

Abstract
Ebola virus (EBOV) infects several cell types and while viral entry is known to be pH dependent; the exact entry pathway(s) remains unknown. To gain insights into EBOV entry, the role of several inhibitors of clathrin-mediated endocytosis in blocking infection mediated by HIV pseudotyped with the EBOV envelope glycoprotein (EbGP) was examined. Wild type HIV and envelope-minus HIV pseudotyped with Vesicular Stomatitis Virus glycoprotein (VSVg) were used as controls to assess cell viability after inhibiting clathrin pathway. Inhibition of clathrin pathway using dominant-negative Eps15; siRNA-mediated knockdown of clathrin heavy chain; chlorpromazine and sucrose blocked EbGP pseudotyped HIV infection. Also, both chlorpromazine and Bafilomycin A1 inhibited entry of infectious EBOV. Sensitivity of EbGP pseudotyped HIV as well as infectious EBOV to inhibitors of clathrin suggests that EBOV uses clathrin-mediated endocytosis as an entry pathway. Furthermore, since chlorpromazine inhibits EBOV infection, novel therapeutic modalities could be designed based on this lead compound.
Keywords: Ebola virus, Entry, Clathrin endocytic pathway, Chlorpromazine, Sucrose, Eps15
Introduction
Ebola virus (EBOV) causes a severe, fatal hemorrhagic fever with mortality rates as high as 90 % (Feldmann et al., 2003). There is currently no effective anti-viral therapy available for EBOV infection. EBOV is an enveloped, negative-sense, single stranded RNA virus (Regnery, Johnson, and Kiley, 1980) with a broad species and cell-type tropism (Wool-Lewis and Bates, 1998). The envelope glycoprotein (GP) is responsible for entry (Elliott, Kiley, and McCormick, 1985). Very little is known about the pathway used by EBOV to enter target cells and its receptor(s) remains unidentified. It is known that EBOV entry in the context of pseudotypes is pH dependent (Chazal et al., 2001). Depletion of membrane cholesterol by β–methyl cyclodextrin inhibits EBOV infection in a dose dependant manner (Yonezawa, Cavrois, and Greene, 2005) and (Bavari et al., 2002). There have been conflicting reports on the endocytic pathway(s) used by EBOV to enter target cells. One group suggested that Ebola enters cells via caveolar uptake (Empig and Goldsmith, 2002) and uses the folate receptor alpha as a cofactor for EbGP mediated fusion (Chan et al., 2001). Another study has suggested that EBOV uses clathrin as well as caveolae mediated endocytosis for entry using various chemical inhibitors of these pathways (Sanchez, 2007). However, most of the inhibitors caused considerable cell detachment in this study and it was conducted only in Vero cells so cell type specific effects could not be ruled out. In contrast, two reports had previously shown that neither caveolae nor folate receptor alpha are required for EbGP fusion function (Simmons et al., 2003) and (Sinn et al., 2003). In light of these controversies more analysis is required.
The major endocytic pathways include clathrin-mediated endocytosis, uptake by caveolae, macropinocytosis and phagocytosis. Some non-clathrin (Nemerow and Cooper, 1984), non-caveolae mediated viral endocytic pathways have also been reported (Sanchez-San Martin et al., 2004) but they are not well described.
Among all the known pathways of viral endocytosis, clathrin-mediated endocytosis is predominant. Key proteins involved in clathrin-mediated endocytosis include clathrin, Adaptor Protein Complex-2 (AP-2) and epidermal growth factor receptor (EGFR) pathway substrate clone 15 (Eps15). Clathrin assembles into a polyhedral lattice on the inner side of the plasma membrane to form the coated pit. Eps15 acts as an adaptor between the clathrin coats and AP-2 (Keen, 1987). An AP-2 independent pathway for clathrin-mediated endocytosis has also been reported (Lakadamyali, Rust, and Zhuang, 2006). Eps15 comprises of three structural domains: an N-terminal region (DI) composed of three Eps15 homology regions, a central coiled-coil domain (DII) involved in oligomerization and a C-terminal region (DIII) which contains the AP-2 binding sites. DI is required for correct coated pit targeting of Eps15. It was shown that in cells expressing mutants lacking Eps15 homology domains, punctuate distribution of AP-2 and clathrin is lost and endocytosis of transferrin, which is a specific marker of clathrin-mediated endocytosis (Hanover, Willingham, and Pastan, 1984) is inhibited (Benmerah et al., 1999). Certain chemical agents such as chlorpromazine (Wang, Rothberg, and Anderson, 1993) and sucrose (Heuser and Anderson, 1989) are also known to prevent recycling of clathrin to the plasma membrane thereby inhibiting clathrin-mediated endocytosis.
In this study we examined whether EBOV requires the clathrin-mediated endocytic pathway for entry. Our results demonstrate that EbGP pseudotyped HIV as well as replication-competent EBOV use clathrin-mediated endocytosis as an entry pathway. We have also shown that chlorpromazine significantly inhibits replication-competent EBOV infection and hence is a potential lead candidate for development of anti-viral therapy against EBOV infection.
Materials and methods
Cell lines, plasmids and reagents
HEK293T, HOS-CD4, HeLa and Vero cells were maintained in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10 % fetal bovine serum, penicillin-streptomycin-L-glutamine solution and 10 μg/ml ciprofloxacin. Human microvascular endothelial cells (HMEC) were obtained from American Type Culture Collection (ATCC) and grown in MCDB131 medium (Gibco) supplemented with 0.7 % FBS, 10 ng/ml EGF, 10 mM L-glutamine, 1 μg/ml hydrocortisone and 50 units/ml penicillin-streptomycin.
The plasmid encoding Ebola Zaire GP (pCB6-EbGP) was obtained from Dr. Paul Bates (Wool-Lewis and Bates, 1998). The VSVg and the HIV provirus (R7ΔEnvGFP) vectors were obtained from the NIH AIDS Research and Reference Reagent Program. The R73X4EnvGFP plasmid was provided by Dr. Mark Muesing (Fisher et al., 1986). The eGFP-clathrin plasmid was received from Dr. Lois Greene (Wu et al., 2001). The GFP dominant-negative Eps15 (DIII) and GFP control Eps15 (D3Δ2) constructs were made by Alexandre Benmerah (Benmerah et al., 1999) and received from Dr. John Young.
The GFP-Eps15 plasmids were digested with AgeI and BsrGI restriction enzymes to remove the GFP fragment and mRFP was inserted using the rapid ligation kit (Roche).
Polyethylenimine (PEI), Linear, MW 25,000 (Polysciences, Inc.) was dissolved in distilled water to make 1 mg/ml stock solution. Chlorpromazine hydrochloride (Sigma) and Sucrose (Fisher) were dissolved in distilled water to make 1 mg/ml and 5 M stock solutions respectively. Bafilomycin A1 (Baf A1) (Sigma) was dissolved in DMSO to make 20 μM stock solution. Texas Red (TR) and Fluorescein conjugated transferrin (Molecular Probes) were reconstituted to 5 mg/ml solutions in phosphate-buffered saline (PBS). Mouse monoclonal anti-human transferrin receptor antibody (Cat # 13-6800) was purchased from Zymed Laboratories and Cy3 and Cy5 conjugated anti-mouse secondary antibodies were purchased from Jackson ImmunoResearch Laboratories, Inc.
Pseudotyped virus production
For preparing EbGP pseudotyped virus, HEK293T cells in a 15 cm plate were transiently co-transfected with 20 μg of pCB6-EbGP and 30 μg of R7ΔEnvGFP HIV plasmids using 112 μl of PEI. 48 h post-transfection, the supernatant was collected, centrifuged at 1500 rpm for 5 min and clarified by filtration through a 0.45-μm pore-size filter. VSVg pseudotyped GFP reporter virus was prepared similarly. For making replication-competent HIV, the cells were transfected with 45 μg of R73X4EnvGFP plasmid. Viral content was measured using the p24 ELISA kit (Perkin Elmer Lifesciences) according to the manufacturer’s guidelines.
Infection with Ebola Zaire GFP virus and quantification of infectivity
GFP-expressing replication competent EBOV Zaire (ZEBOV-GFP) was received from Dr. Jason Paragas (USAMRIID) (Towner et al., 2005). Vero E6 and HeLa cells were plated on black-wall 96 well plates at a density of 50,000 cells/well. Inhibitory compounds were added to the wells 2 h prior to infection. Virus at indicated MOI was diluted in EMEM medium and added to the wells. After 1 h attachment, the excess virus was removed by washing twice with PBS and medium containing the experimental compounds was added for the duration of the culture. Cells were then fixed by submerging the whole plate in 10 % neutral-buffered formalin for 3 days. The percentage of cells expressing GFP and the average fluorescence intensity of those cells were measured with a Discovery-1 high content screening device (Molecular Devices Corp., Downingtown, PA) for 9 regions per well. These experiments were performed inside a Biosafety Level 4 (BSL-4) laboratory.
Flow cytometric analysis to measure viral transduction following various inhibitor treatments
HOS cells in 12 well plates were pre-treated with 50 nM Baf A1 for 30 min followed by overnight incubation with virus in the presence of the drug. To block clathrin-mediated endocytosis, HOS, HeLa, Vero, 293T and HMEC cells in 12 well plates were pre-treated with either 0.45 M sucrose for 10 min or 10 μg/ml chlorpromazine for 45 min or a combination of 10 μg/ml chlorpromazine and 0.45 M sucrose for 45 min. The cells were incubated with virus for 4 h in the presence of the drug. After 4 h, the cells were washed extensively with PBS followed by overnight incubation with the drug. Three independent infection samples were analyzed for each virus. GFP fluorescence (as a marker of infectivity) was measured 48 h post-infection by flow cytometry using FACS Calibur (Becton Dickinson). Cells were gated on forward and side scatter and the same gates were used for treated and control samples. 10,000 gated events were accrued and analyzed for each sample. Each experiment was repeated three times and similar results were obtained each time. The data from one representative experiment is shown in the figures.
Transfection with eGFP-clathrin followed by chlorpromazine or sucrose treatment
HOS cells were plated on coverslips and transiently transfected with eGFP-clathrin using Effectene transfection reagent (Qiagen) as per the manufacturer’s guidelines. 24 h post-transfection, cells were treated with 0.45 M sucrose for 10 min or 10 μg/ml chlorpromazine for 45 min or a combination of 10 μg/ml chlorpromazine and 0.45 M sucrose for 45 min. Cells were fixed with 3.7 % formaldehyde (Polysciences Inc.) in PIPES buffer (0.1 M PIPES (pH 6.8), 2 mM MgCl2, 1 mM EGTA). Cells were then rinsed with PBS and the DNA was stained using Hoechst. Finally, the coverslips were mounted on glass slides with Gel Mount (Biomeda) containing antifade agents. Dried slides were imaged with an Olympus IX70 epifluorescent microscope fitted with an automated stage (Applied Precision Inc.) and images were captured in z-series on a CCD digital camera. Out of focus light was digitally removed using the Softworks deconvolution software (Applied Precision Inc.).
Immunofluorescence analysis of viral transduction after transfection with mRFP-Eps15 plasmids
HOS, HeLa, Vero and 293T cells were plated on coverslips followed by transient transfection with dominant-negative (DN) or control mRFP-Eps15 plasmids using Effectene transfection reagent (Qiagen). 24 h post-transfection, the cells were incubated with virus for 4 h. 48 h later, the cells were fixed and the DNA was stained with Hoechst. The coverslips were imaged as described in the transfection with eGFP-clathrin followed by chlorpromazine or sucrose treatment section. Several panels of images were collected from different sections of the coverslips and the number of transfected cells and transfected and infected cells in each panel were counted. A minimum of 100 transfected cells was counted in each case. The experiment was repeated three times and the percentage of transfected and infected cells was determined in every population for each individual experiment.
Immunofluorescence studies with TR or Fluorescein transferrin
HOS cells were pre-incubated with 10 μg/ml of chlorpromazine for 2 h or 0.45 M sucrose for 45 min or a combination of 10 μg/ml of chlorpromazine and 0.45 M sucrose for 2 h. This was followed by incubation with 14 μg/ml of TR-transferrin for various time points. In another experiment, HOS cells were transiently transfected with dominant-negative or control mRFP-Eps15 plasmids. 24 h post-transfection, the cells were incubated with 14 μg/ml of Fluorescein-transferrin for various time points. In all the experiments, the cells were rinsed extensively to remove any unbound transferrin, fixed and the DNA was stained with Hoechst. The coverslips were imaged as described in the transfection with eGFP-clathrin followed by chlorpromazine or sucrose treatment section. The fluorescence intensity settings for transferrin were equalized in the treated and control images.
Intracellular detection of transferrin receptor by Immunofluorescence
HOS cells were plated on coverslips and incubated with 10 μg/ml of chlorpromazine for 45 min or 0.45 M sucrose for 10 min. The cells were fixed and stained for transferrin receptor using mouse monoclonal antibody against human transferrin receptor and Cy3-conjugated secondary antibody. The DNA was stained with Hoechst. The coverslips were imaged as described in the transfection with eGFP-clathrin followed by chlorpromazine or sucrose treatment section. The fluorescence intensity settings for transferrin receptor were equalized in the treated and control images.
Immunofluorescence study to examine transferrin endocytosis following siRNA-mediated knockdown of clathrin
The siRNA duplex was synthesized as a 21-mer with UU overhangs (Dharmacon). The clathrin heavy chain target sequence was GCAAUGAGCUGUUUGAAGA. The siRNA duplex was designed and synthesized exactly as described previously (Huang et al., 2004). The siRNA duplex was resuspended in 1X siRNA universal buffer (Dharmacon) to 20 μM before transfection.
HOS cells were seeded on coverslips and incubated in DMEM containing 10 % FBS without antibiotics for 24 h. The cells were transiently transfected twice at 24 h intervals with 4 μl of siRNA duplex, 1 μg/ml of eGFP (as a transfection marker) and 3 μl of Lipofectamine 2000 reagent (Invitrogen) in 100 μl of Opti-MEM I medium. 4 h later, the transfection media was replaced with regular DMEM supplemented with antibiotics. 48 h following the second transfection, the cells were incubated with 14 μg/ml of TR-transferrin for 30 min. Cells were rinsed extensively to remove any unbound transferrin, fixed and stained for clathrin using mouse monoclonal antibody against clathrin heavy chain (Affinity Bioreagents) and Cy5-conjugated anti-mouse secondary antibody. The coverslips were mounted onto glass slides and imaged as described in the transfection with eGFP-clathrin followed by chlorpromazine or sucrose treatment section.
Western blotting to detect siRNA-mediated knockdown of clathrin
HOS cells were seeded in 12-well plates and incubated in 1 ml media containing 10 % FBS without antibiotics for 24 h. Cells were transfected twice at 24 h intervals with 4 μl of siRNA duplex and 3 μl of Lipofectamine 2000 reagent (Invitrogen) in 100μl of Opti-MEM I medium. 4 h later, the transfection media was removed and regular media was added to the cells. 48 h after the second transfection, the cells were lysed by adding IVKA lysis buffer (50 mM HEPES, 150 mM NaCl, 10 % glycerol, 1 % Triton X-100, 1 mM EGTA, 1.5 mM MgCl2, 1 mM Na-orthovanadate, 1 mM phenylmethylsulfonyl fluoride, 1X protease inhibitor cocktail (Roche)) and then scraped off from the plate with a polyethylene cell lifter. The protein concentration in each lysate was measured using the DC protein assay kit (Bio-Rad). Equal amounts of lysates were loaded onto 8 % Tris-Glycine Sodium dodecyl sulphate, polyacrylamide gel electrophoresis (SDS PAGE) gel, the proteins were separated by SDS PAGE and then transferred to a PVDF membrane. The blot was probed for clathrin using mouse monoclonal antibody against clathrin heavy chain followed by detection using enhanced chemiluminescence system (Pierce). The blots were also probed for GAPDH as a loading control.
Immunofluorescence analysis of viral transduction following siRNA-mediated knockdown of clathrin
HOS cells were seeded on coverslips and incubated in 1 ml DMEM containing 10 % FBS without antibiotics for 24 h. The cells were then transfected twice at 24 h intervals with 4 μl of siRNA duplex, 0.25 μg/ml mCherry plasmid (as a transfection marker) and 3 μl of Lipofectamine 2000 reagent (Invitrogen) in 100 μl of Opti-MEM I medium. 4 h later, the transfection media was replaced with regular DMEM supplemented with antibiotics. 48 h following the second transfection, the cells were incubated with virus for 4 h. Control cells were transfected with 0.25 μg/ml mCherry plasmid and 3 μl of Lipofectamine 2000 reagent (Invitrogen) in 100 μl of Opti-MEM I medium. 48 h post-infection, the cells were fixed and the DNA was stained with Hoechst. The coverslips were imaged and analyzed as described in the immunofluorescence analysis of viral transduction after transfection with mRFP-Eps15 plasmids section. The experiment was repeated three times and fold change in viral infectivity in siRNA-transfected cells compared to the control cells was calculated.
Statistical analysis of experimental data
p values were determined by comparing treated versus control samples using a paired student t test with GraphPad InStat3 software. For the Eps15 and clathrin siRNA experiments, p values were calculated by comparing the fold decrease in EbGP mediated infectivity with VSVg and HIV infectivity using the one way analysis of variance (ANOVA) test.
Results
EbGP mediated entry of the HIV pseudotype is pH-dependent
To analyze the pathway of Ebola virus entry, we used an envelope-minus HIV pseudotyped with the Ebola virus envelope glycoprotein (EbGP). For this analysis we utilized two control viruses, HIV using its native envelope and envelope-minus HIV pseudotyped with VSVg. HIV entry is pH-independent (Stein et al., 1987), while entry mediated by VSVg pseudotyped HIV is pH-dependent (Matlin et al., 1982). The control viruses were used to demonstrate target cell viability and specificity of the inhibitory disruption of the clathrin pathway. In all cases, the viral genomes contained the GFP gene located in the Nef position within the HIV genome. Therefore, infected cells could be readily identified for GFP expression using flow cytometry or microscopy. The initial analysis was designed to confirm the previously reported role of acidification in the function of EbGP, VSVg and HIV envelope. Vacuolar ATPases hydrolyze ATP, creating a proton gradient that causes acidification of endosomes. Bafilomycin A1 (Baf A1) is a potent inhibitor of vacuolar ATPases and thereby prevents acidification of endosomes (Bowman, Siebers, and Altendorf, 1988). For this analysis, the titer of the different viral stocks was determined using infectivity studies. In this study, the different stocks were added at amounts that gave comparable levels of infectivity. When normalized by p24 content, the VSVg pseudotyped virus was typically 25 times more infectious than replication-competent HIV and 79 times more infectious than the EbGP pseudotyped HIV after 4 h of exposure to target cells.
Baf A1 treatment reduced both EbGP and VSVg mediated infectivity to near background levels, while HIV infectivity showed a minor increase compared to the untreated control (Fig. 1). These results validated the functionality of the pseudotyping system and infectivity assays since a previous study has also reported that treatment with Baf A1 significantly inhibited EbGP and VSVg pseudotyped viral infection, while enhancing HIV infection (Chazal et al., 2001).
Fig. 1.
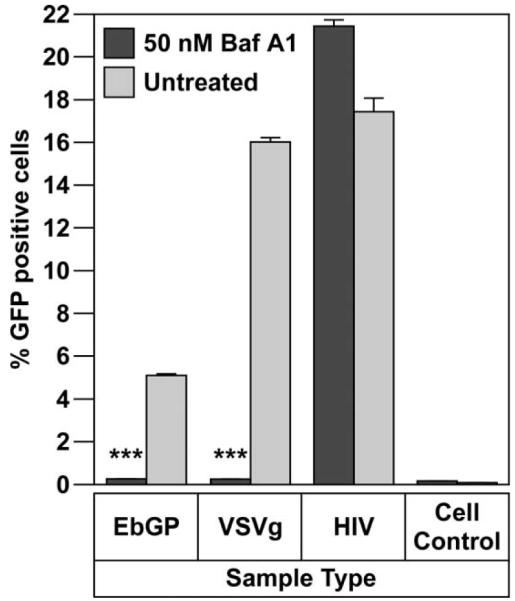
Importance of acidification on Ebola entry.
Infectivity of EbGP, VSVg pseudotyped virus and HIV on HOS cells in the presence or absence of 50 nM Bafilomycin A1 (Baf A1). Error bars represent SEM for three independent infection samples. * p value for EbGP = 0.0003 (extremely significant) and p value for VSVg = 0.0003 (extremely significant).
Inhibition of EbGP mediated entry by chlorpromazine
To investigate the potential role of clathrin-mediated endocytosis, we first determined the ability of the drug chlorpromazine to inhibit EbGP mediated viral entry. Chlorpromazine is known to sequester clathrin, causing it to assemble around endosomal membranes thereby preventing recycling of clathrin to the plasma membrane (Wang, Rothberg, and Anderson, 1993). A previous study used 10 μg/ml of chlorpromazine to determine the involvement of clathrin pathway in Influenza entry (Sieczkarski and Whittaker, 2002).
To determine the effect of chlorpromazine treatment on clathrin localization, cells were transiently transfected with eGFP-clathrin. After 24 h, the cells were treated with 10 μg/ml of chlorpromazine for 45 min, fixed and analyzed by fluorescent deconvolution microscopy. In the treated cells, clathrin was seen in random clusters in the cytoplasm, while in the untreated cells, a distinct punctate distribution of clathrin was seen throughout the cell (Fig. 2A). To determine the functional consequences of chlorpromazine treatment, we examined if the endocytosis of transferrin, a known marker for clathrin-mediated endocytosis (Hanover, Willingham, and Pastan, 1984), was altered. Fluorescently labeled transferrin was added to HOS cells either pre-treated with 10 μg/ml chlorpromazine for 2 h or untreated cells. After 5 min of incubation, the cells were rinsed to remove any unbound transferrin and fixed for fluorescent deconvolution microscopy analysis. As shown in Fig. 2B, fluorescent transferrin was localized in a perinuclear compartment, most likely recycling endosomes, in untreated cells. In contrast, no internalized transferrin was detected in the chlorpromazine treated cells. These studies demonstrate that 10 μg/ml chlorpromazine potently inhibits clathrin-mediated endocytosis. We also examined the effect of chlorpromazine on the recycling of transferrin receptor and found that transferrin receptor is predominantly localized in the perinuclear region following treatment with chlorpromazine (Fig. 2C), while in the untreated cells, transferrin receptor is uniformly distributed all over the cell. This result explained the absence of any transferrin on the cell surface after chlorpromazine treatment in Fig. 2B.
Fig. 2.

Chlorpromazine (CPZ) strongly inhibits EbGP mediated viral infection.
(A) Transfection of HOS cells with eGFP-clathrin followed by treatment with 10 μg/ml chlorpromazine for 45 min. Left panel shows an untreated cell while the right panel shows a chlorpromazine treated cell. Scale bars represent 15 μm. (B) Treatment of HOS cells with 10 μg/ml chlorpromazine for 2 h followed by incubation with 14 μg/ml of TR-transferrin for 5 min. Image on the left represents untreated cells, while the image on the right depicts chlorpromazine treated cells. Scale bars represent 15 μm. (C) Treatment of HOS cells with 10 μg/ml chlorpromazine for 45 min followed by fixing and staining for transferrin receptor using mouse monoclonal antibody against human transferrin receptor and Cy3-conjugated anti-mouse secondary antibody. Image on the left represents untreated cells, while the image on the right depicts chlorpromazine treated cells. Scale bars represent 30 μm. (D) Infectivity of EbGP, VSVg pseudotyped virus and HIV on HOS cells was measured in the presence or absence of 10 μg/ml chlorpromazine. Error bars represent SEM for three independent infection samples. * p value = 0.0449 (significant). (E) Infectivity of EbGP pseudotyped virus on HMEC cells was measured in the presence or absence of 10 μg/ml chlorpromazine. Error bars represent SEM for three independent infection samples. * p value = 0.0009 (extremely significant). (F) Comparison of infectivity of EbGP pseudotyped virus in HOS, HeLa, Vero and 293T cells measured in the presence or absence of 10 μg/ml chlorpromazine. Error bars represent SEM for three independent infection samples. * p value for HOS = 0.0071 (very significant), p value for HeLa = 0.0017 (very significant), p value for Vero = 0.0037 (very significant) and p value for 293T = 0.0001 (extremely significant). (G) XY plot depicting dose response effect of chlorpromazine treatment on EbGP mediated infectivity in HOS cells. # represents complete cytotoxicity meaning no cells were left on the plate.
Next, we determined the effect of chlorpromazine treatment on infection mediated by the EbGP, VSVg and HIV envelopes in HOS cells. As seen in Fig. 2D, treatment with 10 μg/ml chlorpromazine potently inhibited EbGP pseudotyped HIV infection without having any effect on infection with either wt HIV or VSVg pseudotyped HIV. The normal infectivity by wt HIV and VSVg pseudotyped HIV demonstrated that the chlorpromazine treatment was not toxic to the target cells. The effect of chlorpromazine on EbGP mediated infectivity was also tested in HeLa, Vero, 293T and HMEC cell lines to determine whether the effect was cell-type specific for HOS cells or could be seen in other cell types including endothelial cells, which are one of the target cells for filoviral infection. Chlorpromazine severely inhibited EbGP mediated infectivity in HMEC (Fig. 2E) and also HeLa, Vero and 293T cells (Fig. 2F), which suggests that inhibition of EbGP mediated infectivity by chlorpromazine is not restricted to a certain cell type but is prevalent among different cell types. The inhibitory effect of chlorpromazine on EbGP mediated infectivity was found to be dose dependent for doses of 10 μg/ml or lower with maximum inhibitory effect seen at 10 μg/ml (Fig. 2G).
Inhibition of EbGP mediated entry by sucrose
To further explore the observed involvement of clathrin-mediated endocytosis on entry mediated by EbGP; we inhibited clathrin-mediated endocytosis using 0.45 M sucrose. A previous study has shown that treatment with 0.45 M sucrose led to formation of clathrin microcages on the inner surface of the plasma membrane thereby depleting the cytoplasmic pools of clathrin necessary for normal coated pit formation (Heuser and Anderson, 1989).
When cells were transfected with eGFP-clathrin followed by treatment with 0.45 M sucrose for 10 min, there was an increased accumulation of clathrin in the perinuclear region, while in untreated cells, clathrin exhibited a uniform punctuate distribution throughout the cell (Fig. 3A). An increased accumulation of clathrin in the perinuclear region has been previously reported in sucrose treated cells (Heuser and Anderson, 1989). Next, we determined the functional consequences of sucrose treatment on clathrin-mediated endocytosis. For this, we determined the ability of sucrose treatment to inhibit the internalization of fluorescently labeled transferrin. Pre-treatment of HOS cells with 0.45 M sucrose for 45 min inhibited entry of TR-transferrin, whereas in untreated cells, transferrin was endocytosed within 5 min (Fig. 3B). The effect of sucrose treatment on recycling of transferrin receptor was also examined and it was found that sucrose causes accumulation of transferrin receptor in the perinuclear region (Fig. 3C) and this explains why transferrin is not present on the cell surface upon sucrose treatment in Fig. 3B.
Fig. 3.

EbGP mediated viral infection is inhibited by sucrose.
(A) Transient transfection of HOS cells with eGFP-clathrin followed by treatment with 0.45 M sucrose for 10 min. Left panel shows an untreated cell while the right panel shows sucrose treated cells. Scale bars represent 15 μm. (B) Treatment of HOS cells with 0.45 M sucrose for 45 min followed by incubation with 14 μg/ml of TR-transferrin for 5 min. Image on the left represents untreated cells, while the image on the right shows sucrose treated cells. Scale bars represent 15 μm. (C) Treatment of HOS cells with 0.45 M sucrose for 10 min followed by fixing and staining for transferrin receptor using mouse monoclonal antibody against human transferrin receptor and Cy3-conjugated anti-mouse secondary antibody. Image on the left represents untreated cells, while the image on the right depicts sucrose treated cells. Scale bars represent 30 μm. (D) Infectivity of EbGP, VSVg pseudotyped virus and HIV on HOS cells was measured in the presence or absence of 0.45 M sucrose. Error bars represent SEM for three independent infection samples. * p value = 0.0047 (very significant). (E) Infectivity of EbGP pseudotyped virus on HMEC cells measured in the presence or absence of 0.45 M sucrose. Error bars represent SEM for three independent infection samples. * p value = 0.0001 (extremely significant). (F) Comparison of infectivity of EbGP pseudotyped virus on HOS, HeLa, Vero and 293T cells measured in the presence or absence of 0.45 M sucrose. Error bars represent SEM for three independent infection samples. * p value for HOS = 0.0091 (very significant), p value for HeLa = 0.0018 (very significant), p value for Vero = 0.0014 (very significant) and p value for 293T = 0.0012 (very significant). (G) Effect of combination of chlorpromazine and sucrose on viral entry. Viral infectivity was measured following pre-treatment with a combination of 10 μg/ml chlorpromazine and 0.45 M sucrose for 45 min. Error bars represent SEM for three independent infection samples. * p value for EbGP = 0.0125 (significant) and p value for HIV = 0.0309 (significant).
Next, we determined the effect of sucrose treatment on infection mediated by the EbGP, VSVg, and HIV envelopes. A potent decrease in EbGP mediated infectivity was observed in sucrose treated cells; while VSVg and HIV infectivity remained unaffected when compared to untreated controls (Fig. 3D). Since VSVg and HIV infectivity were unaltered, it suggests that the sucrose treatment was not toxic to the cells. Furthermore, we investigated the effect of sucrose treatment on EbGP mediated infectivity in HMEC cells and as seen in Fig. 3E, sucrose severely inhibited EbGP mediated infectivity in endothelial cells, which are one of the target cells of filoviral infection. We also compared the effect of sucrose treatment on EbGP mediated infectivity in HOS, HeLa, Vero and 293T cells and as seen in Fig. 3F, sucrose significantly inhibited EbGP mediated infectivity in all these cell lines indicating that the inhibitory effect of sucrose was not restricted to HOS cells.
One surprising observation was that VSVg pseudotyped viral infection was not inhibited by either chlorpromazine or sucrose treatment since it has been previously reported that VSV entry is mediated by clathrin-mediated endocytosis (Sun et al., 2005). To further investigate this process, we evaluated a combination treatment with chlorpromazine and sucrose. Treatment with a combination of 10 μg/ml chlorpromazine and 0.45 M sucrose caused a relocalization of eGFP-clathrin to a perinuclear region and inhibited the endocytosis of fluorescently labeled transferrin (data not shown). In cells treated with a combination of 10 μg/ml chlorpromazine and 0.45 M sucrose, EbGP mediated infectivity was reduced to background levels; VSVg infectivity remained unchanged; while HIV infectivity was increased in treated cells when compared to untreated controls (Fig. 3G). We also examined the effect of increasing concentrations of chlorpromazine on VSVg mediated infectivity in HOS cells and found that concentrations between 50 μg/ml to 150 μg/ml were completely cytotoxic, while 10 μg/ml or lower concentrations didn’t have any effect on infectivity (data not shown).
EbGP mediated entry is inhibited by expression of dominant-negative Eps15
Having established the effect of two chemical inhibitors of clathrin-mediated endocytosis in inhibiting EbGP mediated entry; we next examined the effect of a molecular inhibitor of the clathrin pathway to see if it could also block EbGP mediated entry. For this, we transfected HOS cells with a dominant-negative Eps15; DIII, which has deletion of all three Eps15 homology domains (Benmerah et al., 2000) or a control Eps15 plasmid D3Δ2, which is a 760 nucleotides insert analogous to DIII but lacking the AP-2 binding sites (Benmerah et al., 2000). First, we examined the functional effect of these constructs on transferrin endocytosis and found that in cells transfected with mRFP-DIII, transferrin entry was completely inhibited, while in D3Δ2 transfected cells, transferrin was endocytosed at similar levels compared to adjoining untransfected cells (Fig. 4A).
Fig. 4.

Dominant-negative Eps15 inhibits EbGP mediated viral entry.
(A) Transfection of HOS cells with mRFP-D3Δ2 control Eps15 plasmid (left panel) or mRFP-DIII dominant-negative (DN) Eps15 plasmid (right panel) followed by incubation with Fluorescein-transferrin for 30 min. The nuclei were stained with Hoechst. Red represents Eps15 transfected cells, green represents transferrin and blue represents nuclei. Arrows indicate transferrin endocytosis in the D3Δ2 control Eps15 cells, which is comparable to neighboring untransfected cells. Scale bar represents 30 μm. (B) Fold decrease in viral infectivity in HOS cells transfected with DIII DN Eps15 when compared to D3Δ2 control Eps15 plasmid. Error bars represent SEM for three independent experiments. * p value for fold change in EbGP mediated infectivity compared to either VSVg or HIV = 0.0021 (very significant). (C) Comparison of fold decrease in EbGP mediated infectivity in HeLa, 293T and Vero cells transfected with DIII DN Eps15 when compared to D3Δ2 control Eps15 plasmid. Error bars represent SEM for three independent experiments. Typically, there was 20-30 % transfected and infected cells (double positive cells) in cells transfected with the D3Δ2 control plasmid and infected with EbGP virus in all four cell lines.
Next, we examined the effect of dominant-negative and control Eps15 on viral entry mediated by EbGP, VSVg and wt HIV. There was a greater than 5 fold decrease in EbGP mediated infectivity in cells transfected with dominant-negative DIII plasmid when compared to cells transfected with the D3Δ2 control plasmid over three independent experiments, while VSVg and HIV infectivity levels were comparable in cells transfected with DIII or D3Δ2 plasmids (Fig. 4B). This result confirmed that EbGP facilitates infection via clathrin-mediated endocytosis. Further, we compared the role of Eps15 in EbGP mediated infectivity in HeLa, 293T and Vero cells and as seen in Fig. 4C, there was a considerable inhibition of EbGP mediated infectivity in all three cell types transfected with dominant-negative Eps15 when compared to cells transfected with control Eps15.
siRNA-mediated knockdown of clathrin inhibits EbGP mediated viral infectivity
In order to further confirm the involvement of clathrin in EbGP mediated viral infectivity, we compared the effect of knockdown of clathrin heavy chain on EbGP and VSVg pseudotyped virus and wt HIV entry in HOS cells. We used a previously well characterized siRNA duplex (Huang et al., 2004), which was shown to specifically knockdown clathrin by 90-95 % in HeLa cells without causing any off-target effects.
First, we performed a functional study to examine transferrin endocytosis upon clathrin knockdown. For this, HOS cells were transiently co-transfected with siRNA against clathrin heavy chain and eGFP plasmid (as a transfection marker) followed by incubation with TR-transferrin for 30 min. In the siRNA-transfected cells, transferrin entry was severely inhibited in cells expressing eGFP and these cells also showed very low levels of clathrin thereby confirming efficient knockdown of clathrin (Fig. 5A). In contrast, transferrin was endocytosed in cells where clathrin expression was not downregulated (cells not expressing GFP). The efficiency of clathrin knockdown was further confirmed by western blotting, which showed a protein band of approximately 180 kD representing the clathrin heavy chain in the control sample, while there was no clathrin protein detected in the siRNA sample (Fig. 5B). The blots were also probed for GAPDH as a loading control and similar amounts of GAPDH were detected in the siRNA-treated and control samples.
Fig. 5.

EbGP mediated viral infection is inhibited by siRNA-mediated knockdown of clathrin heavy chain.
(A) Co-transfection of HOS cells with eGFP and siRNA against clathrin heavy chain followed by incubation with 14 μg/ml of TR-transferrin for 30 min. Cells were fixed and stained for clathrin (shown in blue) using mouse monoclonal antibody against clathrin heavy chain and Cy5-conjugated anti-mouse secondary antibody. Arrows indicate transferrin endocytosis in the neighboring untransfected cells. Scale bar represents 15 μm. The side panels show individual channels in a single siRNA transfected cell (co-transfected with eGFP) and demonstrate that there is very little clathrin and no internalized transferrin in that GFP expressing cell. (B) HOS cells in 12 well plates were transfected twice with siRNA against clathrin followed by western blot detection of clathrin using mouse monoclonal antibody against clathrin heavy chain. GAPDH was measured as a loading control. (C) HOS cells on coverslips were transfected twice with siRNA against clathrin heavy chain and mCherry plasmid as a transfection marker. Control cells were transfected with mCherry plasmid alone. 48 h following the second transfection, the cells were incubated with virus for 4 h. 48 h post-infection, the cells were fixed and the DNA was stained with Hoechst. Several panels of images were collected from each sample and the number of transfected cells and transfected and infected cells were counted in each panel. Graph represents the fold decrease in viral infectivity in siRNA-transfected cells when compared to control cells. Error bars represent SEM for three independent experiments. * p value for fold change in EbGP mediated infectivity compared to either VSVg or HIV = 0.0006 (extremely significant). Typically, there was 20 % transfected and infected cells (double positive cells) in control cells infected with EbGP virus and 25 % transfected and infected cells in siRNA-treated as well as control cells for VSVg and HIV infection.
Next, we determined viral infectivity following siRNA-mediated knockdown of clathrin. For this, we co-transfected HOS cells with the siRNA against clathrin heavy chain and mCherry plasmid (as a transfection marker) followed by infection with EbGP, VSVg and replication-competent HIV GFP reporter viruses. Control cells were transfected with mCherry plasmid alone. The total number of transfected alone and transfected and infected cells were counted in each population after imaging several panels of cells from each coverslip. As seen in Fig. 5C, there was a 4.8 fold decrease in EbGP mediated infectivity in cells transfected with siRNA against clathrin when compared to control cells. VSVg and wt HIV infectivity remained unchanged in siRNA transfected versus control cells. This result further confirmed that entry mediated by EbGP is clathrin dependent.
Bafilomycin A1 and chlorpromazine inhibit infection by replication-competent Ebola virus
Given the significant difference in the morphology of the EbGP pseudotyped HIV and the authentic Ebola virus, it was important to verify the results of the experiments with EbGP pseudotyped HIV in the context of real EBOV infection. For this purpose, we used a recombinant virus ZEBOV-GFP in which the GFP gene has been inserted downstream of the gene for Ebola nucleoprotein (NP) (Towner et al., 2005). Infection of cells with this virus can be monitored by visualization of GFP expression using microscopy or flow cytometry.
First, we examined if infectious EBOV entry requires acidification of endosomes. For this, Vero E6 cells plated in triplicate in a 96 well plate were treated with or without Baf A1 for 2 h followed by addition of ZEBOV-GFP at an MOI of 1. Cells were incubated with the virus for 1 h then washed to remove the excess virus and the medium without drug was replenished. After 48 h, the cells were fixed on the plate, nuclei were stained with Hoechst and the percentage of infected cells was determined with a Discovery 1 screening device. As shown in Fig. 6A, 10 nM Baf A1 effectively blocked EBOV entry. This data supports the findings with the pseudotyped virus system that acidification of endosomes is required for the entry of infectious EBOV.
Fig. 6.

Baf A1 and chlorpromazine inhibit replication-competent EBOV infection.
(A) Vero E6 cells were seeded in 96 well plates and pretreated with 10 nM Baf A1 before infection with ZEBOV-GFP. After 1 h attachment of virus, cells were washed and incubated with medium alone for 48 h, fixed and the infected cells visualized and quantified by Discovery 1 microscope for 9 regions per well. Error bars represent SEM for three independent infection samples. * p value = 0.0028 (very significant). (B) Effect of chlorpromazine on viability of Vero E6 cells. Cells were treated with increasing concentrations of chlorpromazine for 48 h and cell viability was measured by Sytox green staining and quantification of cells using Discovery 1 microscope. (C) Vero cells were plated in 96 well plates and treated with different concentrations of chlorpromazine and infected with ZEBOV-GFP at an MOI of 1. After virus attachment and washing the excess virus, media containing the same concentrations of drug were replenished. After 48 h, the cells were fixed and percent infection determined using Discovery 1 microscope for 9 regions per well. Error bars represent SEM for three independent infection samples. (D) Images captured with Discovery 1 microscope from infected cells treated with medium alone (Upper panel) or 10 μg/ml chlorpromazine (Lower panel). Green represents infected cells and blue represents nuclear staining with Hoechst. Scale bar represents 50 μm. (E) HeLa cells were treated with different concentrations of chlorpromazine and infected with ZEBOV-GFP as described above in (C). 48 h later, the cells were fixed and percent infection determined using Discovery 1 microscope for 9 regions per well. Error bars represent SEM for three independent infection samples.
Next, we examined the effect of chlorpromazine on ZEBOV-GFP infection of Vero-E6 cells in a similar fashion as the above experiment, with the exception that the drug was kept in the culture for the entire incubation period. To ensure lack of cytotoxicity, we first treated cells with increasing concentrations of chlorpromazine for 48 h and measured cell viability by Sytox green exclusion assay using the Discovery 1 microscope. No significant cytotoxicity was observed at concentrations up to 10 μg/ml whereas 30-40 % decline in viability was observed at 20 μg/ml (Fig. 6B). We then examined the effect of chlorpromazine on infection of ZEBOV-GFP. As shown in Fig. 6C, chlorpromazine caused a dose dependent inhibition of EBOV infection with almost complete inhibition at 10 μg/ml. Representative images from untreated and drug treated cells are shown in Fig. 6D. Next, we examined the effect of various concentrations of chlorpromazine on ZEBOV-GFP infectivity in HeLa cells and similar to Vero-E6 cells, there was a dose dependent decline and viral infectivity dropped to background level at 10 μg/ml concentration (Fig. 6E). This data indicated that the inhibitory effect of chlorpromazine on EBOV infectivity was not restricted to Vero-E6 cells. Since chlorpromazine was shown to block entry of transferrin, which is a known marker for clathrin-mediated endocytosis, inhibition of ZEBOV-GFP infection following treatment with chlorpromazine in Vero-E6 and HeLa cells supports the results seen with EbGP pseudotyped virus to demonstrate that Ebola virus uses clathrin-mediated endocytosis as an entry pathway.
Discussion
In this study, we present two lines of investigation that reveal that EBOV utilizes the clathrin-mediated endocytic pathway to infect cells. First, using envelope-minus HIV pseudotyped with EbGP, we find that viral infection is blocked by chemical and dominant-negative inhibitors of clathrin endocytic pathway. Likewise, siRNA-mediated knockdown of clathrin heavy chain also inhibited infection with EbGP pseudotyped HIV. Importantly, the inhibition of clathrin-mediated endocytosis was specific because chlorpromazine, sucrose, dominant-negative Eps15 and siRNA-mediated knockdown of clathrin had no inhibitory effect on infection mediated by either VSVg or HIV envelope. This specificity demonstrated that the inhibition of clathrin-mediated endocytosis was not cytotoxic. Secondly, chlorpromazine could potently inhibit infection with replication-competent EBOV. These studies validate the results obtained with the HIV pseudotype system and demonstrate that clathrin-mediated endocytosis is necessary for infection with the filamentous particles characteristic of the filovirus Ebola.
The potential role of clathrin-mediated endocytosis may seem surprising when considering the known small and spherical nature of clathrin coated pits. Typically, they are on the order of 120 nm (Conner and Schmid, 2003). Ebola virus on the other hand has a diameter of 80 nm, but is filamentous in nature. It has previously been reported that maximal infectivity of EBOV is associated with filamentous particles 970 nm long and EBOV particles can reach up to 14 μm in length (Kiley et al., 1982). However, a previous report reveals that clathrin-mediated endocytosis is not restricted to small spherical coated pits. Listeria has been shown to utilize clathrin-mediated endocytosis to enter cells (Veiga and Cossart, 2005). The large size of the Listeria, which extends to 2 μm (Giardini and Theriot, 2001), suggests that clathrin coated pits should be able to readily accommodate the filamentous particles of EBOV.
Inhibition of clathrin-mediated endocytosis was previously shown to block wild type VSV infection (Sun et al., 2005). So a surprising finding from our study is that infection mediated by VSVg pseudotyped HIV remained unaffected after disrupting the clathrin endocytic pathway using four different approaches. However, VSVg infection was inhibited by Baf A1, which agreed with the pH-dependence previously reported for VSVg pseudotyped viruses and wild type VSV (Matlin et al., 1982). There could be several reasons why VSVg mediated entry was unaffected by inhibitors of the clathrin pathway in our study. Perhaps, there are differences in the context of wild type virus compared to the function of VSVg in the context of pseudotyped HIV. Alternatively, the use of certain entry pathways may be cell-type dependent. For example, in the case of influenza virus it has been shown that the virus can enter via several pathways including clathrin (Rust et al., 2004), caveolae (Nunes-Correia et al., 2004) and a clathrin and caveolae independent pathway (Sieczkarski and Whittaker, 2002) and (Rust et al., 2004). Likewise, similar results have been reported for adenovirus (Varga, Weibull, and Everitt, 1991); (Svensson, 1985); (Yoshimura, 1985) and (Meier et al., 2002). Therefore, past data and findings from this study put together suggests that like influenza virus, VSV could perhaps use multiple endocytic pathways for entry.
Like influenza virus and adenovirus, it is also possible that EBOV could utilize other entry pathways in addition to clathrin-mediated endocytosis under different circumstances. However, there appears to be an important role for clathrin-mediated endocytosis in the infection of HOS, HeLa, Vero, 293T and primary human endothelial cells with EbGP pseudotypes and Vero and HeLa cells with replication-competent EBOV as presented in this study.
Recent studies indicate that the viral endocytic pathways are not as well defined as was previously believed. Several studies have shown that although some viruses such as HIV (Popik, Alce, and Au, 2002), Vaccinia virus (Chung, Huang, and Chang, 2005), Coxsackievirus (Triantafilou and Triantafilou, 2003) and filoviruses (Bavari et al., 2002) could use lipid rafts as sites of entry and cholesterol depletion inhibits viral entry, yet these viruses may not necessarily enter via caveolae-mediated endocytosis. Moreover, some studies have shown that clathrin-mediated endocytosis may occur in the absence of AP-2 (Lakadamyali, Rust, and Zhuang, 2006) and (Motley et al., 2003), which was initially considered to be a critical adaptor for this pathway. These data indicate that although a virus may use a certain pathway for entry, it could use proteins or adaptors that are different from what have been previously described as essential and hence the use of certain inhibitors of a pathway may not necessarily block viral entry.
There are currently no therapeutic modalities available for EBOV infection. It is intriguing that chlorpromazine was highly effective in blocking EBOV infection. Chlorpromazine is a well-known psychotropic drug approved by the Food and Drug Administration (Adams et al., 2005), which has also been shown to possess antimicrobial activities (Kristiansen and Mortensen, 1987). Our findings suggest that this drug could be further investigated as a lead for chemoinformatics-based design and development of a series of potential inhibitors of EBOV replication. In this regard, it would be advantageous to examine other phenothiazine derivatives with lower psychotropic effects. Although chlorpromazine is shown to disrupt the assembly of clathrin (Wang, Rothberg, and Anderson, 1993), the detailed mechanism of action of chlorpromazine is not entirely understood. Chlorpromazine is also known to inhibit calmodulin (Marshak, Lukas, and Watterson, 1985) and (Wrenn et al., 1981) and can bind to phospholipid components of the plasma membrane of endothelial cells (Hueck et al., 2000). Therefore, it is possible that modulation of other cellular pathways may also contribute to the inhibitory effect of chlorpromazine on EBOV replication.
Using EbGP pseudotyped HIV as well as replication-competent EBOV, the results of this study have provided extensive evidence that EBOV uses clathrin-mediated endocytosis as an entry pathway. Another novel observation is the abrogation of EBOV infection by chlorpromazine, which could have important therapeutic implications. The role of chlorpromazine in inhibiting EBOV infection in animal models is under investigation.
Acknowledgements
We thank Lois Greene (National Institutes of Health), John Young (Salk Institute for Biological Studies), Paul Bates (University of Pennsylvania), Mark Muesing (Aaron Diamond AIDS Research Center), David Rekosh and Marie-Louise Hammarskjold (University of Virginia), and the NIH AIDS reagent repository for providing plasmids and Jason Paragas (USAMRIID) for providing ZEBOV-GFP. This work was supported by National Institutes of Health grant R21 AI054495 to T.J.H. T.J.H is an Elizabeth Glaser Scientist. Work performed at USAMRIID was supported by a grant from Defense Threat Reduction Agency (F_X012_04_RD_B).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Opinions, interpretations, conclusions, and recommendations are those of the authors and are not necessarily endorsed by the U.S. Army.
References
- Adams CE, Rathbone J, Thornley B, Clarke M, Borrill J, Wahlbeck K, Awad AG. Chlorpromazine for schizophrenia: a Cochrane systematic review of 50 years of randomised controlled trials. BMC Med. 2005;3:15. doi: 10.1186/1741-7015-3-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bavari S, Bosio CM, Wiegand E, Ruthel G, Will AB, Geisbert TW, Hevey M, Schmaljohn C, Schmaljohn A, Aman MJ. Lipid raft microdomains: a gateway for compartmentalized trafficking of Ebola and Marburg viruses. J Exp Med. 2002;195(5):593–602. doi: 10.1084/jem.20011500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benmerah A, Bayrou M, Cerf-Bensussan N, Dautry-Varsat A. Inhibition of clathrin-coated pit assembly by an Eps15 mutant. J Cell Sci. 1999;112(Pt 9):1303–11. doi: 10.1242/jcs.112.9.1303. [DOI] [PubMed] [Google Scholar]
- Benmerah A, Poupon V, Cerf-Bensussan N, Dautry-Varsat A. Mapping of Eps15 domains involved in its targeting to clathrin-coated pits. J Biol Chem. 2000;275(5):3288–95. doi: 10.1074/jbc.275.5.3288. [DOI] [PubMed] [Google Scholar]
- Bowman EJ, Siebers A, Altendorf K. Bafilomycins: a class of inhibitors of membrane ATPases from microorganisms, animal cells, and plant cells. Proc Natl Acad Sci U S A. 1988;85(21):7972–6. doi: 10.1073/pnas.85.21.7972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SY, Empig CJ, Welte FJ, Speck RF, Schmaljohn A, Kreisberg JF, Goldsmith MA. Folate receptor-alpha is a cofactor for cellular entry by Marburg and Ebola viruses. Cell. 2001;106(1):117–26. doi: 10.1016/s0092-8674(01)00418-4. [DOI] [PubMed] [Google Scholar]
- Chazal N, Singer G, Aiken C, Hammarskjold ML, Rekosh D. Human immunodeficiency virus type 1 particles pseudotyped with envelope proteins that fuse at low pH no longer require Nef for optimal infectivity. J Virol. 2001;75(8):4014–8. doi: 10.1128/JVI.75.8.4014-4018.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung CS, Huang CY, Chang W. Vaccinia virus penetration requires cholesterol and results in specific viral envelope proteins associated with lipid rafts. J Virol. 2005;79(3):1623–34. doi: 10.1128/JVI.79.3.1623-1634.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conner SD, Schmid SL. Regulated portals of entry into the cell. Nature. 2003;422(6927):37–44. doi: 10.1038/nature01451. [DOI] [PubMed] [Google Scholar]
- Elliott LH, Kiley MP, McCormick JB. Descriptive analysis of Ebola virus proteins. Virology. 1985;147(1):169–76. doi: 10.1016/0042-6822(85)90236-3. [DOI] [PubMed] [Google Scholar]
- Empig CJ, Goldsmith MA. Association of the caveola vesicular system with cellular entry by filoviruses. J Virol. 2002;76(10):5266–70. doi: 10.1128/JVI.76.10.5266-5270.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmann H, Jones S, Klenk HD, Schnittler HJ. Ebola virus: from discovery to vaccine. Nat Rev Immunol. 2003;3(8):677–85. doi: 10.1038/nri1154. [DOI] [PubMed] [Google Scholar]
- Fisher AG, Feinberg MB, Josephs SF, Harper ME, Marselle LM, Reyes G, Gonda MA, Aldovini A, Debouk C, Gallo RC, et al. The trans-activator gene of HTLV-III is essential for virus replication. Nature. 1986;320(6060):367–71. doi: 10.1038/320367a0. [DOI] [PubMed] [Google Scholar]
- Giardini PA, Theriot JA. Effects of intermediate filaments on actin-based motility of Listeria monocytogenes. Biophys J. 2001;81(6):3193–203. doi: 10.1016/S0006-3495(01)75955-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanover JA, Willingham MC, Pastan I. Kinetics of transit of transferrin and epidermal growth factor through clathrin-coated membranes. Cell. 1984;39(2 Pt 1):283–93. doi: 10.1016/0092-8674(84)90006-0. [DOI] [PubMed] [Google Scholar]
- Heuser JE, Anderson RG. Hypertonic media inhibit receptor-mediated endocytosis by blocking clathrin-coated pit formation. J Cell Biol. 1989;108(2):389–400. doi: 10.1083/jcb.108.2.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang F, Khvorova A, Marshall W, Sorkin A. Analysis of clathrin-mediated endocytosis of epidermal growth factor receptor by RNA interference. J Biol Chem. 2004;279(16):16657–61. doi: 10.1074/jbc.C400046200. [DOI] [PubMed] [Google Scholar]
- Hueck IS, Hollweg HG, Schmid-Schonbein GW, Artmann GM. Chlorpromazine modulates the morphological macro- and microstructure of endothelial cells. Am J Physiol Cell Physiol. 2000;278(5):C873–8. doi: 10.1152/ajpcell.2000.278.5.C873. [DOI] [PubMed] [Google Scholar]
- Keen JH. Clathrin assembly proteins: affinity purification and a model for coat assembly. J Cell Biol. 1987;105(5):1989–98. doi: 10.1083/jcb.105.5.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiley MP, Bowen ET, Eddy GA, Isaacson M, Johnson KM, McCormick JB, Murphy FA, Pattyn SR, Peters D, Prozesky OW, Regnery RL, Simpson DI, Slenczka W, Sureau P, van der Groen G, Webb PA, Wulff H. Filoviridae: a taxonomic home for Marburg and Ebola viruses? Intervirology. 1982;18(1-2):24–32. doi: 10.1159/000149300. [DOI] [PubMed] [Google Scholar]
- Kristiansen JE, Mortensen I. Antibacterial effect of four phenothiazines. Pharmacol Toxicol. 1987;60(2):100–3. doi: 10.1111/j.1600-0773.1987.tb01504.x. [DOI] [PubMed] [Google Scholar]
- Lakadamyali M, Rust MJ, Zhuang X. Ligands for clathrin-mediated endocytosis are differentially sorted into distinct populations of early endosomes. Cell. 2006;124(5):997–1009. doi: 10.1016/j.cell.2005.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshak DR, Lukas TJ, Watterson DM. Drug-protein interactions: binding of chlorpromazine to calmodulin, calmodulin fragments, and related calcium binding proteins. Biochemistry. 1985;24(1):144–50. doi: 10.1021/bi00322a020. [DOI] [PubMed] [Google Scholar]
- Matlin KS, Reggio H, Helenius A, Simons K. Pathway of vesicular stomatitis virus entry leading to infection. J Mol Biol. 1982;156(3):609–31. doi: 10.1016/0022-2836(82)90269-8. [DOI] [PubMed] [Google Scholar]
- Meier O, Boucke K, Hammer SV, Keller S, Stidwill RP, Hemmi S, Greber UF. Adenovirus triggers macropinocytosis and endosomal leakage together with its clathrin-mediated uptake. J Cell Biol. 2002;158(6):1119–31. doi: 10.1083/jcb.200112067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motley A, Bright NA, Seaman MN, Robinson MS. Clathrin-mediated endocytosis in AP-2-depleted cells. J Cell Biol. 2003;162(5):909–18. doi: 10.1083/jcb.200305145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemerow GR, Cooper NR. Early events in the infection of human B lymphocytes by Epstein-Barr virus: the internalization process. Virology. 1984;132(1):186–98. doi: 10.1016/0042-6822(84)90102-8. [DOI] [PubMed] [Google Scholar]
- Nunes-Correia I, Eulalio A, Nir S, de Lima M. C. Pedroso. Caveolae as an additional route for influenza virus endocytosis in MDCK cells. Cell Mol Biol Lett. 2004;9(1):47–60. [PubMed] [Google Scholar]
- Popik W, Alce TM, Au WC. Human immunodeficiency virus type 1 uses lipid raft-colocalized CD4 and chemokine receptors for productive entry into CD4(+) T cells. J Virol. 2002;76(10):4709–22. doi: 10.1128/JVI.76.10.4709-4722.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regnery RL, Johnson KM, Kiley MP. Virion nucleic acid of Ebola virus. J Virol. 1980;36(2):465–9. doi: 10.1128/jvi.36.2.465-469.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rust MJ, Lakadamyali M, Zhang F, Zhuang X. Assembly of endocytic machinery around individual influenza viruses during viral entry. Nat Struct Mol Biol. 2004;11(6):567–73. doi: 10.1038/nsmb769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez A. Analysis of filovirus entry into vero e6 cells, using inhibitors of endocytosis, endosomal acidification, structural integrity, and cathepsin (B and L) activity. J Infect Dis. 2007;196(Suppl 2):S251–8. doi: 10.1086/520597. [DOI] [PubMed] [Google Scholar]
- Sanchez-San Martin C, Lopez T, Arias CF, Lopez S. Characterization of rotavirus cell entry. J Virol. 2004;78(5):2310–8. doi: 10.1128/JVI.78.5.2310-2318.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieczkarski SB, Whittaker GR. Influenza virus can enter and infect cells in the absence of clathrin-mediated endocytosis. J Virol. 2002;76(20):10455–64. doi: 10.1128/JVI.76.20.10455-10464.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons G, Rennekamp AJ, Chai N, Vandenberghe LH, Riley JL, Bates P. Folate receptor alpha and caveolae are not required for Ebola virus glycoprotein-mediated viral infection. J Virol. 2003;77(24):13433–8. doi: 10.1128/JVI.77.24.13433-13438.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinn PL, Hickey MA, Staber PD, Dylla DE, Jeffers SA, Davidson BL, Sanders DA, McCray PB., Jr. Lentivirus vectors pseudotyped with filoviral envelope glycoproteins transduce airway epithelia from the apical surface independently of folate receptor alpha. J Virol. 2003;77(10):5902–10. doi: 10.1128/JVI.77.10.5902-5910.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein BS, Gowda SD, Lifson JD, Penhallow RC, Bensch KG, Engleman EG. pH-independent HIV entry into CD4-positive T cells via virus envelope fusion to the plasma membrane. Cell. 1987;49(5):659–68. doi: 10.1016/0092-8674(87)90542-3. [DOI] [PubMed] [Google Scholar]
- Sun X, Yau VK, Briggs BJ, Whittaker GR. Role of clathrin-mediated endocytosis during vesicular stomatitis virus entry into host cells. Virology. 2005;338(1):53–60. doi: 10.1016/j.virol.2005.05.006. [DOI] [PubMed] [Google Scholar]
- Svensson U. Role of vesicles during adenovirus 2 internalization into HeLa cells. J Virol. 1985;55(2):442–9. doi: 10.1128/jvi.55.2.442-449.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towner JS, Paragas J, Dover JE, Gupta M, Goldsmith CS, Huggins JW, Nichol ST. Generation of eGFP expressing recombinant Zaire ebolavirus for analysis of early pathogenesis events and high-throughput antiviral drug screening. Virology. 2005;332(1):20–7. doi: 10.1016/j.virol.2004.10.048. [DOI] [PubMed] [Google Scholar]
- Triantafilou K, Triantafilou M. Lipid raft microdomains: key sites for Coxsackievirus A9 infectious cycle. Virology. 2003;317(1):128–35. doi: 10.1016/j.virol.2003.08.036. [DOI] [PubMed] [Google Scholar]
- Varga MJ, Weibull C, Everitt E. Infectious entry pathway of adenovirus type 2. J Virol. 1991;65(11):6061–70. doi: 10.1128/jvi.65.11.6061-6070.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veiga E, Cossart P. Listeria hijacks the clathrin-dependent endocytic machinery to invade mammalian cells. Nat Cell Biol. 2005;7(9):894–900. doi: 10.1038/ncb1292. [DOI] [PubMed] [Google Scholar]
- Wang LH, Rothberg KG, Anderson RG. Mis-assembly of clathrin lattices on endosomes reveals a regulatory switch for coated pit formation. J Cell Biol. 1993;123(5):1107–17. doi: 10.1083/jcb.123.5.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wool-Lewis RJ, Bates P. Characterization of Ebola virus entry by using pseudotyped viruses: identification of receptor-deficient cell lines. J Virol. 1998;72(4):3155–60. doi: 10.1128/jvi.72.4.3155-3160.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrenn RW, Katoh N, Schatzman RC, Kuo JF. Inhibition by phenothiazine antipsychotic drugs of calcium-dependent phosphorylation of cerebral cortex proteins regulated by phospholipid or calmodulin. Life Sci. 1981;29(7):725–33. doi: 10.1016/0024-3205(81)90026-6. [DOI] [PubMed] [Google Scholar]
- Wu X, Zhao X, Baylor L, Kaushal S, Eisenberg E, Greene LE. Clathrin exchange during clathrin-mediated endocytosis. J Cell Biol. 2001;155(2):291–300. doi: 10.1083/jcb.200104085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonezawa A, Cavrois M, Greene WC. Studies of ebola virus glycoprotein-mediated entry and fusion by using pseudotyped human immunodeficiency virus type 1 virions: involvement of cytoskeletal proteins and enhancement by tumor necrosis factor alpha. J Virol. 2005;79(2):918–26. doi: 10.1128/JVI.79.2.918-926.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura A. Adenovirus-induced leakage of co-endocytosed macromolecules into the cytosol. Cell Struct Funct. 1985;10(4):391–404. doi: 10.1247/csf.10.391. [DOI] [PubMed] [Google Scholar]