrosequencing product, and the Pearson correlation coefficient (RSQ) between the qPCR data and the pyrosequencing data in the 20 time courses measured.
Discussion
By adapting previously published procedures of LM-PCR and 454 pyrosequencing (Kustikova et al., 2009b; Maetzig et al., 2011), we have established a method to analyze multiple samples in a single 454 pyrosequencing run, which allowed us to retrieve up to 2700 sequences per sample. The clonal repertoire reflected at this sequencing depth, however, is not exhaustive, partly because of the limitations imposed by the use of a given specific restriction enzyme (Harkey et al., 2007; Gabriel et al., 2009; Wu et al., 2013) and partly because of sampling issues. Moreover, the LM-PCR method can be skewed, as can any method that employs PCR amplification of intermediate products, toward amplification of smaller PCR products, among other limitations (Berry et al., 2012; Bystrykh et al., 2012), although we could not show that small amplicons had an effect on the extent to which the pyrosequencing reads correlate with the qPCR data (Supplementary Fig. S3). If the size of the LM-PCR amplicons indeed has an impact on the frequency with which a sequence is retrieved by deep sequencing (Berry et al., 2012), it will become necessary to use methods that randomly shear or amplify DNA rather than the restriction enzyme-based methods.
Experimental (Cornils et al., 2013) as well as clinical studies (Cartier et al., 2009; Boztug et al., 2010; Cavazzana-Calvo et al., 2010; Wang et al., 2010) have used the number of reads acquired from deep sequencing experiments as a quantitative measure of abundance. However, our analysis revealed that for quantification purposes, the simple analysis of read numbers seems to be insufficient to determine the abundance of a clone, because our locus-specific qPCR experiments showed that only 55% (11 of 20) of the traced insertions showed a positive correlation and a p<0.05 (Fig. 2D and Supplementary Table S4). In addition, the two spike-in experiments demonstrated that the amount of reads varies for each specific insertion site, even when the contribution of each of these insertions was equal, because the insertions were derived from the same clone. We observed that the method of insertion amplification, LM-PCR or LAM-PCR, might influence the estimates of the size of a clone, but this effect was small when compared with the differences seen between the different insertions in a clone.
Our PCR product quantification by density measurements after electrophoretic separation showed only a moderate concordance with site-specific qPCR for individual insertions (Fig. 2B). These differences are also observed when comparing site-specific qPCR and pyrosequencing reads (Fig. 2C and D). It therefore seems likely that the LM-PCR procedure already introduces differences, with possible additional errors introduced by the pyrosequencing. The presence of the errors introduced by LM-PCR together with pyrosequencing means that tracking insertions in mixed populations of clones after LM-PCR is inadvisable unless the source of variation can be defined and methods are developed to overcome this limitation. We thus suggest appropriate controls such as site-specific qPCR on insertions of interest (Ott et al., 2006; Kustikova et al., 2009b). One study looking into chemoselection of hematopoietic clones also discussed whether pyrosequencing reads could be used for quantification of clones (Giordano et al., 2011). The authors of this study concluded that abundances estimated by qPCR and pyrosequencing were similar. Closer inspection of the data, however, shows that although the three investigated insertions tended to show similar estimates of abundance, the variation between the methods was considerable, which was also reflected in our data.
The current protocol, combining LM-PCR with 454 pyrosequencing, was set up to establish repertoires of insertions in samples from mouse experiments and aimed to obtain a digital equivalent of the LM-PCR gel pictures. If the main focus of pyrosequencing experiments is on quantification, it would be advisable to reduce the number of PCR cycles and perhaps take samples for pyrosequencing after fewer PCR cycles in the LM-PCR procedure (Fig. 1), because less amplification might result in fewer artifacts introduced by PCR. The 454 pyrosequencing protocol does, however, contain a PCR step as well, which might introduce differences in abundance measurements similar to those observed here. A similar method, which uses sequencing of both ends of the vector integration, was developed; however, this methodology showed good correlation only for some of the insertions tested (Kim et al., 2010). Several theoretical constraints that complicate insertion analysis and quantification, such as genome coverage and PCR bias, have been summarized in a review (Bystrykh et al., 2012).
Two further complications introduced by the pyrosequencing approach need to be taken into account. First, with an increasing number of multiplexed samples, the amount of reads per sample decreases, because of the upper limit of sequences that can be retrieved. In our spike-in experiments, this was already apparent in the range of 2373–29,010 sequences per sample, because we were not able to retrieve all insertions below the 10% spike-in sample. For quantification purposes it would be advisable to maximize the number of reads per sample rather than the number of samples per sequencing run. Second, there is an ongoing debate as to which statistical method is best to describe abundance and variation in deep sequencing of insertion sites. In high-throughput RNA sequencing (RNA-Seq), several parameters for abundance have been investigated with their associated error models (Robinson et al., 2007; Marioni et al., 2008), yet studies on the quantification of insertion sites use the ratio of specific reads over total sample reads. It has yet to be determined which model is best suited for the quantification of insertion site sequences.
Newly developed techniques, such as nonrestrictive LAM-PCR (Gabriel et al., 2009), a phage Mu transposition-based method (Brady et al., 2011), shear-splink (Koudijs et al., 2011), and Re-free LAM-PCR (Wu et al., 2013), eliminate the restriction enzyme bias and may thus also temper PCR-associated bias related to GC content, secondary structure, and amplicon length. As an alternative to the vector mark, vectors might be supplied with DNA barcodes (Gerrits et al., 2010; Lu et al., 2011). Because these barcodes vary only in a small number of bases, the effect of these differences on PCRs used in pyrosequencing should be minimal. The design of the barcoded vector does, however, introduce a problem for therapeutic gene therapy vectors, because the barcode, by its very nature, causes sequence inhomogeneity within the vector product and thereby fails to comply with regulations for product consistency (European Medicines Agency, 2012). As with the LM-PCR method described here, such newly developed techniques should be experimentally validated. Methodological biases in the quantitative description of clonal inventories could thus be minimized, greatly improving the usefulness of high-throughput polyclonal tracking in stem cell biology, cancer models, and clinical monitoring of gene therapy patients.
Supplementary Material
Acknowledgments
The authors thank Thomas Neumann and Maike Stahlhut for excellent technical support, and Sabrina Woltemate (Department of Medicinal Microbiology, Hannover Medical School) for performing the 454 pyrosequencing runs. This work was supported by grants from the DFG (SPP1230 DF BA1837/7-2), the BMBF (iGene), and the EU (Clinigene, Persist, Cell-Pid).
Author Disclosure Statement
All authors have read the manuscript and agree with its submission. The authors have no competing financial interests.
References
- Altschul S.F. Gish W. Miller W., et al. Basic Local Alignment Search Tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Barese C.N. Dunbar C.E. Contributions of gene marking to cell and gene therapies. Hum. Gene Ther. 2011;22:659–668. doi: 10.1089/hum.2010.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beard B.C. Keyser K.A. Trobridge G.D., et al. Unique integration profiles in a canine model of long-term repopulating cells transduced with gammaretrovirus, lentivirus, or foamy virus. Hum. Gene Ther. 2007;18:423–434. doi: 10.1089/hum.2007.011. [DOI] [PubMed] [Google Scholar]
- Berry C.C. Gillet N.A. Melamed A., et al. Estimating abundances of retroviral insertion sites from DNA fragment length data. Bioinformatics. 2012;28:755–762. doi: 10.1093/bioinformatics/bts004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biasco L. Ambrosi A. Pellin D., et al. Integration profile of retroviral vector in gene therapy treated patients is cell-specific according to gene expression and chromatin conformation of target cell. EMBO Mol. Med. 2011;3:89–101. doi: 10.1002/emmm.201000108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozorgmehr F. Laufs S. Sellers S.E., et al. No evidence of clonal dominance in primates up to 4 years following transplantation of multidrug resistance 1 retrovirally transduced long-term repopulating cells. Stem Cells. 2007;25:2610–2618. doi: 10.1634/stemcells.2007-0017. [DOI] [PubMed] [Google Scholar]
- Boztug K. Schmidt M. Schwarzer A., et al. Stem-cell gene therapy for the Wiskott-Aldrich syndrome. N. Engl. J. Med. 2010;363:1918–1927. doi: 10.1056/NEJMoa1003548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady T. Roth S.L. Malani N., et al. A method to sequence and quantify DNA integration for monitoring outcome in gene therapy. Nucleic Acids Res. 2011;39:e72. doi: 10.1093/nar/gkr140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bystrykh L.V. Generalized DNA barcode design based on Hamming codes. PLoS One. 2012;7:e36852. doi: 10.1371/journal.pone.0036852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bystrykh L.V. Verovskaya E. Zwart E., et al. Counting stem cells: Methodological constraints. Nat. Methods. 2012;9:567–574. doi: 10.1038/nmeth.2043. [DOI] [PubMed] [Google Scholar]
- Cartier N. Hacein-Bey-Abina S. Bartholomae C.C., et al. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science. 2009;326:818–823. doi: 10.1126/science.1171242. [DOI] [PubMed] [Google Scholar]
- Cavazzana-Calvo M. Payen E. Negre O., et al. Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia. Nature. 2010;467:318–322. doi: 10.1038/nature09328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornils K. Bartholomae C.C. Thielecke L., et al. Comparative clonal analysis of reconstitution kinetics after transplantation of hematopoietic stem cells gene marked with a lentiviral SIN or a γ-retroviral LTR vector. Exp. Hematol. 2013;41:28–38. doi: 10.1016/j.exphem.2012.09.003. e3. [DOI] [PubMed] [Google Scholar]
- European Medicines Agency. Reflection paper on design modifications of gene therapy medicinal products during development. 2012. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/02/WC500122743.pdf http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/02/WC500122743.pdf
- Gabriel R. Eckenberg R. Paruzynski A., et al. Comprehensive genomic access to vector integration in clinical gene therapy. Nat. Med. 2009;15:1431–1436. doi: 10.1038/nm.2057. [DOI] [PubMed] [Google Scholar]
- Gerrits A. Dykstra B. Kalmykowa O.J., et al. Cellular barcoding tool for clonal analysis in the hematopoietic system. Blood. 2010;115:2610–2618. doi: 10.1182/blood-2009-06-229757. [DOI] [PubMed] [Google Scholar]
- Giordano F.A. Sorg U.R. Appelt J.-U., et al. Clonal inventory screens uncover monoclonality following serial transplantation of MGMT P140K-transduced stem cells and dose-intense chemotherapy. Hum. Gene Ther. 2011;22:697–710. doi: 10.1089/hum.2010.088. [DOI] [PubMed] [Google Scholar]
- Hacein-Bey-Abina S. von Kalle C. Schmidt M., et al. A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. N. Engl. J. Med. 2003;348:255–256. doi: 10.1056/NEJM200301163480314. [DOI] [PubMed] [Google Scholar]
- Hacein-Bey-Abina S. Garrigue A. Wang G.P., et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J. Clin. Invest. 2008;118:3132–3142. doi: 10.1172/JCI35700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacein-Bey-Abina S. Hauer J. Lim A., et al. Efficacy of gene therapy for X-linked severe combined immunodeficiency. N. Engl. J. Med. 2010;363:355–364. doi: 10.1056/NEJMoa1000164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamady M. Walker J.J. Harris J.K., et al. Error-correcting barcoded primers for pyrosequencing hundreds of samples in multiplex. Nat. Methods. 2008;5:235–237. doi: 10.1038/nmeth.1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harkey M.A. Kaul R. Jacobs M.A., et al. Multiarm high-throughput integration site detection: Limitations of LAM-PCR technology and optimization for clonal analysis. Stem Cells Dev. 2007;16:381–392. doi: 10.1089/scd.2007.0015. [DOI] [PubMed] [Google Scholar]
- Howe S.J. Mansour M.R. Schwarzwaelder K., et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J. Clin. Invest. 2008;118:3143–3150. doi: 10.1172/JCI35798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S. Kim N. Presson A.P., et al. High-throughput, sensitive quantification of repopulating hematopoietic stem cell clones. J. Virol. 2010;84:11771–11780. doi: 10.1128/JVI.01355-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koudijs M.J. Klijn C. van der Weyden L., et al. High-throughput semiquantitative analysis of insertional mutations in heterogeneous tumors. Genome Res. 2011;21:2181–2189. doi: 10.1101/gr.112763.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kustikova O.S. Geiger H. Li Z., et al. Retroviral vector insertion sites associated with dominant hematopoietic clones mark “stemness” pathways. Blood. 2007;109:1897–907. doi: 10.1182/blood-2006-08-044156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kustikova O.S. Modlich U. Fehse B. Retroviral insertion site analysis in dominant haematopoietic clones. Methods Mol. Biol. 2009a;506:373–390. doi: 10.1007/978-1-59745-409-4_25. [DOI] [PubMed] [Google Scholar]
- Kustikova O.S. Schiedlmeier B. Brugman M.H., et al. Cell-intrinsic and vector-related properties cooperate to determine the incidence and consequences of insertional mutagenesis. Mol. Ther. 2009b;17:1537–1547. doi: 10.1038/mt.2009.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W. Godzik A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- Li Z. Dullmann J. Schiedlmeier B., et al. Murine leukemia induced by retroviral gene marking. Science. 2002;296:497. doi: 10.1126/science.1068893. [DOI] [PubMed] [Google Scholar]
- Lu R. Neff N.F. Quake S.R. Weissman I.L. Tracking single hematopoietic stem cells in vivo using high-throughput sequencing in conjunction with viral genetic barcoding. Nat. Biotechnol. 2011;29:928–933. doi: 10.1038/nbt.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund A.H. Turner G. Trubetskoy A., et al. Genome-wide retroviral insertional tagging of genes involved in cancer in Cdkn2a-deficient mice. Nat. Genet. 2002;32:160–165. doi: 10.1038/ng956. [DOI] [PubMed] [Google Scholar]
- Maetzig T. Brugman M.H. Bartels S., et al. Polyclonal fluctuation of lentiviral vector-transduced and expanded murine hematopoietic stem cells. Blood. 2011;117:3053–3064. doi: 10.1182/blood-2010-08-303222. [DOI] [PubMed] [Google Scholar]
- Marioni J.C. Mason C.E. Mane S.M., et al. RNA-seq: An assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008;18:1509–1517. doi: 10.1101/gr.079558.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall H.M. Ronen K. Berry C., et al. Role of PSIP1/LEDGF/p75 in lentiviral infectivity and integration targeting. PLoS One. 2007;2:e1340. doi: 10.1371/journal.pone.0001340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell R.S. Beitzel B.F. Schroder A.R., et al. Retroviral DNA integration: ASLV, HIV, and MLV show distinct target site preferences. PLoS Biol. 2004;2:E234. doi: 10.1371/journal.pbio.0020234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modlich U. Bohne J. Schmidt M., et al. Cell-culture assays reveal the importance of retroviral vector design for insertional genotoxicity. Blood. 2006;108:2545–2553. doi: 10.1182/blood-2005-08-024976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott M.G. Schmidt M. Schwarzwaelder K., et al. Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nat. Med. 2006;12:401–409. doi: 10.1038/nm1393. [DOI] [PubMed] [Google Scholar]
- Pfaffl M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasband W.S. ImageJ. U.S. National Institutes of Health; Bethesda, MD: 1997–2012. [Google Scholar]
- R Foundation for Statistical Computing. R: A Language and Environment for Statistical Computing. Vienna, Austria: 2012. [Google Scholar]
- Robinson M.D. Smyth G.K. Moderated statistical tests for assessing differences in tag abundance. Bioinformatics. 2007;23:2881–2887. doi: 10.1093/bioinformatics/btm453. [DOI] [PubMed] [Google Scholar]
- Rothe M. Rittelmeyer I. Iken M., et al. Epidermal growth factor improves lentivirus vector gene transfer into primary mouse hepatocytes. Gene Ther. 2012;19:425–434. doi: 10.1038/gt.2011.117. [DOI] [PubMed] [Google Scholar]
- Schambach A. Bohne J. Baum C., et al. Woodchuck hepatitis virus post-transcriptional regulatory element deleted from X protein and promoter sequences enhances retroviral vector titer and expression. Gene Ther. 2006a;13:641–645. doi: 10.1038/sj.gt.3302698. [DOI] [PubMed] [Google Scholar]
- Schambach A. Galla M. Modlich U., et al. Lentiviral vectors pseudotyped with murine ecotropic envelope: Increased biosafety and convenience in preclinical research. Exp. Hematol. 2006b;34:588–592. doi: 10.1016/j.exphem.2006.02.005. [DOI] [PubMed] [Google Scholar]
- Schmidt M. Hoffmann G. Wissler M., et al. Detection and direct genomic sequencing of multiple rare unknown flanking DNA in highly complex samples. Hum. Gene Ther. 2001;12:743–749. doi: 10.1089/104303401750148649. [DOI] [PubMed] [Google Scholar]
- Schmidt M. Schwarzwaelder K. Bartholomae C., et al. High-resolution insertion-site analysis by linear amplification-mediated PCR (LAM-PCR) Nat. Methods. 2007;4:1051–1057. doi: 10.1038/nmeth1103. [DOI] [PubMed] [Google Scholar]
- Schroder A.R. Shinn P. Chen H., et al. HIV-1 integration in the human genome favors active genes and local hotspots. Cell. 2002;110:521–529. doi: 10.1016/s0092-8674(02)00864-4. [DOI] [PubMed] [Google Scholar]
- Seglen P.O. Hepatocyte suspensions and cultures as tools in experimental carcinogenesis. J. Toxicol. Environ. Health. 1979;5:551–560. doi: 10.1080/15287397909529766. [DOI] [PubMed] [Google Scholar]
- Stein S. Ott M.G. Schultze-Strasser S., et al. Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat. Med. 2010;16:198–204. doi: 10.1038/nm.2088. [DOI] [PubMed] [Google Scholar]
- Suerth J.D. Maetzig T. Brugman M.H., et al. Alpharetroviral self-inactivating vectors: Long-term transgene expression in murine hematopoietic cells and low genotoxicity. Mol. Ther. 2012;20:1022–1032. doi: 10.1038/mt.2011.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uren A.G. Mikkers H. Kool J., et al. A high-throughput splinkerette-PCR method for the isolation and sequencing of retroviral insertion sites. Nat. Protocols. 2009;4:789–798. doi: 10.1038/nprot.2009.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G.P. Garrigue A. Ciuffi A., et al. DNA bar coding and pyrosequencing to analyze adverse events in therapeutic gene transfer. Nucleic Acids Res. 2008;36:e49. doi: 10.1093/nar/gkn125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G.P. Berry C.C. Malani N., et al. Dynamics of gene-modified progenitor cells analyzed by tracking retroviral integration sites in a human SCID-X1 gene therapy trial. Blood. 2010;115:4356–4366. doi: 10.1182/blood-2009-12-257352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C. Jares A. Winkler T., et al. High efficiency restriction enzyme-free linear amplification-mediated polymerase chain reaction approach for tracking lentiviral integration sites does not abrogate retrieval bias. Hum. Gene Ther. 2013;24:38–47. doi: 10.1089/hum.2012.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.