

Abstract
Molecular dynamics (MD) simulations were carried out to compare the free and bound structures of wild type U1A protein with several Phe56 mutant U1A proteins that bind the target stem loop 2 (SL2) RNA with a range of affinities. The simulations indicate the free U1A protein is more flexible than the U1A-RNA complex for both wild type and Phe56 mutant systems. A complete analysis of the hydrogen bonding (HB) and non-bonded (VDW) interactions over the course of the MD simulations suggested that changes in the interactions in the free U1A protein caused by the Phe56Ala and Phe56Leu mutations may stabilize the helical character in loop 3, and contribute to the weak binding of these proteins to SL2 RNA. Compared to wild type, changes in HB and VDW interactions in Phe56 mutants of the free U1A protein are global, and include differences in β-sheet, loop 1 and loop 3 interactions. In the U1A-RNA complex, the Phe56Ala mutation leads to a series of differences in interactions that resonate through the complex, while the Phe56Leu and Phe56Trp mutations cause local differences around the site of mutation. The long-range networks of interactions identified in the simulations suggest that direct interactions and dynamic processes in both the free and bound forms contribute to complex stability.
Introduction
One of the most common eukaryotic RNA binding domains is the RNA recognition motif (RRM), also known as the RNA binding domain (RBD) or the ribonucleoprotein (RNP) domain.1, 2 Proteins containing this particular motif bind single-stranded RNA and are involved in post-transcriptional regulation of gene expression.3 The prototype RRM protein, U1A, is a component of the spliceosome, which excises introns from pre-mRNA prior to gene expression in eukaryotes.4–6 Extensive structural and experimental studies have been performed on the protein U1A,7–28 though a general understanding of RNA binding and recognition at the molecular level remains elusive.
The N-terminal RRM of U1A binds with high affinity and specificity to stem loop 2 (SL2) of U1 snRNA as well as two adjacent internal loops in the 3'-untranslated region of its own pre-mRNA.29–31 These target sites contain nearly identical sequences in the loop, AUUGCAC closed by a CG base pair. The structure of the N-terminal RRM of U1A (residues 2–117) has been determined by NMR spectroscopy32 and X-ray crystallography (residues 3–92),33 and the complex of the N-terminal RRM of U1A (residues 2–97) with SL2 RNA (21 bases) (U1A-RNA) has been determined by X-ray crystallography,34 albeit with two surface mutations. The structure of U1A is shown in Figure 1a and the structure of the U1A-RNA complex is shown in Figure 1b, with the secondary structure of SL2 RNA inset.
Figure 1.

(a) Structure of the N-terminal RRM of U1A. (b) Structure of the N-terminal RRM of U1A bound to SL2 RNA. Inset: Nucleic acid sequence of SL2 RNA with nucleotides recognized by U1A for binding highlighted in red.
There are two highly conserved aromatic amino acids in U1A, Tyr13 and Phe56, which participate in stacking interactions with C5 and A6, respectively, in the RNA target site. The contribution of the conserved aromatic amino acid Phe56 to the affinity and specificity of U1A for SL2 RNA has been previously investigated.35 Mutation of Phe56 to Ala reduced the binding affinity by 5.5 kcal/mol, while mutation of Phe56 to Leu also destabilized the complex, by 4.1 kcal/mol. In contrast, mutation to Trp had little effect on complex stability.35 It was determined that the aromatic group is a stronger contributor to binding affinity in U1A-RNA than individual hydrogen bond donors and acceptors.35, 36 Double mutant studies suggested that Phe56 plays a complex role in target site recognition by contributing to binding affinity by stacking with A6 and participating in energetically coupled interactions with the functional groups on A6.36 Non-polar base isosteres were substituted for A6 with little loss of binding affinity, which is evidence that increased hydrophobicity can compensate for the loss of hydrogen-bonding interactions.37 Altogether, this data points to an important role for the conserved Phe56 residue in U1A-RNA binding.
In order to interpret the experimental results on the effect of mutations on U1A-RNA binding, it is necessary to know the structures of the various mutant species in solution. In the absence of experimental structure determination, molecular dynamics (MD) simulations can be used to predict solution structures with considerable accuracy. A number of theoretical and computational studies on the U1A-RNA system have been reported previously; 17–19, 21, 22, 25–27, 38–45 however, none have provided an overall analysis of the contributions of specific interactions to binding. To investigate this problem further, MD simulations were performed on U1A and the Phe56Ala, Phe56Leu and Phe56Trp mutants free and bound to SL2 RNA, followed by a complete analysis of the hydrogen bonding and non-bonded interactions for each system. Analysis of the results indicates that a comparison of the mutant interactions to those in the wild type system provides insight into U1A-RNA recognition.
Methods
Molecular Dynamics Simulations
The initial structure for the MD simulation of free wild type U1A was based on the NMR solution structure of the unbound protein,32 PDBID:46 1FHT. Model 5 was selected as the representative structure based on analysis of the 43 NMR structures by NMRCLUST 2.1.47 The U1A NMR structure was truncated from residues 2 – 117 to residues 2 – 102 to match the construct used in the binding affinity experiments. Point mutations were introduced at the Phe56 position in this structure to create the Phe56Ala, Phe56Leu and Phe56Trp mutant forms of free U1A.
The initial structure for the MD simulation of wild type U1A-RNA complex reported earlier was based on the X-ray cocrystal structure of the N-terminal RRM of U1A bound to SL2 of U1 snRNA solved at 1.92 Å resolution,34 PDB ID:46 1URN. Biological unit 2 was chosen for the initial structure as it contains the most complete structural information for SL2 RNA. The U1A protein was extended from the crystal structure construct to obtain a structure containing residues 2 – 102 to match that used in the binding affinity experiments, and two point mutations (His31Tyr and Arg36Gln) were introduced to revert the protein to the wild type sequence. Details of these procedures have been described previously.48 The question naturally arises if the previous MD simulation was stable and converged. To investigate this issue, we carried out 10 additional MD simulations on wild type U1A, each beginning with a different distribution of initial velocities. A superposition of the 10 average structures formed from the equilibrated regions of the trajectories showed that the simulations were stable within an RMSD of 1.5±0.6 Å. In addition, we extended our original simulation to 100 ns. The RMSD from the initial structure is stable at ~ 2.1 Å RMSD, consistent with that found in our earlier MD (2.3 Å). We divided this 100ns trajectory into tracts of 10 ns each and found them all to agree within an RMSD of 0.9±0.2 Å. The results of all of these additional simulations, a principal component analysis of the 100 ns trajectory, as well as the overlays of structural snapshots are included in the supplementary material available on the Biopolymers Website.
Point mutations were introduced at the Phe56 position in this structure to create the Phe56Ala, Phe56Leu and Phe56Trp mutant complexes and MD simulations were performed using the same protocol as above, AMBER 949 with the force field ff99SB, the latest modification to ff99 backbone torsions by Simmerling and coworkers50 for the protein and the parmbsc0 force field51 for RNA. The systems were solvated in a box of explicit TIP3P52 water molecules that extended a minimum distance of 12 Å from the solute atoms. Neutralizing Na+ or Cl− ions were added to each system, after which Na+ and Cl− ions were added to each system to obtain a salt concentration of 250 mM, in accordance with the conditions of binding affinity experiments. Additional details of the minimization and MD procedures are described in Pitici et al.48 The results presented here, as in our previous MD simulations on wild type U1A, are based on production runs of 10 ns.
Hydrogen Bond and Non-bonded Interactions
Snapshots were collected from each MD trajectory for analysis at 5 ps intervals for a total of 2000 structures each. The program HBPLUS53 in NUCPLOT54 was used to calculate the hydrogen bonds and non-bonded contacts present in each snapshot, enabling the calculation of a percentage of each interaction over the 2000 snapshots. HBPLUS calculates all hydrogen atom (H) positions and counts the interaction as a hydrogen bond if specific criteria are satisfied with respect to donor (D) and acceptor (A) atom positions. The criteria for a hydrogen bonding interaction used in this study include a H-A distance < 2.7 Å, a D-A distance < 3.35 Å, a D-H-A angle > 90°, and a H-A-AA angle > 90°, where AA is the atom attached to the acceptor. The criterion for a VDW interaction in this study included atoms within 3.9 Å of each other.
Results
RMSD Plots
The MD trajectories computed for this study include free wild type U1A and the wild type U1A-RNA complex, as well as those of three U1A mutants, Phe56Ala, Phe56Leu and Phe56Trp. MD simulations were performed with explicit water and counterions. The root-mean-square deviations (RMSD) of the protein backbone residues Phe8 – Lys98 and RNA backbone atoms from the equilibrated structures were calculated over the course of the trajectories and are plotted in Figure 2. N- and C-terminal protein residues were omitted from the RMSD analysis due to their high flexibility throughout the MD simulations, which causes high RMSD values that are not indicative of significant structural changes of interest in the proteins or protein-RNA complexes.
Figure 2.

Plots of the RMSD of protein backbone residues Phe8 – Lys98 and RNA backbone atoms from the equilibrated structures calculated over the course of MD trajectories. (a) Free U1A protein and Phe56 mutants. (b) U1A-RNA complex and Phe56 mutants.
The RMSD plots of the free Phe56Ala and Phe56Leu U1A proteins stabilize after 0.5 ns, while the wild type and Phe56Trp mutants take 4 ns to stabilize (Figure 2a). The higher RMSD of the wild type system after 4 ns is due to a change in the position of helix C compared to the initial structure. The helix positions itself more horizontally across the β-sheet (relative to the orientation of helix C in Figure 1a). The slightly smaller RMSD of the Phe56Trp system after 4 ns is due to a small change in the position of helix C, moving further away from loop 3, and due to changes in the positions of the loop 3 residues. Comparison to previous MD studies on this system48, 55 indicate good agreement with an average RMSD value of 2.3 Å for the wild type U1A protein.
The RMSD plots of the Phe56Leu and Phe56Trp U1A-RNA mutant complexes (Figure 2b) are quite stable over the 10 ns trajectories. The wild type U1A-RNA complex stabilizes after 5 ns, and has larger deviations from the equilibrated structure after that point. Comparing the average structures over these two time frames, the main difference is due to the positions of the dynamic U8, C9 and C10 bases in the RNA loop. The Phe56Ala U1A-RNA mutant complex stabilizes after 2 ns, but also has larger deviations from the equilibrated structure throughout the course of the rest of the trajectory. These larger deviations are due to more fluctuations in the RNA stem. During this trajectory, A15 flips out of the RNA stem, which enables A16 to stack with A-5 from the opposite strand. Similar fluctuations of the RNA stem are seen in the trajectory of the free SL2 RNA (results not shown), but are not observed in any of the other trajectories of the wild type and mutant U1A-RNA complexes. Further analysis of RMSF and average structures (Figures 3 and 5, detailed below) indicate these RNA base fluctuations in the stem do not affect the backbone conformation of the RNA stem or loop 1 residues, and should not impact the results or conclusions of this paper. Validation of the U1A-RNA MD trajectory for the wild type is provided by comparison to previous MD studies on this system.56–60 The average RMSD of 1.9 Å is consistent with these prior studies. The average RMSD of the Phe56Ala mutant complex from the initial structure is 2.4 Å, larger than wild type and the other mutants (Phe56Leu = 1.6 Å, Phe56Trp = 1.6 Å).
Figure 3.

Root-mean-square fluctuations (RMSF) computed on a residue or nucleotide basis with reference to the average structure calculated over the 10 ns simulations of (a) free U1A protein and Phe56 mutants and (b) U1A-RNA complex and Phe56 mutants.
Figure 5.

Overlay of time averaged structures: (a) free U1A protein and Phe56 mutants. (b) U1A-RNA complex and Phe56 mutants. White: NMR structure of the free protein and X-ray crystal structure of the U1A-RNA complex, respectively; Black: Wild Type; Green: Phe56Ala; Blue: Phe56Leu; Red: Phe56Trp.
RMSF Plots
Root-mean-square fluctuations (RMSF) on a per residue or per nucleotide basis were computed for free wild type and Phe56 mutant U1A protein and U1A-RNA complex trajectories with reference to their respective average structures (Figures 3a and b, respectively). The largest fluctuations in the free proteins occur in the flexible N- and C-terminal regions of the U1A protein, and in the flexible loop regions. The fluctuations of the helix C residues in wild type and Phe56Leu mutant U1A proteins are larger than those of the Phe56Ala and Phe56Trp mutants, indicating greater fluctuations of these residues from their average structure positions. Of the residues involved in loop regions, those in loop 3 have the greatest fluctuations. The largest fluctuations in the U1A-RNA complexes again occur in the N- and C-terminal regions of the protein, as well as in the 5′ and 3′ ends of the SL2 RNA stem and the U8/C9/C10 loop nucleotides, which do not interact with the protein.
Secondary Structure Plots
Secondary structures were computed over the course of each 10 ns trajectory for free wild type and Phe56 mutant U1A proteins and U1A-RNA complexes (Figures 4a and b, respectively) using the program STRIDE61 in the timeline extension of VMD.62 In the free proteins, Figure 4a, the four β-strands that form the β-sheet and the three α-helices are readily apparent throughout the course of the wild type and Phe56 mutant trajectories. Loop 3 residues, which connect β2-β3, fluctuate between turn and 310 helix character throughout the simulations of the Phe56Ala and Phe56Leu mutants. Within loop 3, the Leu49-Lys50-Met51 residues have an average of 64% and 49% 310 helix character throughout the Phe56Ala and Phe56Leu simulations, respectively, while wild type and Phe56Trp have an average of 0.2% and 5%, respectively.63 Loop 5 in the Phe56Trp mutant, which connects helix B to β4, exhibits different secondary structure behavior than the other free U1A proteins. The residues in loop 5 exhibit both turn and antiparallel β-sheet characteristics. The Phe77-Tyr78-Asp79-Lys80 residues in Phe56Trp exhibit a very high average percentage of turn character (> 85%), while in the other systems, only the Tyr78-Asp79 residues exhibit > 85% of turn character and the Phe77 and Lys80 residues exhibit antiparallel β-sheet characteristics. The residues in Loop 6 in the Phe56Trp mutant also exhibit higher percentage of turn character than the other free U1A proteins.
Figure 4.

Secondary structure of U1A and Phe56 mutant U1A proteins calculated over the course of each 10 ns trajectory:  = α-helix,
= α-helix,  = 310 helix,
= 310 helix,  = extended conformation (β-strand),
= extended conformation (β-strand),  = turn, and
= turn, and  = random coil. (a) Free U1A protein and Phe56 mutants. (b) U1A-RNA complex and Phe56 mutants.
= random coil. (a) Free U1A protein and Phe56 mutants. (b) U1A-RNA complex and Phe56 mutants.
In the U1A-RNA complexes, Figure 4b, the β-sheet and three α-helices are again readily apparent. The N-terminal residues, though flexible as seen in the RMSF plot (Figure 3b), have an average of 46% 310 helical character and 53% turn character across the trajectories. Helix C, however, loses its stability in both the wild type and Phe56Leu simulations. This loss of stability occurs at approximately 7 ns in the wild type system and at approximately 4.5 ns in the Phe56Leu system. This behavior has not been observed in other wild type or mutant simulations of these systems.16–29
Average Structure Overlays
Average structures were computed for all systems over the stable portion of the trajectories, as determined by the RMSD plots. The free U1A protein and U1A-RNA complex average structures were superimposed and can be found in Figure 5a and b, respectively. In the overlay of the free proteins, which includes the wild type and Phe56 mutants as well as the NMR solution structure,32 the βαββαβ motif superimposes quite well. The regions of the protein that differ significantly in the superposition correspond to those regions of the protein that have the largest RMSFs in Figure 3a: the N- and C-terminal residues, loop 3, and helix C. The differences in position of the helix C residues, which have been rendered in a tubular form for clarity, are especially striking. In the average structures of all of the systems, helix C has relaxed from its initial position in the NMR structure. The NMR structure that was chosen for the starting structure, model 5, was one of 43 structures in the 1FHT PDB file, which show helix C in a variety of positions. These observations indicate significant flexibility of helix C in the U1A protein in solution. The loop 5 residues in the Phe56Trp mutant, shown in red, are also in a different position than is observed in the rest of the systems. These are the residues that had different secondary structure characteristics in the Phe56Trp mutant compared to the other systems.
In the overlay of the U1A-RNA complexes (Figure 5b), which includes the wild type and Phe56 mutants as well as the X-ray crystal structure,34 again the βαββαβ motif superimposes quite well. The regions of the protein and SL2 RNA that differ significantly in the superposition correspond to those regions that have the largest RMSFs in Figure 3b: the C-terminal residues, helix C, the 5′ and 3′ ends of the RNA stem, and the U8/C9/C10 loop nucleotides, which do not interact with the protein. In contrast to the overlay of the average structures of the free proteins, the N-terminal and loop 3 residues superimpose very well in the overlay of the complexes. For the N-terminal residues, this is due to the turn and helical character induced upon SL2 RNA binding, observed in the secondary structure plots (Figure 4b), and for loop 3, this is due to the interactions between several loop 3 residues and SL2 RNA. For the systems in which helix C remains intact throughout the simulation, Phe56Ala and Phe56Trp, the position of this helix does not change from the starting position as drastically as is observed in the free protein systems. It is also obvious from the overlay of these average structures that helix C in the wild type and Phe56Leu systems does not maintain its helical character, as was noted in the secondary structure analysis.
Evaluation of Hydrogen Bonding and Non-bonding Interactions
The previous analyses have indicated quite a number of differences between the simulations of the wild type and mutant systems. To determine whether these differences in the average structures of the wild type and Phe56 mutant U1A proteins and U1A-RNA complexes are representative of differences observed throughout the 10 ns simulations and if they are caused by the mutation at position 56 or due to normal fluctuations throughout the simulations, an analysis of the hydrogen-bonding and non-bonded interactions was carried out.
Residues Phe8 – Lys98 and the RNA nucleotides were the focus of the following analysis of H-bond and VDW interactions. The N- and C-terminal residues were neglected in this analysis due to their significant flexibility throughout the simulation, which is indicated in the preceding results of the RMSF. The flexibility of these terminal residues leads to large differences in interactions between the systems that are not of particular interest to this study. In the analyses of the U1A-RNA complex systems, the helix C residues Ile93 – Lys98 were not included in the Phe56Leu analysis since they are unstructured through most of the trajectory (Figure 4). The helix C residues Met97 – Lys98 were not included in Phe56Ala and Phe56Trp U1A-RNA complex analyses if they were interacting with other helix C residues since these residues are less structured in the trajectory of the wild type system.
Wild type MD interactions compared to experimental structures
The percentage of hydrogen-bonding (HB) and non-bonded (VDW) interactions were computed for 2000 structures extracted from the 10 ns MD simulation of wild type U1A and compared to those computed for the 43 structures obtained from the NMR structure of the free protein,32 PDB ID: 1FHT. The top left half of Figure 6 illustrates the differences in interactions between the experimental NMR and MD structures, with crosses and blue dots indicating greater and fewer than 50% differences, respectively. There are a large number of differences as may be expected due merely to the differences in sampling or because the free U1A protein has several regions that are less well-defined due to their flexibility, including the N- and C-terminal residues, loops 1 and 3, and helix C.32, 48, 55 Most of the differences in interactions occur in the flexible loop regions of the protein, in helix C, or are due to flexible side chain orientations, detailed in the supplementary material, Tables S1 and S2. These differences tend to be compensatory. For instance, if the percent difference between the total number of HB and VDW interactions present in the NMR structures and MD simulation is averaged, there are 2% more HB interactions and 4% fewer VDW interactions in the MD simulation compared to the NMR structures.
Figure 6.
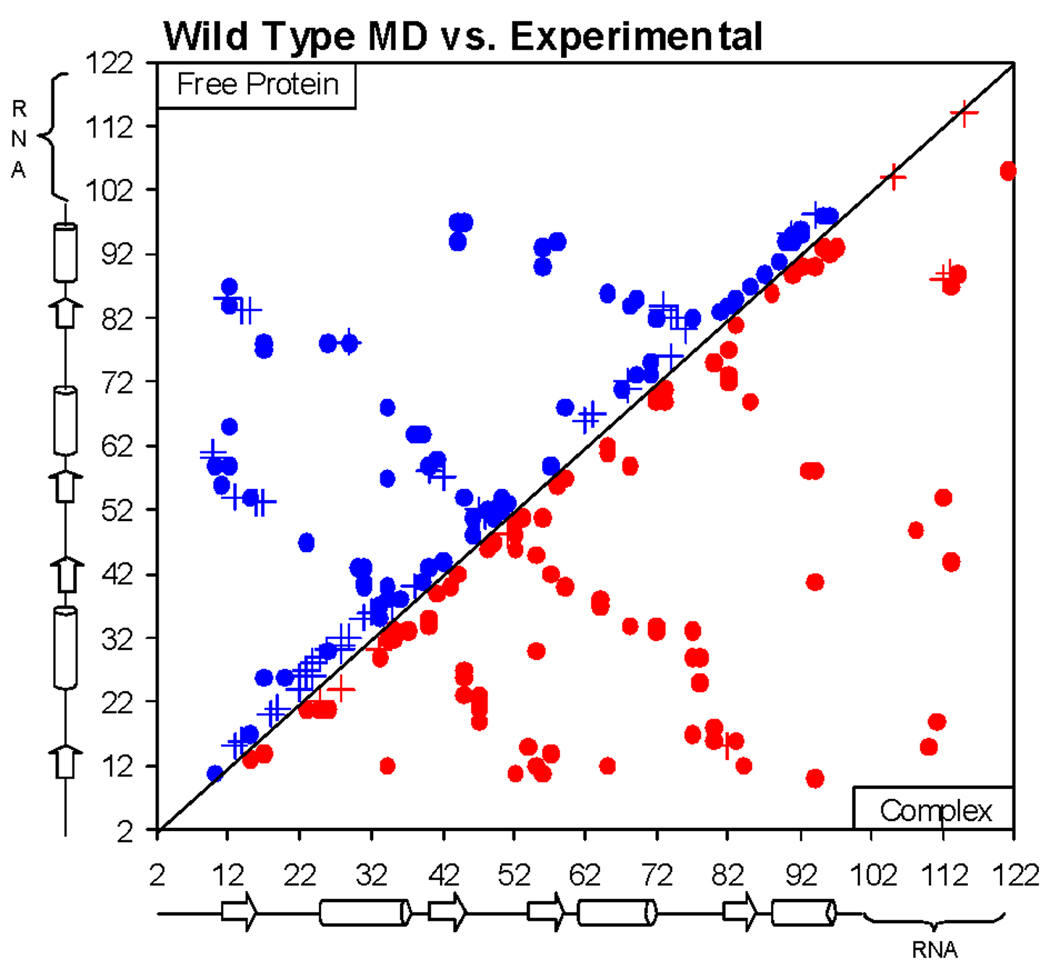
Plot of differences in HB and VDW interactions in MD simulations compared to experimental structures. Crosses and solid dots indicate greater and fewer than 50% differences, respectively. Upper left: NMR structures compared to MD simulation of free wild-type U1A protein. Bottom right: X-ray crystal structures compared to MD simulation of wild-type U1A-RNA complex.
The percentage of HB and VDW interactions were also compared between the 2000 structures extracted from the 10 ns MD simulation of wild type U1A and for the three structures obtained from the asymmetric unit of the X-ray co-crystal structure of the U1A-RNA complex,34 PDB ID: 1URN. Of these three structures, pdb2 contains the most structural information. Nucleotides C14 and U21 in SL2 RNA were unresolved in pdb1 and pdb3 due to disorder, and only residues Ala2 – Met97 were well-defined in the protein part in all three complexes. In addition, two point mutations were made in 1URN to aid in crystallization, Tyr31His and Gln36Arg. These differences were all taken into account upon comparing differences in interactions between the X-ray crystal structures and the MD structures.
As with the free protein, there are a number of differences in interactions between the MD simulation and the X-ray crystal structures. The bottom right half of Figure 6 illustrates the differences in interactions between the experimental and MD structures, with crosses and red dots indicating greater and fewer than 50% differences, respectively, in the MD simulation compared to the X-ray crystal structure. Averaging the percent difference between the total number of HB and VDW interactions present in the X-ray crystal structures and the MD simulation indicates 2% more HB interactions and 26% fewer VDW interactions in the MD simulation compared to the X-ray crystal structures. In actuality, the difference in VDW interactions is not as large as it seems. The majority of the VDW interactions in the X-ray crystal structures are very close to the cutoff distance of 3.9 Å, so upon relaxing in the MD simulation, the distances increase to just over the 3.9 Å cutoff. Other differences are due to the flexibility of residue side chains and nucleotide bases through the MD simulation. These interactions are detailed in the supplementary material, Tables S3 and S4. This analysis suggests that the MD model obtained for the solution structure compares well with the experimental structures. Most force field issues will cancel out between the mutants since only relevant differences are revealed.
Unique Phe56 U1A mutant interactions compared to wild type
Phe56Ala free protein
Table 1 lists the HB and VDW interactions that are more than 50% different in the 2000 snapshots sampled from the free Phe56Ala mutant U1A trajectory compared to the wild type system. The residues involved in these interactions are illustrated in Figure 7a. The main differences are clustered around the Phe56Ala mutation site on β1, β2 and β3, and extend to loop 1, loop 3, helix A and helix C. There are fewer interactions between Tyr13 and Ala56 due to the size difference between the much smaller Ala amino acid compared to the wild type Phe at position 56. There are different interactions between helix C and the β-sheet, specifically Val45 and Ala55 with Met97. Since the mutant has a smaller amino acid at position 56, helix C is able to move closer to the β-sheet, enabling these interactions. This can also be seen in the overlay of the average structures (Figure 5a), where helix C is in a different position in the average structure of the Phe56Ala mutant with the C-terminal residues of the helix closer to the β-sheet. These differences in the interactions of β2, β3 and helix C residues could contribute to the differences in the interactions of the loop 3 residues, including their H-bonding patterns: Ser46, Arg47, Met51, Arg52 and Gln54. Loop 3 is more structured and less flexible in the Phe56Ala mutant (Figure 3a) and has more helical content than is seen in the wild type (Figure 4a).
Table 1.
Differences in interactions (>50%) observed in free Phe56Ala U1A simulation compared to wild type
| Residues | Positions | Type of interaction | Difference |
|---|---|---|---|
| Tyr13 – Ala56 | β1 to β3 | VDW | − 85% |
| Ala55 – Met97 | β3 to αC | VDW | + 74% |
| Val45 – Met97 | β2 to αC | VDW | + 63% |
| Ser46 – Gln54 | loop 3 | H-bond (N of Ser46 – O of Gln54) | + 87% |
| Ser46 – Met51 | loop 3 | H-bond (OG of Ser46 – O of Met51) | + 95% |
| Ser46 – Met51 | loop 3 | VDW | + 53% |
| Arg47 – Arg52 | loop 3 | VDW | − 69% |
| Lys23 – Ser46 | αA to loop 3 | H-bond (NZ of Lys23 – O of Ser46) | + 52% |
| Lys23 – Ser46 | αA to loop 3 | VDW | + 58% |
| Asn18 – Lys20 | loop 1 | H-bond (N of Lys20 – OD1 of Asn18) | − 63% |
| Asn18 – Ile21 | loop 1 | VDW | − 53% |
Figure 7.

Positions of residues in Phe56Ala mutant U1A trajectories that differ in H-bond and VDW interactions (> 50%) compared to wild type. Residues are colored according to location in the protein for clarity. (a) Free protein. (b) U1A-RNA complex.
Phe56Ala complex
Table 2 lists the H-bonding and VDW interactions that are more than 50% different in the 2000 snapshots sampled from the Phe56Ala U1A-RNA complex trajectory compared to the wild type system. The residues involved in these interactions are illustrated in Figure 7b. Table 2 indicates the net loss of a series of interactions that cascade through the Phe56Ala U1A-RNA complex resulting from the single Phe56Ala mutation starting with Ala56, through Tyr13 and SL2 RNA to specific loop 1 and loop 3 residues. The gain in H-bonding interactions between Ile33 and Gln36 in helix A does not appear to be caused by the Phe56Ala mutation because Ile33 and Gln36 are far removed spatially in the protein from the other residues that are affected by the Ala mutation, and there is no cascade through differences in interactions originating from the Ala mutation to these residues. The bottom half of Table 2 consists of SL2 RNA stem residues in the Phe56Ala U1A-RNA complex simulation that differ from the wild type simulation by greater than 50%. The terminal SL2 RNA stem bases fluctuate more than the wild type during this simulation. A15 flips out from the double stranded helical stem, which enables the dangling A16 base to stack with A-5 from the other strand. It is difficult to attribute these differences in the simulation to the Phe56Ala mutation since they are removed from the residues and nucleotides in the top of Table 2 that showed differences in interactions compared to the wild type system. The RMSF and average structures (Figures 3 and 5) indicate these RNA base fluctuations in the stem do not affect the backbone conformation of the RNA stem or loop 1 residues and should not affect the fluctuations of the residues in Table 2.
Table 2.
Differences in interactions (>50%) observed in Phe56Ala U1A-RNA complex simulation compared to wild type
| Residues | Positions | Type of interaction | Difference |
|---|---|---|---|
| Tyr13 – Ala56 | β1 to β3 | VDW | − 86% |
| Ala56 – C5 | β3 to RNA loop | VDW | − 73% |
| Ala56 – A6 | β3 to RNA loop | VDW | − 70% |
| Lys50 – A6 | loop 3 to RNA loop | VDW | − 52 % |
| U2 – G4 | RNA loop | H-bond (N2 of G4 – O2 of U2) | − 61% |
| Glu19 – U2 | loop 1 to RNA loop | VDW | − 74% |
| Arg52 – U2 | loop 3 to RNA loop | VDW | − 51% |
| Arg52 – A1 | loop 3 to RNA loop | VDW | − 58% |
| Ile33 – Gln36 | helix A | H-bond (NE2 of Gln36 – O of Ile33) | + 52% |
| A-4 – U15 | RNA stem | VDW | − 64% |
| A-5 – U15 | RNA stem | H-bond (N3 of U15 – N1 of A-5) | − 80% |
| A-5 – U15 | RNA stem | H-bond (N6 of A-5 – O4 of U15) | − 82% |
| A-5 – U15 | RNA stem | VDW | − 83% |
| A-5 – U16 | RNA stem | H-bond (N6 of A-5 – O2* of U16) | + 64% |
| A-5 – U16 | RNA stem | VDW | + 60% |
| U14 – U15 | RNA stem | H-bond (O2* of U14 – O5* of U15) | + 72% |
| U14 – U16 | RNA stem | VDW | + 66% |
Phe56Leu free protein
Table 3 lists the HB and VDW interactions that are more than 50% different in the 2000 snapshots sampled from the free Phe56Leu mutant U1A trajectory compared to the wild type system. The residues involved in these interactions are illustrated in Figure 8a. The Phe56Leu mutation affects interactions with Tyr13 due to its smaller size and may also affect other β-sheet interactions seen by disruption of the Thr11 – Ile58 interaction, as well as the interaction of His10 with the loop 4 residue, Lys60, and the helix B residue, Val62. Alternatively, His10 has a different side chain orientation in the Phe56Leu mutant that could be caused by the flexibility of the N-terminal residues. The Phe56Leu mutation may also directly contribute to the differences observed in the interactions of loop 3 and, to a lesser extent, loop 1 residue. The loss in interactions between Tyr13 – Leu56 may lead to an increase in H-bonding interactions of 41% between Tyr13 and Gln54 in this system (not shown in the table because the increase is less than 50%). This interaction optimally aligns Gln54 for a H-bonding interaction with Met51, which contributes to the helical character of loop 3 in this system (Figure 4a), altering the interactions of the other loop 3 residues: Ser46, Arg47, Ser48, and Arg52. This difference in the structure of loop 3 compared to wild type causes a different orientation of the Arg47 side chain, increasing its interactions with the loop 1 residues Glu19 and Ile21.
Table 3.
Differences in interactions (>50%) observed in free Phe56Leu U1A simulation compared to wild type
| Residues | Positions | Type of interaction | Difference |
|---|---|---|---|
| Tyr13 – Leu56 | β1 to β3 | VDW | − 82% |
| Thr11 – Ile58 | β1 to β3 | VDW | − 56% |
| His10 – Lys60 | β1 to loop 4 | VDW | − 63% |
| His10 – Val62 | β1 to helix B | VDW | − 64% |
| Met51 – Gln54 | loop 3 | H-bond (N of Gln54 – O of Met51) | + 55% |
| Ser48 – Arg52 | loop 3 | H-bond (N of Arg52 – O of Ser48) | + 66% |
| Ser48 – Arg52 | loop 3 | VDW | + 76% |
| Ser46 – Ser48 | loop 3 | VDW | + 79% |
| Ile21 – Arg47 | loop 1 to loop 3 | VDW | + 55% |
| Glu19 – Arg47 | loop 1 to loop 3 | H-bond (NH1 of Arg47 – O of Glu19) | + 61% |
Figure 8.

Positions of residues in Phe56Leu mutant U1A trajectories that differ in H-bond and VDW interactions (> 50%) compared to wild type. Residues are colored according to location in the protein for clarity. (a) Free protein. (b) U1A-RNA complex.
Phe56Leu complex
Table 4 lists the HB and VDW interactions that are more than 50% different in the 2000 snapshots sampled from the Phe56Leu U1A-RNA complex trajectory compared to the wild type system. The residues involved in these interactions are illustrated in Figure 8b. The Phe56Leu mutation has more of a local affect on the interactions of the U1A-RNA complex compared to the Phe56Ala system. Leu56 has fewer interactions with Tyr13 and the C5 loop nucleotide of SL2 RNA compared to wild type. Surprisingly, the Leu56 VDW interactions with A6 are not altered significantly compared to the wild type system, though the stacking interactions are absent since the aromaticity has been removed by the Phe56Leu mutation. The increase in interactions between U8 and C9, as well as Ser48 and C10 is due to the flexibility of the RNA backbone throughout the simulation. This flexibility is evidenced by the difference in average structure of the RNA backbone at that site in the Phe56Leu mutant (Figure 5b). Similarly, the loss in interactions between Lys22 and A-4 is due to the flexibility in the ends of the RNA stem throughout the simulation, which can also be seen in Figure 5b. There is no obvious connection between the Phe56Leu mutation and these differences in interactions.
Table 4.
Differences in interactions (>50%) observed in Phe56Leu U1A-RNA complex simulation compared to wild type
| Residues | Positions | Type of interaction | Difference |
|---|---|---|---|
| Tyr13 – Leu56 | β1 to β3 | VDW | − 77% |
| Leu56 – C5 | β3 to RNA loop | VDW | − 72% |
| Ser48 – C10 | loop 3 to RNA loop | VDW | + 61% |
| U8 – C9 | RNA loop | H-bond (O2* of U8 – O4* of C9) | + 54 % |
| U8 – C9 | RNA loop | H-bond (O2* of U8 – O5* of C9) | + 55% |
| Lys22 – A-4 | loop 1 to RNA stem | VDW | − 50 % |
Phe56Trp free protein
Table 5 lists the HB and VDW interactions that are more than 50% different in the 2000 snapshots sampled from the free Phe56Trp mutant U1A trajectory compared to the wild type system. The residues involved in these interactions are illustrated in Figure 9a. The Phe56Trp mutation causes an increase in interactions between Trp56 and the helix C residues Asp90 and Ile94 which, in turn, increases interactions of helix C with β2 and loop 6 residues: Lys98 to Asp42, Asp90 to Ala87 and Lys88, and Ser91 to Thr89. These differences can be visualized by the overlay of average structures in Figure 5a in which the orientation of helix C in the free Phe56Trp U1A mutant is different compared to wild type. The loop 6 residues are in close enough proximity to His10 to cause a change in its side chain orientation and its interactions with Phe59 and Glu61; alternatively, this may be due to the flexibility of the N-terminal residues. The changes in interactions in loop 3 residues Ser46, Arg47, Ser48, and Gln54 may be due to the different dynamics of helix C residues, though there is no direct link between these differences and the Phe56Trp mutation. There is not an obvious explanation for the differences in the loop 5 interactions between Pro76 and Phe77 with Lys80, or the different side chain orientation of Tyr78 leading to fewer interactions with Ser29 based on the Phe56Trp mutation. As seen in the secondary structure plot of free Phe56Trp U1A protein (Figure 4a), loop 5 is less structured compared to the other simulations with Phe77 and Lys80 exhibiting turn character rather than β-sheet character, which would affect the interactions of these residues.
Table 5.
Differences in interactions (>50%) observed in free Phe56Trp U1A simulation compared to wild type
| Residues | Positions | Type of interaction | Difference |
|---|---|---|---|
| Trp56 – Asp90 | β3 to helix C | VDW | + 78% |
| Trp56 – Ile94 | β3 to helix C | VDW | + 69% |
| Asp42 – Lys98 | β2 to helix C | VDW | + 60% |
| Ala87 – Asp90 | loop 6 to helix C | VDW | + 84% |
| Lys88 – Asp90 | loop 6 to helix C | H-bond (N of Lys88 – OD2 of Asp90) | + 97% |
| Thr89 – Ser91 | loop 6 to helix C | VDW | + 63% |
| His10 – Phe59 | β1 to β3 | H-bond (ND1 of His10 – O of Phe59) | + 56% |
| His10 – Glu61 | β1 to loop 4 | VDW | − 54% |
| Ser46 – Gln54 | loop 3 | H-bond (NE2 of Gln54 – O of Ser46) | + 56% |
| Ser48 – Gln54 | loop 3 | H-bond (NE2 of Gln54 – OG of Ser48) | + 57% |
| Ser48 – Gln54 | loop 3 | VDW | + 65% |
| Arg47 – Ser48 | loop 3 | H-bond (NH2 of Arg47 – O of Ser48) | + 62% |
| Pro76 – Lys80 | loop 5 | VDW | − 71% |
| Phe77 – Lys80 | loop 5 | H-bond (N of Phe77 – O of Lys80) | − 66% |
| Ser29 – Tyr78 | helix A to loop 5 | VDW | − 65% |
Figure 9.

Positions of residues in Phe56Trp mutant U1A trajectories that differ in H-bond and VDW interactions (> 50%) compared to wild type. Residues are colored according to location in the protein for clarity. (a) Free protein. (b) U1A-RNA complex.
Phe56Trp complex
Table 6 lists the H-bonding and VDW interactions that are more than 50% different in the 2000 snapshots sampled from the Phe56Trp U1A-RNA complex trajectory compared to the wild type system. The residues involved in these interactions are illustrated in Figure 9b. Surprisingly, the average structure of the Phe56Trp U1A-RNA complex indicates that Trp56 does not stack with A6 to the extent that Phe56 does in the wild type structure. This is likely due to a HB interaction between NE1 of Trp56 and N3 of A6, not indicated in Table 6 since the interaction is only 34% more than seen in the wild type system. This different side chain orientation of Trp56 causes a loss of VDW interactions with Tyr13 and C5. The loss of interactions between Met51 and A6 are due to a different side chain orientation of Met51 in the Phe56Trp mutant. There is a greater distance between A6 in the RNA loop and the Met51 side chain (~ 1 Å measured from SD of Met51 to C4* of A6) due to the different interactions between A6 and Trp56, perhaps leaving more room for the Met51 side chain to fluctuate. The remaining differences in protein interactions are all with the Lys98 residue. This is mainly due to the difference in the Lys98 side chain orientation, which is caused by more helical character in helix C at this residue (Figure 4b) compared to the wild type system. The increase in interactions between C10 and G11 are due to the flexibility of C10 in the RNA loop.
Table 6.
Differences in interactions (>50%) observed in Phe56Trp U1A-RNA complex simulation compared to wild type
| Residues | Positions | Type of interaction | Difference |
|---|---|---|---|
| Tyr13 – Trp56 | β1 to β3 | VDW | − 61% |
| Trp56 – C5 | β3 to RNA loop | VDW | − 72% |
| Met51 – A6 | loop 3 to RNA loop | VDW | − 65% |
| His10 – Lys98 | β1 to helix C | H-bond (NZ of Lys98 – NE2 of His10) | + 56% |
| His10 – Lys98 | β1 to helix C | VDW | + 62% |
| Lys60 – Lys98 | loop 4 to helix C | H-bond (NZ of Lys98 – O of Lys60) | + 56% |
| Glu61 – Lys98 | loop 4 to helix C | VDW | + 57% |
| C10 – G11 | RNA loop to RNA stem | H-bond (O2* of C10 – O5* of G11) | + 51% |
Common differences in Phe56 U1A mutant interactions compared to wild type
Free U1A proteins
Differences in interactions (> 50%) that are common between either two or all three of the free Phe56 U1A mutants compared to wild type are listed in Table 7. Other than the interactions in Table 7 explicitly mentioned below, the differences that are common between the Phe56 mutants compared to the wild type system are primarily due to different side chain orientations that can be attributed to natural fluctuations throughout the simulations and not to the Phe56 mutation. The difference in interactions between Ser46 and Met97 are due to different side chain orientations, as well, but are also due to different helix C positions compared to wild type in the Phe56Ala and Phe56Trp systems. All of the common differences in loop 3 interactions, including Arg47, Ser48 and Leu49 with Met51 and Leu49 with Arg52, are due to the flexibility of loop 3 throughout the simulations and the helical character in loop 3 that is present in the Phe56Ala and Phe56Leu systems (Figure 4a). Note that the differences in interactions for these residues are very similar in the Phe56Ala and Phe56Leu systems compared to wild type and the Phe56Trp system.
Table 7.
Differences in interactions common to Phe56 mutants in free U1A protein simulations compared to wild type (>50% in at least two of the mutants)
| Residues | Positions | Type of interaction | Difference | ||
|---|---|---|---|---|---|
| Phe56Ala | Phe56Leu | Phe56Trp | |||
| Asn16 – Met82 | β1 to β4 | VDW | + 66% | + 50% | + 67% |
| Asn16 – Arg83 | β1 to β4 | H-bond (N of Arg83 – O of Asn16) | + 77% | + 40% | + 51% |
| Asn16 – Arg83 | β1 to β4 | VDW | + 81% | + 52% | + 66% |
| Glu19 – Gly53 | loop 1 to loop 3 | VDW | + 52% | + 51% | |
| Ser29 – Tyr78 | helix A to loop 5 | H-bond (OH of Tyr78 – OG of Ser29) | − 65% | − 62% | |
| Ala32 – Gln36 | helix A | H-bond (NE2 of Gln36 – O of Ala32) | − 81% | − 80% | − 82% |
| Ala32 – Gln36 | helix A | VDW | − 82% | − 79% | − 83% |
| Ile33 – Gln36 | helix A | H-bond (NE2 of Gln36 – O of Ile33) | + 79% | + 77% | + 76% |
| Ser46 – Gln54 | loop 3 | VDW | + 95% | + 54% | |
| Ser46 – Met97 | loop 3 to helix C | VDW | + 62% | + 62% | |
| Arg47 – Met51 | loop 3 | VDW | − 94% | − 93% | − 40% |
| Ser48 – Met51 | loop 3 | H-bond (N of Ser48 – O of Met51) | − 85% | − 85 % | − 29% |
| Leu49 – Met51 | loop 3 | VDW | + 82% | + 83% | |
| Leu49 – Arg52 | loop 3 | VDW | + 83% | + 75% | |
| Thr66 – Tyr86 | helix B to β4 | H-bond (OH of Tyr86 – OG1 of Thr66) | + 92% | + 63% | + 97% |
U1A-RNA Complexes
Differences in interactions (> 50%) that are common between either two or all three of the Phe56 U1A-RNA mutant complexes compared to wild type are listed in Table 8. The differences seen in the first two entries in Table 8 are due solely to the orientation of the Ser48 side chain. In the wild type system, the hydroxyl group is oriented to H-bond with the N of Met51, and in the mutant systems, it is oriented to H-bond with the phosphate group. This increase in interactions between Ser48 and C10 may cause this flexible area of the RNA loop to remain closer to loop 3 in the mutants, as is seen in the average structure overlay in Figure 5b. The interactions between the C7 and U8 nucleotides in the loop region of the RNA causes a bend in the RNA backbone that is stabilized in the wild type system by this H-bond. The mutant systems have a bend in the RNA backbone at this point, as well, which can be seen in Figure 5b.
Table 8.
Differences in interactions common to Phe56 mutants in U1A-RNA complex simulations compared to wild type (>50% in at least two of the mutants)
| Residues | Positions | Type of interaction | Difference | ||
|---|---|---|---|---|---|
| Phe56Ala | Phe56Leu | Phe56Trp | |||
| Ser48 – Met51 | loop 3 | H-bond (N of Met51 – OG of Ser48) | − 48% | − 50% | − 52% |
| Ser48 – C10 | loop 3 to RNA loop | H-bond (OG of Ser48 – O1P of C10) | + 70% | + 62% | + 55% |
| C7 – U8 | RNA loop | H-bond (O2* of U8 – O2P of C7) | − 73% | − 60 % | − 76% |
Discussion
The MD simulations of the free U1A protein in this and other studies17, 21, 22, 25–28, 39 indicate large fluctuations and significant flexibility in loop 3 and helix C residues (Figures 3 and 5). These same secondary structure elements undergo large changes upon binding SL2 RNA, and are also important for specificity in U1A-RNA binding.32–34, 64–68 The flexibility of these regions may play a role in recognition by enabling the protein to sample various subtle conformations of which the appropriate ones can then bind SL2 RNA.28 The MD simulations presented here suggest that mutation of Phe56 may alter the helical character of loop 3, which occurs transiently in the MD simulations of Phe56Ala and Phe56Leu U1A, but not in the wild type and Phe56Trp simulations (Figure 4a). Loop 3 helical character has been observed in one of the two molecules in the asymmetric unit of the crystal structure of the free U1A protein33 and, according to the Avis, et al.,32 in the NMR structure, though a DSSP calculation of the 43 models in 1FHT indicates the loop 3 residues Lys50-Met51-Arg52 have an average 310 helix character of only 8.5%. The helical character of this loop may be transient on the nanosecond (or longer) timescale as evidenced by disappearance in some trajectories and reappearance in others. The flexibility of this loop has been suggested to be necessary for binding,11, 17, 18 and therefore, the more permanent nature of the helical form in the Phe56Ala and Phe56Leu systems could be detrimental for binding.
The differences in interactions observed in the MD of free Phe56 U1A mutant proteins compared to wild type are not localized around the mutation site, but are distributed throughout the protein. These results are consistent with previously performed MM-GBSA calculations of the energetics of these systems, which indicated that the different U1A mutants obtain overall binding energies by very different combinations of energetic contributions.55 A breakdown of MM-GBSA energetics by residue in the wild type and Phe56 mutant systems indicated that differences in binding energy between the mutant and wild type proteins occur throughout the proteins, especially in the β3 – loop 3 and loop 6 – helix C regions. Similarly, many of the differences in hydrogen bonds and non-bonded interactions between the free wild type and mutant proteins occur in the β3 – loop 3 and loop 6 – helix C regions.
The number of differences in interactions in the free Phe56 U1A mutants far exceeds those in the Phe56 U1A-RNA complexes. This is a consequence of the greater flexibility and dynamical nature of the free U1A proteins, which become more rigid upon binding RNA. The analysis of the Phe56Ala U1A-RNA complex indicates a net loss of interactions through the complex that originates at the site of mutation. The differences observed in the Phe56Ala mutant complex are much more extensive compared to the localized perturbations in the Phe56Leu and Phe56Trp U1A-RNA complexes (if the differences in the Phe56Trp complex due to the Lys98 side chain orientation are not considered). The interactions that are lost in the Phe56Ala U1A-RNA complex are predominantly located between the RNA and protein (Table 2). The loss of these interactions only in the Phe56Ala U1A-RNA complex is consistent with the significantly greater loss in binding affinity observed experimentally upon mutation of Phe56 to Ala compared to Leu and Trp.20
Many of the residues identified to have altered interactions in the free or bound mutant systems have previously been reported to be important contributors to the stability of the complex. For example, the mutation of the amino acids in loop 3, including Lys50, Met51, Arg52, and Gln54, resulted in at least a 5-fold decrease in binding affinity.11, 12, 69 Mutation of amino acids in the N-terminal region, such as Thr11 or Asn18, and in the C-terminal helix, such as Asp90 and Ser91 also destabilized the complex by at least 5-fold.24, 28, 69 Finally, mutation of the charged amino acids Lys23, Lys50, and Lys80, has been observed to result in a greater than 5-fold decrease in binding affinity.9, 11, 13 Thus the computational predictions are consistent with previously reported experimental binding investigations.
Conclusions
The MD simulations reported here are predictions of the solution structures for the wild type and various mutants of U1A in the RNA bound and unbound forms. Analysis of the results indicates that the free U1A protein is more flexible and has greater thermal fluctuations than the U1A-RNA complex for both wild type and Phe56 mutant systems. Regions of the protein with the greatest flexibility include N- and C-termini, loops 1 and 3, and helix C. Changes in hydrogen bonding and non-bonded interactions in the free U1A protein caused by the Phe56Ala and Phe56Leu mutations may contribute to stabilization of loop 3, which could be detrimental to SL2 RNA binding. Compared to the wild type protein, changes in hydrogen bonds and non-bonded interactions in the Phe56 mutants of the free U1A protein are globally dispersed away from the site of mutation, and include differences in β-sheet, loop 1 and loop 3 interactions. The Phe56Ala mutation in the U1A-RNA complex leads to a series of differences in hydrogen bonding and non-bonded interactions that cascade through the complex compared to wild type and especially target interactions with the RNA, while the Phe56Leu and Phe56Trp mutations cause differences that are more localized around the site of mutation Taken together with a previous analysis of the energetics of these systems using MM-GBSA,27 these results suggest that the binding of the Phe56Ala mutant U1A protein to RNA is destabilized by both the changes in the free protein structure that stabilize the structure of loop 3 and the reduced interactions between the RNA and the protein compared to the wild type complex. The long-range networks of interactions identified in these simulations suggest a complex interplay between the contributions of direct interactions and dynamic processes in both the free and bound components to the stability of the U1A-RNA complex.
Supplementary Material
Acknowledgements
Funding was provided by the NSF to AMB (MCB-0843728) and by the NIH to AMB (GM-56857) and to DLB (GM-076490). BLK was supported by a NIH Postdoctoral Fellowship (F32 GM072345). This work was partially supported by the National Science Foundation through TeraGrid resources provided by the National Center for Supercomputing Applications under LRAC/NRAC grant number MCA94P011 utilizing the Xeon Linux Cluster and by the Texas Advanced Computing Center utilizing the Cray-Dell PowerEdge Xeon Linux Cluster.
References
- 1.Maris C, Dominguez C, Allain FH-T. FEBS J. 2005;272:2118–2131. doi: 10.1111/j.1742-4658.2005.04653.x. [DOI] [PubMed] [Google Scholar]
- 2.Varani G. Acc. of Chem. Res. 1997;30:189–195. [Google Scholar]
- 3.Varani G, Nagai K. Annu Rev Biophys Biomol Struct. 1998;27:407–445. doi: 10.1146/annurev.biophys.27.1.407. [DOI] [PubMed] [Google Scholar]
- 4.Green MR. Annu. Rev. Cell Biol. 1991;7:559–599. doi: 10.1146/annurev.cb.07.110191.003015. [DOI] [PubMed] [Google Scholar]
- 5.Stark H, Dube P, Lührmann R, Kastner B. Nature. 2001;409:539–542. doi: 10.1038/35054102. [DOI] [PubMed] [Google Scholar]
- 6.Stark H, Luhrmann R. Ann. Rev. Biophys. Biomol. Struct. 2006;35:435–457. doi: 10.1146/annurev.biophys.35.040405.101953. [DOI] [PubMed] [Google Scholar]
- 7.Scherly D, Boelens W, Dathan NA, van Venrooij WJ, Mattaj IW. Nature. 1990;345:502–506. doi: 10.1038/345502a0. [DOI] [PubMed] [Google Scholar]
- 8.Avis JM, Allain FH-T, Howe PWA, Varani G, Nagai K, Neuhaus D. J. Mol. Biol. 1996;257:398–411. doi: 10.1006/jmbi.1996.0171. [DOI] [PubMed] [Google Scholar]
- 9.Nagai K, Oubridge C, Jessen TH, Li J, Evans PR. Nature. 1990;348:515–520. doi: 10.1038/348515a0. [DOI] [PubMed] [Google Scholar]
- 10.Oubridge C, Ito N, Evans PR, Teo CH, Nagai K. Nature. 1994;372:432–438. doi: 10.1038/372432a0. [DOI] [PubMed] [Google Scholar]
- 11.Katsamba PS, Bayramyan M, Haworth IS, Myszka DG, Laird-Offringa IA. J. Biol. Chem. 2002;277:33267–33274. doi: 10.1074/jbc.M200304200. [DOI] [PubMed] [Google Scholar]
- 12.Law MJ, Chambers EJ, Katsamba PS, Haworth IS, Laird-Offringa IA. Nucleic Acids Res. 2005;33:2917–2928. doi: 10.1093/nar/gki602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Law MJ, Linde ME, Chambers EJ, Oubridge C, Katsamba PS, Nilsson L, Haworth IS, Laird-Offringa IA. Nucleic Acids Res. 2006;34:275–285. doi: 10.1093/nar/gkj436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Law MJ, Rice AJ, Lin P, Laird-Offringa IA. RNA. 2006;12:1168–1178. doi: 10.1261/rna.75206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zeng Q, Hall KB. RNA. 1997;3:303–314. [PMC free article] [PubMed] [Google Scholar]
- 16.Kranz JK, Hall KB. J. Mol. Biol. 1998;275:465–481. doi: 10.1006/jmbi.1997.1441. [DOI] [PubMed] [Google Scholar]
- 17.Showalter SA, Hall KB. J. Mol. Biol. 2002;322:533–542. doi: 10.1016/s0022-2836(02)00804-5. [DOI] [PubMed] [Google Scholar]
- 18.Showalter SA, Hall KB. J. Mol. Biol. 2004;335:465–480. doi: 10.1016/j.jmb.2003.10.055. [DOI] [PubMed] [Google Scholar]
- 19.Showalter SA, Hall KB. Biophys. J. 2005;89:2046–2058. doi: 10.1529/biophysj.104.058032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nolan SJ, Shiels JC, Tuite JB, Cecere KL, Baranger AM. J. Am. Chem. Soc. 1999;121:8951–8952. [Google Scholar]
- 21.Blakaj DM, McConnell KJ, Beveridge DL, Baranger AM. J. Am. Chem. Soc. 2001;123:2548–2551. doi: 10.1021/ja005538j. [DOI] [PubMed] [Google Scholar]
- 22.Pitici F, Beveridge DL, Baranger AM. Biopolymers. 2002;65:424–435. doi: 10.1002/bip.10251. [DOI] [PubMed] [Google Scholar]
- 23.Shiels JC, Tuite JB, Nolan SJ, Baranger AM. Nucleic Acids Res. 2002;30:550–558. doi: 10.1093/nar/30.2.550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tuite JB, Shiels JC, Baranger AM. Nucleic Acids Res. 2002;30:5269–5275. doi: 10.1093/nar/gkf636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kormos BL, Baranger AM, Beveridge DL. J. Am. Chem. Soc. 2006;128:8992–8993. doi: 10.1021/ja0606071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kormos BL, Baranger AM, Beveridge DL. J. Struct. Biol. 2007;157:500–513. doi: 10.1016/j.jsb.2006.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kormos BL, Benitex Y, Baranger AM, Beveridge DL. J. Mol. Biol. 2007;371:1405–1419. doi: 10.1016/j.jmb.2007.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anunciado D, Agumeh M, Kormos BL, Beveridge DL, Knee JL, Baranger AM. J. Phys. Chem. B. 2008;112:6122–6130. doi: 10.1021/jp076896c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hall KB. Biochemistry. 1994;33:10076–10088. doi: 10.1021/bi00199a035. [DOI] [PubMed] [Google Scholar]
- 30.Tsai DE, Harper DS, Keene JD. Nuc. Acids Res. 1991;19:4931–4936. doi: 10.1093/nar/19.18.4931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boelens WC, Jansen EJ, van Venrooij WJ, Stripecke R, Mattaj IW, Gunderson SI. Cell. 1993;72:881–892. doi: 10.1016/0092-8674(93)90577-d. [DOI] [PubMed] [Google Scholar]
- 32.Avis JM, Allain FHT, Howe PWA, Varani G, Nagai K, Neuhaus D. J. Mol. Biol. 1996;257:398–411. doi: 10.1006/jmbi.1996.0171. [DOI] [PubMed] [Google Scholar]
- 33.Nagai K, Oubridge C, Jessen TH, Li J, Evans PR. Nature. 1990;348:515–520. doi: 10.1038/348515a0. [DOI] [PubMed] [Google Scholar]
- 34.Oubridge C, Ito N, Evans PR, Teo CH, Nagai K. Nature. 1994;372:432–438. doi: 10.1038/372432a0. [DOI] [PubMed] [Google Scholar]
- 35.Nolan SJ, Shiels JC, Tuite JB, Cecere KL, Baranger AM. J. Amer. Chem. Soc. 1999;121:8951–8952. [Google Scholar]
- 36.Shiels JC, Tuite JB, Nolan SJ, Baranger AM. Nuc. Acids Res. 2002;30:550–558. doi: 10.1093/nar/30.2.550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tuite JB, Shiels JC, Baranger AM. Nuc. Acids Res. 2002;30:5269–5275. doi: 10.1093/nar/gkf636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reyes CM, Kollman PA. RNA. 1999;5:235–244. doi: 10.1017/s1355838299981657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reyes CM, Kollman PA. J. Mol. Biol. 2000;297:1145–1158. doi: 10.1006/jmbi.2000.3629. [DOI] [PubMed] [Google Scholar]
- 40.Reyes CM, Kollman PA. J. Mol. Biol. 2000;295:1–6. doi: 10.1006/jmbi.1999.3319. [DOI] [PubMed] [Google Scholar]
- 41.Hermann T, Westhof E. Nature Struct. Biol. 1999;6:540–544. doi: 10.1038/9310. [DOI] [PubMed] [Google Scholar]
- 42.Olson MA. Biophys. J. 2001;81:1841–1853. doi: 10.1016/S0006-3495(01)75836-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tang Y, Nilsson L. Biophys. J. 1999;77:1284–1305. doi: 10.1016/S0006-3495(99)76979-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhao Y, Kormos BL, Beveridge DL, Baranger AM. Biopolymers. 2006;81:256. doi: 10.1002/bip.20408. [DOI] [PubMed] [Google Scholar]
- 45.Qin S, Zhou H-X. J. Phys. Chem. B. 2008;112:5955–5960. doi: 10.1021/jp075919k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. Nucl. Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kelley LA, Gardner SP, Sutcliffe M. J. Protein Eng. 1996;9:1063–1065. doi: 10.1093/protein/9.11.1063. [DOI] [PubMed] [Google Scholar]
- 48.Pitici F, Beveridge DL, Baranger AM. Biopolymers. 2002;65:424–435. doi: 10.1002/bip.10251. [DOI] [PubMed] [Google Scholar]
- 49.Case DA, Darden TA, Cheatham TE, III, Simmerling CL, Wang J, Duke RE, Luo R, Merz KM, Pearlman DA, Crowley M, Walker R, Zhang W, Wang B, Hayik S, Roitberg A, Seabra G, Wong K, Paesani F, Wu X, Brozell S, Tsui V, Gohlke H, Yang L, Tan C, Mongan J, Hornak V, Cui G, Beroza P, Mathews D, Schafmeister C, Ross WS, Kollman P. AMBER 9. San Francisco: University of California; 2006. [Google Scholar]
- 50.Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. Proteins: Struct., Funct. Bioinform. 2006;65:712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Peréz A, Marchán I, Svozil D, Sponer J, Cheatham TE, III, Laughton CA, Orozco M. Biophys. J. 2007;92:3817–3829. doi: 10.1529/biophysj.106.097782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. J. Chem. Phys. 1983;79:926–936. [Google Scholar]
- 53.McDonald IK, Thornton JM. J. Mol. Biol. 1994;238:777–793. doi: 10.1006/jmbi.1994.1334. [DOI] [PubMed] [Google Scholar]
- 54.Luscombe NM, Laskowski RA, Thornton JM. Nucleic Acids Res. 1997;25:4940–4945. doi: 10.1093/nar/25.24.4940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kormos BL, Benitex Y, Baranger A, Beveridge DL. J. Mol. Biol. 2007;371:1405–1419. doi: 10.1016/j.jmb.2007.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Reyes CM, Kollman PA. RNA. 1999;5:235–244. doi: 10.1017/s1355838299981657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tang Y, Nilsson L. Biophys. J. 1999;77:1284–1305. doi: 10.1016/S0006-3495(99)76979-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hermann T, Westhof E. Nat. Struct. Biol. 1999;6:540–544. doi: 10.1038/9310. [DOI] [PubMed] [Google Scholar]
- 59.Blakaj DM, McConnell KJ, Beveridge DL, Baranger AM. J. Am. Chem. Soc. 2001;123:2548–2551. doi: 10.1021/ja005538j. [DOI] [PubMed] [Google Scholar]
- 60.Showalter SA, Hall KB. Biophys. J. 2005;89:2046–2058. doi: 10.1529/biophysj.104.058032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Frishman D, Argos P. Proteins. 1995;23:566–579. doi: 10.1002/prot.340230412. [DOI] [PubMed] [Google Scholar]
- 62.Humphrey W, Dalke A, Schulten K. J. Mol. Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 63.Kabsch W, Sander C. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 64.Bentley RC, Keene JD. Mol. Cell. Biol. 1991;11:1829–1839. doi: 10.1128/mcb.11.4.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kranz JK, Hall KB. J Mol Biol. 1998;275:465–481. doi: 10.1006/jmbi.1997.1441. [DOI] [PubMed] [Google Scholar]
- 66.Kranz JK, Hall KB. J Mol Biol. 1999;285:215–231. doi: 10.1006/jmbi.1998.2296. [DOI] [PubMed] [Google Scholar]
- 67.Laird-Offringa IA, Belasco JG. Proc Natl Acad Sci. 1995;92:11859–11863. doi: 10.1073/pnas.92.25.11859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Scherly D, Boelens W, Dathan NA, van Venrooij WJ, Mattaj IW. Nature. 1990;345:502–506. doi: 10.1038/345502a0. [DOI] [PubMed] [Google Scholar]
- 69.Jessen T-H, Oubridge C, Teo CH, Pritchard C, Nagai K. EMBO J. 1991;10:3447–3456. doi: 10.1002/j.1460-2075.1991.tb04909.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.