

Abstract
A series of new Gd(III) hydroxypyridonate complexes featuring a mesitylene (ME)-derived ligand cap has been prepared. Relaxometric characterization reveals that the complexes tend to form large aggregates in solution with slow tumbling rates, as estimated from NMRD analysis, and unique pH-dependent relaxivities. The solution behavior and relaxometric properties are compared to those observed for analogous TREN-capped compounds, and the potential for use of these new ME-capped complexes as pH-responsive MRI contrast agents is explored.
Keywords: hydroxypyridinone (HOPO), mesityl, gadolinium, MRI contrast agent, relaxivity, aggregation
Introduction
Magnetic Resonance Imaging (MRI) has become a valuable diagnostic technique by enabling the acquisition of detailed soft tissue images without requiring harmful ionizing radiation. Signal intensity in an MRI scan can be enhanced by administering a paramagnetic contrast agent that functions by increasing the nuclear relaxation rate of in vivo water protons. Gadolinium (III) remains the most widely used paramagnetic metal for such applications, combining a long electron spin relaxation time with a high magnetic moment to affect water proton relaxation times.[2–5] Due to the high toxicity of free Gd(III), the ion must be complexed by an organic ligand and administered in the form of a stable chelate for in vivo use. Consequently, an active research area in lanthanide coordination chemistry remains the design and synthesis of ligands for Gd(III) to produce stable complexes suitable for imaging applications.
Over the last decade, we have developed a family of hydroxypyridinone (HOPO)-based Gd(III) complexes which possess favorable properties for imaging, including a higher inner-sphere water coordination number (q = 2 or 3) and fast water exchange rate as compared with commercial aminocarboxylate-based agents.[6, 7] These properties combine to yield relaxivities more than twice that of commercial agents and which remain high at high magnetic field strengths, making these chelates unique as prospective clinical agents. While the effect of varying the chelator and substituents present in the ligand motif on stability and relaxometric properties has been extensively explored, an evaluation of HOPO-based Gd(III) complexes employing different ligand capping structures has been limited. The majority of HOPO complexes assessed for contrast agent applications has utilized the tris-(2-aminoethyl)-amine (TREN) platform as a tripodal ligand cap (1, Figure 1). Assessment of those derivatives which do incorporate capping structures differing slightly from TREN (2–4, Figure 1) has typically revealed lower stabilities and relaxivities as compared with the TREN compounds.[8, 9] One notable exception to this trend arises for TACN-capped HOPO Gd(III) complexes. In one of the few examples of a non-TREN based series of Gd(III) HOPO complexes, triazacyclononane (TACN) was employed as a ligand cap resulting in an increased hydration number, high aqueous solubility, and adequate thermodynamic stability.[10]
Figure 1.

Representative HOPO-based Gd(III) complexes utilizing TREN and other derivative ligand scaffolds.
In efforts to explore other novel ligand architectures for HOPO-based Gd(III) complexes, a mesitylene (ME) derived scaffold (Figure 2) was considered. Past studies by this research group have employed a tris-amino derivative of mesitylene as a ligand capping structure in the synthesis of Fe(III) sequestering agents.[11–14] This so-called ME cap served to link three catechol (CAM) chelators together to produce a tripodal, hexadentate ligand that coordinates Fe(III) via the six catechol oxygens. Interestingly, ME-CAM (5, Figure 2) forms a more stable complex than the TREN-capped analog despite the lack of hydrogen bonding between the tertiary TREN nitrogen and amide protons that helps to preorganize TREN-CAM for metal binding.[13, 15] In more recent work published by Hider and coworkers, the ME cap was used in combination with the 3,4-HOPO isomer to yield tripodal ligands for coordination of Fe(III) as well.[16]
Figure 2.

Fe-ME-CAM complex (previously reported) and three ME-capped Gd(III) HOPO complexes reported in this paper.
Inspired by this work on iron chelation, the ME-cap was chosen as a scaffold for a new class of HOPO-based Gd(III) complexes discussed here. To ensure adequate water solubility, the bis-HOPO-TAM motif was chosen (Figure 2) and allows for the attachment of solubilizing substituents such as polyethylene glycol chains and amine derivatives to the terephthalamide (TAM) chelator as in past work with TREN-capped complexes.[17] The resultant Gd(III) complexes exhibit solution behavior unlike other HOPO complexes previously reported in that they form aggregates of high molecular weight and long rotational correlation times. The unique pH dependency of the relaxivity and the consequences for tumor mapping will also be discussed.
Results and Discussion
Ligand Design and Synthesis
Utilization of the ME-CAM ligand shown in Figure 2 would likely fail in forming a stable lanthanide complex at physiological pH due to the strong basicity of the CAM podand. The catechol binding unit is not fully deprotonated at pH 7.4, and the resultant competition for protons versus the metal ion makes CAM a weak lanthanide binder. The more acidic nature of the 3,2-HOPO chelator is a better match for the lanthanides,[18] and most entries into the HOPO family of Gd(III) complexes have incorporated either the 1- or 6-methyl HOPO isomer. It has also been demonstrated for tripodal TREN-based ligands that thermodynamic stability of the Gd(III) complex is maximized by replacing one HOPO chelator with a terephthalamide (TAM) chelator in the TREN-bis-HOPO-TAM motif.[19] The favorable stability properties of the bis-HOPO-TAM motif motivated its incorporation into the design of ME-capped complexes 6, 7, and 8 shown in Figure 2. Additionally, the TAM podand allows for derivatization of the complex with solubilizing substituents, as aqueous solubility is expected to decrease upon replacement of TREN with the aromatic mesityl capping moiety.
A significant enhancement in aqueous solubility is expected with increasing polyethylene glycol (PEG) chain length as was observed in past work with TREN-capped complexes.[20, 21] Gd-ME-bis-(1-Me-3,2-HOPO)-TAM-PEG450 (6) was therefore synthesized as shown in Scheme 1. The PEG450 compound (10) (named for its approximate molecular weight) was added under slow addition, high dilution conditions to dithiazolide-activated TAM (9) to give the mono-substituted product 13. ME-bis-1-Me-3,2-HOPO-Bn (14) was then prepared under similar slow addition, high dilution conditions and subsequently coupled with 13. After isolation of the benzyl protected ligand 15, palladium catalyzed high pressure hydrogenation afforded the deprotected ligand 16 in quantitative yield. The Gd(III) complex 6 was then prepared and complexation was verified by MALDI-MS.
Scheme 1.

Synthesis of Gd-ME-bis-(1-Me-3,2-HOPO)-TAM-PEG450.
While 6 is expected to have sufficient water solubility for thorough relaxometric study, the presence of the PEG may interfere with water coordination or exchange. Previous studies with TREN-capped complexes indicated that long PEG chains can limit water access to the metal center.[17, 20, 21] Since the exchange mechanism is associative, the rate of exchange depends on local water concentration and a decrease in [H2O] results in a slower exchange rate.[20] Ethylene glycol chains have also been know to coordinate the lanthanide ion via the ether oxygen and can displace coordinated waters, decreasing relaxivity.[21] Another route towards highly soluble ME-capped derivatives was thus examined by the generation of amine-substituted complexes 7 and 8. Pendant amine groups have been used in our group in recent work and have produced Gd complexes of relatively high aqueous solubility and favorable relaxivity properties.[17, 22] It is also expected that the amine nitrogens will not coordinate the Gd, eliminating the potential for water displacement that may be of concern for the PEG derivative. Consequently, synthesis of Gd-ME-bis-(1-Me-3,2-HOPO)-TAM-N3 (7) was undertaken as shown in Scheme 2. First, TREN-diBOC (18) was prepared following an established procedure[23] and then coupled to di-thiazolide activated TAM. Slow addition, high dilution conditions were employed to increase the yield of the mono-substituted product (19), which was then reacted with 14 to yield the benzyl protected ligand 20. Stirring in HCl/HOAc (50/50) afforded the benzyl/BOC deprotected ligand 21 which was then used to prepare stock solutions of the complex by titrating a solution of ligand with Gd(III) standardized stock solution for relaxivity studies. Also, for comparison with the relaxivity data obtained for the in situ prepared Gd-complex solution, the solid complex Gd-ME-bis-(1-Me-3,2-HOPO)-TAM-N3 (7) was prepared. Measurements for both the in situ prepared solution and the solution of the isolated complex yielded identical relaxivity results.
Scheme 2.

Synthesis of Gd-ME-bis-(1-Me-3,2-HOPO)-TAM-N3.
Synthesis of another amine derivative possessing only one primary amine is shown in Scheme 3. For this synthesis, the ME-bis-(1-Me-3,2-HOPO)-TAM-thiaz (22) intermediate was prepared by reacting the bis-HOPO species (14) with an excess of dithiazolide-activated TAM precursor. Mono-BOC protected ethylenediamine was then used to produce ME-bis-(1-Me-3,2-HOPO-Bn)-TAM-Bn2-N1-BOC (23) in good yield. Deprotection of the benzyl and BOC groups simultaneously was accomplished under acidic conditions to yield the free ligand 24 used to prepare in situ solutions of the Gd-complex of 8 for relaxivity studies.
Scheme 3.

Synthesis of ME-bis-(1-Me-3,2-HOPO)-TAM-N1.
Relaxometric Measurements and Aggregate Characterization
Relaxivity values at 20 MHz and 298 K were determined for 6, 7, and 8, and confirmed by verification of Gd concentration. At pH=7.2 the relaxivities of Gd-ME-N3 (7) and Gd-ME-N1 (8) are 11.9 and 9.8 mM−1s−1, respectively, initially suggesting q = 3 for 7 and q = 2 for 8 based on comparisons with typical relaxivity values determined for HOPO-TAM complexes of low molecular weight. However, upon refinement of the Nuclear Magnetic Relaxation Dispersion (NMRD) profiles for these complexes (Figure 3), it is found that no satisfactory refinement yielding reasonable parameters can be obtained assuming q of 2 or 3. Refinements are only possible with a low, fractional q value close to zero and surprisingly long τR values in the nanosecond regime (values on the order of ~100 ps are expected for small molecules at room temperature). In addition, the relatively large difference between the low and high field relaxivity values as well as the peak centered about 30 MHz are characteristic of a high-MW, slowly rotating species.[4, 24–26] As seen in the parameters obtained from the NMRD fits given in Table 1, τR values of several nanoseconds are employed in the fit suggesting some type of aggregation or supramolecular species in solution.
Figure 3.
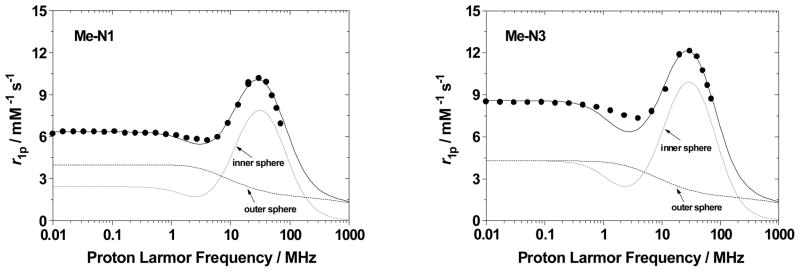
1/T1 NMRD profiles at 25 °C and pH=7.4 of Gd-ME-N1 (8) (left) and Gd-ME-N3 (7) (right).
Table 1.
Best fitting parameters of the NMRD profiles for 7 and 8.
| Parameter | ME-N1 (8) | ME-N3 (7) |
|---|---|---|
| Δ2 (s−2) | 1.2×1019 | 0.8×1019 |
| τV(ps) | 45.7 | 42.6 |
| τR(ns) | 2.2 | 2.5 |
| τM(ns)a | 20 | 20 |
| rGd-H(Å)a | 3.00 | 3.00 |
| q | 0.14 | 0.16 |
fixed in the fitting procedure
The tendency for the ME complexes to exhibit such different solution behavior than what is observed for TREN-capped complexes is evident when compared to the closely related TREN-capped bis-HOPO-TAM derivatives recently reported.[22] These complexes are identical in structure to the N1/N3 compounds reported here, except that they possess the TREN cap instead of ME. For these TREN-capped complexes, relaxivities of 11.1 and 9.0 mM−1s−1 were determined for Gd-TREN-bis-(1-Me-3,2-HOPO)-TAM-N1 and Gd-TREN-bis-(1-Me-3,2-HOPO)-TAM-N3, respectively. Further studies indicated that the N1 substituent in the TREN-capped analog was capable of hydrogen bonding a third water molecule, facilitating its coordination, while the N3 substituted complex exhibited the expected q of 2. Clearly, no such hydrogen bonding interaction is consistent with the relaxivity measurements performed for the Gd-ME-N1 complex, and in fact the relaxivity of Gd-ME-N3 is slightly higher than that of the q = 3 Gd-TREN-N1 compound, suggesting a different structure than a simple q = 2 or q = 3 monomer for the ME complexes. Comparison of the NMRD profiles in Figure 4 illustrates both the different shapes and the more similar low and high-field relaxivity values obtained for the monomer TREN compounds versus what is measured for the aggregate ME species.
Figure 4.
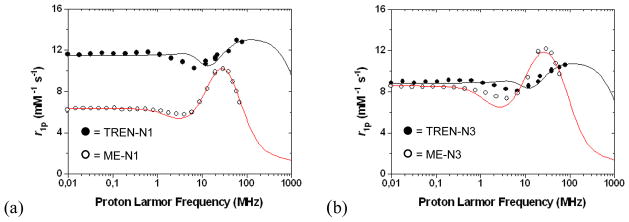
Comparison of the NMRD profiles for (a) Gd-TREN-N1 and Gd-ME-N1 (8) and (b) Gd-TREN-N3 and Gd-ME-N3 (7).
The pH dependency of the relaxivity, shown in Figure 5, is also dramatically different for the ME complexes. In all TREN-capped complexes, the relaxivity peaks at low pH, due to partial dissociation of the complex and increased q, and upon raising the pH the relaxivity decreases to ~9 – 12 mM−1s−1, depending on q, and is constant from about pH 6 – 12. This typical profile shape is not observed for the ME complexes, and in the case of Gd-ME-N3 (7) the relaxivity decreases from 16.3 to 10.3 mM−1s−1 on passing from low (pH ca. 2) to high pH (pH ca. 10). The relaxivity decreases almost linearly (R2=0.99) and the variation is as high as 0.8 (mM−1s−1) per pH unit. Furthermore, this behavior is fully reversible in this pH interval and can be attributed to differences in aggregate size with changes in pH as was seen in past studies of Gd containing fullerenes which showed similar pH-dependent relaxivities.[26, 27]
Figure 5.

pH dependency of the relaxivity (20 MHz, 25 °C for Gd-TREN-N1 and Gd-ME-N1 (8) (top) and Gd-TREN-N3 and Gd-ME-N3 (7) (bottom).
It is worth noting that the unusual pH dependency of the relaxivity of 7 may have implications for the growing interest in pH responsive contrast agents. Studies have shown that the extracellular fluid of tumor cells is more acidic than it is for normal cells due to acid elimination from the tumor.[28, 29] Compounds such as the gadofullerenes noted above, which possess relaxivities with strong pH dependencies similar to that of 7, have been proposed as pH responsive agents suitable for pH mapping of tissues.[26] As the unique relaxivity properties of the ME-capped complexes are attributed to aggregate formation, these large assemblies must persist in solution for use in such in vivo applications. Studies of 7 were thus conducted to determine the effect of increased phosphate concentration on relaxivity, as it has been shown that addition of such biologically available salts may lead to deaggregation and consequent lower relaxivity.[30] In the case of 7, relaxivity is only slightly decreased in the presence of high phosphate concentrations (up to ~200 mM, Figure 6).
Figure 6.

Plot of observed relaxation rate of a solution of Gd-ME-N3 (7) upon progressive addition of phosphate. A slight drop in relaxation rate is seen initially, but the change is minor and no decrease is seen above concentrations of 20 mM phosphate ([Gd] = 0.42 mM, 20 MHz, 25 °C).
While the NMRD profiles have been strongly suggestive of large assemblies of ME complexes in solution as discussed above, separate direct evidence for this unexpected aggregation phenomenon is desirable. In other systems relevant to contrast agent design, dynamic and static light scattering (DLS and SLS) has been employed in the characterization of high molecular weight aggregates and has become a standard technique in understanding aggregation size and behavior under various conditions.[27, 31] The gadofullerenes, for example, have been characterized by DLS as a function of pH, concentration, and temperature to determine their aggregation properties. Dynamic light scattering experiments were performed for solutions of Gd-ME-N3 (7) and Gd-ME-N1 (8) at pH 7.4 and 25 °C. To ensure measurements were not affected by precipitates, the solutions were filtered through 0.2 micron syringe filters immediately before measurement. Average hydrodynamic diameters for 7 and 8 were found to be 47 and 29 nm, respectively. The larger average aggregate size for 7 is consistent with the higher relaxivity measured as compared to 8, suggesting that the higher MW aggregate results in slower rotation and consequent higher relaxivity. However, a contribution to the relaxivity from a different degree of hydration of the species in solution cannot be excluded.
Relaxivity data were also obtained for the highly soluble PEG derivative ME-bis-(1-Me-3,2-HOPO)-TAM-PEG450 (6), and an r1p of 9.5 mM−1s−1 (20 MHz, 298 K, pH 7.4) was determined. This value is higher than the r1p of 7.7 mM−1s−1 measured for the analogous TREN-capped complex.[21] The NMRD profile shown in Figure 7, like those recorded for 7 and 8, shows the characteristic peak at 30 MHz, again consistent with large assemblies. It is noteworthy, however, that in this case the difference between the low and high field relaxivity values is not as great, suggesting aggregation but to a lesser extent than what is observed for the amine-substituted derivatives. This can be explained by the PEG chain partially wrapping itself around the complex, making formation of larger aggregates difficult and favoring formation of the monomer complex. Moreover, the best fit to the experimental NMRD profile shown in Figure 7 yielded a rotational correlation time significantly shorter than those obtained for complexes 7 and 8, as well as a higher q value (see Supporting Information for fit and parameters used). This result, combined with the rather poor NMRD fit at high fields, is again consistent with a combination of monomeric complexes and larger aggregates in solution.
Figure 7.

1/T1 NMRD profile for 6 (25 °C, pH 7.4).
While aggregation of the ME complexes may be attributed to hydrophobic interactions, as was reported in another ligand system employing a similar mesityl cap with pyrazole chelators,[31] it is envisioned that in this case large aggregates may consist of coordination polymers. Such polymeric species may form with bridging HOPO/TAM chelators, where, for example, two bidentate chelate groups coordinate one Gd, and the third coordinates a second Gd ion, and so on resulting in large supramolecular assemblies. The idea of such a supramolecular system is supported by the inability to break apart the aggregates in the presence of phosphate. If the aggregation was due to hydrophobic interactions, as is the case for the gadofullerenes, salt addition would result in significant deaggregation and a decrease in relaxivity. As indicated by the phosphate titrations mentioned above, such a dramatic decrease is not seen for Gd-ME-N3 (7). Also, the hydrogen bonding network involving the central nitrogen in TREN-capped derivatives, known to help lock all three chelators in a predisposed position for metal binding,[32, 33] is absent in the ME-capped complexes. As a result, all three binding groups are not necessarily positioned on the same side of the mesityl ring directing them towards a single metal ion.
Conclusions
The work presented here details the synthesis and relaxometric investigation of a new series of soluble mesityl-capped complexes. The relaxivity values measured for the PEG (6) and N3-substituted (7) complexes are higher than the corresponding TREN-capped analogs, and NMRD analyses of all ME-capped complexes suggests the presence of large assemblies in solution. This is confirmed by dynamic light scattering experiments, which give approximate sizes of about 40 nm for the aggregates, in agreement with the rotational correlation times obtained from the NMRD profile fittings. The unusual pH dependent relaxivities determined for the ME compounds may be explained by the effect of pH on aggregate size and possibly hydration, and may prove useful in tissue pH-mapping. It would be interesting in future studies to perform variable pH/temperature DLS experiments to further probe this unexpected solution behavior. The above data provide insight into the unusual relaxometric behavior of these soluble ME-capped complexes and reveal promising features that may be valuable in future contrast agent applications.
Experimental
Synthesis
General
Unless otherwise noted, starting materials were obtained from commercial suppliers and used without further purification. All organic extracts were dried over MgSO4 and solvents were removed with a rotary evaporator. Flash chromatography was performed on Merck Silica Gel (40-7 Mesh). Unless otherwise noted, 1H NMR spectra were recorded in CDCl3 on a Bruker AMX 300, 400, or DRX 500 instrument at 300, 400, or 500 MHz, and 13C NMR spectra were recorded in CDCl3 on a Bruker DRX 500 instrument at 125 MHz. The residual solvent peak was used as an internal reference. Elemental analyses were performed by the Microanalytical Laboratory at the College of Chemistry at the University of California, Berkeley and mass spectra (LRFAB-MS = low resolution fast atom bombardment mass spectrometry; ES-MS = electrospray mass spectrometry) were performed by the Mass Spectrometry Laboratory at the College of Chemistry at U.C., Berkeley.
ME-bis-1-Me-3,2-HOPO-Bn (14)
To a stirring solution of 1,3,5-tris-aminomethyl-benzene (12)[34, 35] (0.700 g, 4.24 mmol) and NEt3 (3 mL) in CH2Cl2 (250 mL) was added 1-Me-3,2-HOPO-thiaz (11) (2.83 g, 7.84 mmol) in CH2Cl2 (350 mL) dropwise through a narrow capillary over the course of 9 days. TLC indicated the formation of the mono-, bis-, and a small amount of the tris- species so the addition was stopped before complete of addition of the solution of 11. After stirring the reaction mixture under N2 overnight, the solvent was removed in vacuo and the product isolated with a silica column. A gradually increasing gradient of MeOH concentration in CH2Cl2 was used with the pure solid white product eluting with CH2Cl2/MeOH/NEt3 (94:5:1) (1.72 g, 62.5%). 1H NMR (400 MHz): δ 3.59 (s, 6H, HOPO CH3), 3.74 (s, 2H, ME-CH2-NH2), 4.35 (d, 4H, ME-CH2-NH-, J = 5.6), 5.29 (s, 4H, Bn CH2), 6.80 (d, 2H, HOPO Ar-H, J = 7.2), 6.85 (s, 1H, ME Ar-H), 7.01 (s, 2H, ME Ar-H), 7.12 (d, 2H, HOPO Ar-H, J = 7.2) 7.20–7.27 (m, 10H, Bn Ar-H), 8.28 (br t, 2H, NH); LRFAB-MS: m/z 648.5 (MH+).
ME-bis-(1-Me-3,2-HOPO-Bn)-TAM-Bn2-PEG450 (15)
ME-bis-1-Me-3,2-HOPO-Bn (14) (0.793 g, 1.22 mmol) and TAM-Bn2-thiaz-PEG450 (13)[21] (1.20 g, 1.22 mmol) were dissolved in CH2Cl2 (40 mL). NEt3 (0.124 g, 1.22 mmol) was added and the reaction mixture was stirred at room temperature, under N2, and in the dark for 5 days. The solvent was removed under high vacuum and the pure product was obtained after silica column chromatography, eluting with CH2Cl2 and a gradient of increasing MeOH concentration with the product eluting with CH2Cl2/MeOH (92:8) (0.580 g, 31.5%). 1H NMR (400 MHz): δ 3.14 (s, 3H, PEG CH3), 3.32–3.45 (m, 50H, PEG CH2, HOPO CH3), 4.07 (d, 4H, ME CH2, J = 5.6), 4.14 (d, 2H, ME CH2, J = 5.6), 4.81 (s, 2H, TAM Bn CH2), 4.85 (s, 2H, TAM Bn CH2), 5.03 (s, 4H, HOPO Bn CH2), 6.50 (d, 2H, HOPO Ar-H, J = 7.2), 6.65 (s, 1H, ME Ar-H), 6.70 (s, 2H, ME Ar-H), 6.88 (d, 2H, HOPO Ar-H, J = 7.2), 6.95–7.15 (m, 20H, Bn Ar-H), 7.62 (d, 1H, TAM Ar-H, J = 8.4), 7.68 (d, 1H, TAM Ar-H, J = 8.4), 7.83 (br t, 1H, TAM NH), 7.88 (t, 1H, TAM NH, J = 5.6), 8.02 (t, 2H, HOPO NH, J = 5.6); 13C NMR: δ 13.39, 21.84, 30.76, 36.95, 58.23, 69.71, 71.08, 73.77, 103.77, 125.33, 127.77, 127.88, 127.91, 127.94, 127.96, 128.09, 129.60, 131.92, 134.59, 134.81, 135.13, 138.11, 138.31, 145.45, 149.57, 149.80, 158.66, 162.37, 163.26; LRFAB-MS: m/z 1563.3 (M + K+ + H2O, median m/z for 11 ether PEG, additional peaks +/− 44 m/z units corresponding to -CH2CH2O- units).
ME-bis-(1-Me-3,2-HOPO)-TAM-PEG450 (16)
ME-bis-(1-Me-3,2-HOPO-Bn)-TAM-Bn2-PEG450 (15) (0.383 g, 0.254 mmol) was dissolved in glacial HOAc (10 mL), concentrated HCl (2 mL), and H2O (2 mL) in a glass container. Activated Pd/C (10 wt. %) (0.095 g) and a small stir bar was added and the container was placed in a steel bomb. After purging twice with H2, the bomb was filled with H2 to ca. 1,100 psi and the reaction mixture was stirred overnight. TLC indicated the absence of any starting material, and a spot of FeCl3 turned the product black indicating the presence of the Fe complex and the deprotected ligand. The reaction mixture was filtered through a glass fritted filter and the solvent was evaporated under high vacuum with heating to 40°C. The residue was dissolved in MeOH and the solvent removed (×5). The light tan residue was redissolved in MeOH (5 mL) and added dropwise to a rapidly stirring solution of Et2O (300 mL) and the solution was stirred overnight in the dark. The light tan product was filtered and immediately transferred to a vacuum oven and left under high vacuum at 40°C for two days (0.290 g, 100%). 1H NMR (CD3OD, 400 MHz): δ 3.48–3.70 (m, 53H, PEG CH2, CH3, HOPO CH3), 4.50–4.60 (m, 6 H, ME CH2), 6.59 (d, 2H, HOPO Ar-H, J = 7.2), 7.06 (d, 2H, HOPO Ar-H, J = 7.2), 7.15–7.30 (m, 9H, TAM Ar-H, ME Ar-H, NH); LRFAB-MS: m/z 1213.6 (M + Na+, median m/z for 12 ether PEG, additional peaks +/− 44 m/z units corresponding to -CH2CH2O- units).
Gd-ME-bis-(1-Me-3,2-HOPO)-TAM-PEG450 (6)
Me-bis-(1-Me-3,2-HOPO)-TAM-PEG450 (16) (0.0700 g, 0.0589 mmol) was dissolved in MeOH (6 mL) and the solution was purged with N2 for 10 min. A solution of Gd(acac)3·3H2O (0.0284 g, 0.0560 mmol) in MeOH (5 mL) was added, followed by the addition of 4 drops of pyridine. The solution was allowed to reflux under N2 overnight. The solvent was removed in vacuo and the residue was redissolved in MeOH (4 mL) and added to stirring Et2O (350 mL). The solution, with a white suspended precipitate, was stirred in the dark for 2 days. The off-white product was filtered and dried under high vacuum and at 40°C overnight (0.0700 g, 94.8%). MALDI-MS(−): m/z 1253.06 (M−, median m/z for 10 ether PEG, additional peaks +/− 44 m/z units corresponding to -CH2CH2O- units), The isotopic distribution matches closely with the simulated spectrum.
TAM-thiaz-N3-diBOC (19)
A solution of TREN-diBOC (18)[23] (0.538 g, 1.55 mmol) and NEt3 (0.160 g, 1.58 mmol) in CH2Cl2 (200 mL) was slowly added dropwise to a rapidly stirring solution of TAM-Bn2-thiaz2 (7.21 g, 12.4 mmol) in CH2Cl2 (400 mL). Addition was complete after 4 days, and the solvent was removed in vacuo. The residue was redissolved in a minimal amount of CH2Cl2 and loaded onto a silica column packed with CH2Cl2. A gradually increasing gradient of MeOH in CH2Cl2 was used, with the pure solid yellow product eluting with CH2Cl2:MeOH (98:2) (0.992 g, 79.2%). 1H NMR (400 MHz): δ 1.40 (s, 18H, BOC CH3), 2.39 (br. t, 2H, TREN CH2), 2.49 (br. t, 4H, TREN CH2), 2.93 (t, 2H, thiaz CH2, J = 7.2), 3.06 (br. m, 4H, TREN CH2), 3.26 (m, 2H, TREN CH2), 4.37 (t, 2H, thiaz CH2, J = 7.4), 5.12 (s, 2H, Bn CH2), 5.13 (s, 2H, Bn CH2), 7.21 (d, 1H, TAM Ar-H, J = 8.4), 7.30–7.43 (m, 10H, Bn Ar-H), 7.79 (br. t, 1H, amide H), 7.90 (d, 1H, TAM Ar-H, J = 8.4); ES-MS(+): m/z 808.3 (M+H+).
ME-bis-(1-Me-3,2-HOPO-Bn)-TAM-Bn2-N3-diBOC (20)
A solution of TAM-thiaz-N3-diBOC (19) (0.499 g, 0.618 mmol) in CH2Cl2 (10 mL) was added to a solution of ME-bis-(1-Me-3,2-HOPO) (14) (0.400 g, 0.618 mmol) and NEt3 (0.063 g, 0.618 mmol) dissolved in CH2Cl2 (20 mL). After the solution was allowed to stir under N2 and in the dark for 3 days, the persistent yellow color of the solution indicated unreacted TAM-thiaz, and TLC showed the reaction progression to be ca. 50% complete. A few drops of NEt3 were added, and the solution allowed to stir for an additional 2 days. After removal of the solvent in vacuo, the residue was redissolved in a minimal amount of CH2Cl2 and loaded onto a silica column packed with CH2Cl2. A gradually increasing gradient of MeOH in CH2Cl2 was used, with the pure solid white product eluting completely with CH2Cl2:MeOH (95:5) (0.365 g, 44.2%). 1H NMR (500 MHz): δ 1.37 (s, 18H, BOC CH3), 2.40 (br. t, 2H, TREN CH2), 2.48 (t, 4H, TREN CH2, J = 5.8), 3.05 (br. m, 4H, TREN CH2), 3.30 (q, 2H, TREN CH2, J = 6.2), 3.54 (s, 6H, HOPO CH3), 4.27 (d, 4H, ME CH2, J = 5.5), 4.36 (d, 2H, ME CH2, J = 5.5), 5.03 (br. t, 2H, BOC amide H), 5.06 (s, 2H, TAM Bn CH2), 5.24 (s, 4H, HOPO Bn CH2), 5.27 (s, 2H, TAM Bn CH2), 6.73 (d, 2H, HOPO Ar-H, J = 7.0), 6.85 (s, 1H, ME Ar-H), 6.90 (s, 2H, ME Ar-H), 7.07 (d, 2H, HOPO Ar-H, J = 7.0), 7.11–7.39 (m, 20H, Bn Ar-H), 7.79 (t, 1H, TAM amide H, J = 5.5), 7.85 (d, 1H, TAM Ar-H, J = 8.0), 7.92 (d, 1H, TAM Ar-H, J = 8.5), 8.01 (t, 1H, TAM amide H, J = 5.8), 8.20 (t, 2H, HOPO amide H); 13C NMR (500 MHz): δ 14.02, 22.54, 28.30, 31.47, 37.58, 38.33, 43.26, 43.37, 53.36, 53.71, 74.64, 79.02, 104.65, 126.12, 126.24, 126.32, 126.61, 128.43, 128.54, 128.58, 128.69, 128.78, 128.81, 128.90, 130.09, 132.10, 135.30, 135.70, 135.77, 138.79, 138.97, 146.39, 150.27, 150.34, 156.08, 159.37, 162.99, 163.83; LRFAB-MS(+): m/z 1336.7 (M+H+).
ME-bis-(1-Me-3,2-HOPO)-TAM-N3 (21)
To a solution of ME-bis-(1-Me-3,2-HOPO-Bn)-TAM-Bn2-N3-diBOC (20) (0.250 g, 0.187 mmol) in glacial acetic acid (5 mL) was added concentrated HCl (5 mL). The solution was stirred under N2 in the dark for 16 hours. TLC indicated the absence of any starting material, and a spot of FeCl3 turned the product black indicating the presence of the Fe complex and the deprotected ligand. The solvent was evaporated under high vacuum with heating to 40 °C. The residue was suspended in MeOH and the solvent removed (×3) to facilitate removal of HOAc. The residue was suspended in MeOH (7 mL) and added dropwise to a rapidly stirring solution of Et2O (500 mL) and the solution was stirred overnight in the dark. The light purple product was filtered and immediately transferred to a vacuum oven and left under high vacuum at 40°C overnight (0.140, 84.7%). 1H NMR (D2O, 500 MHz): δ 2.75 (br. t, 2H, TREN CH2), 2.81 (br. t, 4H, TREN CH2), 3.04 (br. t, 4H, TREN CH2), 3.38 (s, 6H, HOPO CH3), 3.44 (br. t, 2H, TREN CH2), 4.36 (m, 6H, ME CH2), 6.35 (d, 2H, HOPO Ar-H), 6.89 (d, 2H, HOPO Ar-H), 6.90–7.08 (m, 5H, TAM Ar-H, ME Ar-H); LRFAB-MS(+): m/z 776 (M+H+); Anal. Calcd for 21·3HCl·2H2O·1.5MeOH (Found): C 47.71 (48.03), H 6.03 (5.85), N 13.01 (12.69).
Gd-ME-bis-(1-Me-3,2-HOPO)-TAM-N3 (7)
A solution of ME-bis-(1-Me-3,2-HOPO)-TAM-N3 (21) (34.28 mg, 35.37 μmol) in MeOH (8 mL) was purged with N2 for 15 min. Gd(acac)3 (17.99 mg, 35.37 μmol) dissolved in MeOH (3 mL) was added to the solution of 21, and the solution was purged with N2 for an additional 10 minutes. Pyridine (2 drops) was added and the mixture was stirred at reflux, under N2, for 19 hours. After the solution was allowed to cool to room temperature, the pale yellow solution was added to stirring Et2O to precipitate the complex. The suspension was allowed to stir overnight before collecting the beige solid by filtration. The product was transferred to a vacuum oven and left under high vacuum at 40°C for 2 days (35.10 mg, 95.6%). ES-MS(−): m/z 929.2 (M−); Anal. Calcd for 7·3HCl·4.5H2O·3MeOH (Found): C 39.52 (39.51), H 5.39 (4.68), N 10.37 (9.98).
ME-bis-(1-Me-3,2-HOPO-Bn)-TAM-Bn2-thiaz (22)
A solution of ME-bis-(1-Me-3,2-HOPO-Bn) (14) (0.380 g, 0.587 mmol) and NEt3 (0.060 g) in CH2Cl2 (75 mL) was added dropwise over 24 h to a solution of TAM-Bn2-thiaz2 (0.444 g, 0.765 mmol) in CH2Cl2 (250 mL). After complete addition, an additional 0.5 mL of NEt3 was added and the reaction mixture was stirred for an additional 2 days. The solvent was removed in vacuo and the residue redissolved in a minimal amount of CH2Cl2 and loaded onto a silica column packed with CH2Cl2. An increasing gradient of MeOH was used with the product eluting in 97:3, CH2Cl2:MeOH. Removal of solvents in vacuo afforded the pure solid yellow product (0.335g, 51.4%). 1H NMR (500 MHz): δ 2.91 (t, 2H, thiaz CH2, J = 7.5), 3.52 (s, 6H, HOPO CH3), 4.27 (d, 4H, ME-CH2-HOPO, J = 6.0), 4.29–4.35 (m, 4H, ME-CH2-TAM, thiaz CH2), 4.97 (s, 2H, TAM Bn CH2), 5.01 (s, 2H, TAM Bn CH2), 5.23 (s, 4H, HOPO Bn CH2), 6.72 (d, 2H, HOPO Ar H, J = 7.5), 6.86 (s, 1H, ME Ar H), 6.90 (s, 1H, ME Ar H), 7.03–7.10 (m, 4H, HOPO Ar H, Bn Ar H), 7.13–7.24 (m, 14H, Bn Ar H), 7.29–7.36 (m, 5H, Bn Ar H, TAM Ar H), 7.89 (d, 1H, TAM Ar H, J = 8.5), 8.04 (t, 1H, TAM amide H, J = 5.8), 8.21 (t, 2H, HOPO amide H, J = 5.8); 13C NMR (125 MHz): δ 28.51, 37.51, 43.21, 43.31, 55.36, 74.53, 75.88, 104.55, 124.22, 126.05, 126.28, 126.69, 127.75, 128.24, 128.32, 128.44, 128.51, 128.54, 128.62, 128.66, 128.74, 129.52, 130.06, 132.08, 133.48, 135.21, 135.70, 136.72, 138.69, 138.94, 146.28, 149.12, 149.85, 159.30, 162.93, 163.75, 166.53, 201.19; LRFAB-MS(+): m/z 1109.7 (M+H+).
ME-bis-(1-Me-3,2-HOPO-Bn)-TAM-Bn2-N1-BOC (23)
To a solution of ME-bis-(1-Me-3,2-HOPO-Bn)-TAM-Bn2-thiaz (22) (0.111 g, 0.100 mmol) in CH2Cl2 (10 mL) was added a solution of (2-Amino-ethyl)-carbamic acid tert-butyl ester (0.016 g, 0.100 mmol) and NEt3 (0.010 g) in CH2Cl2 (5 mL). The solution was stirred under N2 until color change of the solution from yellow to colorless, as well as TLC, indicated complete reaction of the amine with the thiazolide of 22 after two days. Solvents were removed in vacuo, and the residue was redissolved in a minimal amount of CH2Cl2 and loaded onto a silica column packed in CH2Cl2. An increasing gradient of MeOH was used with the product eluting in 97:3–96:4, CH2Cl2:MeOH. Pure fractions were combined and solvents removed in vacuo to afford a white, waxy solid (0.110 g, 95.6%). 1H NMR (500 MHz): δ 1.41 (s, 9H, BOC CH3), 3.57 (s, 6H, HOPO CH3), 4.30 (d, 4H, ME-CH2-HOPO, J = 6.0), 4.39 (d, 2H, ME-CH2-TAM, J = 5.5), 5.04 (s, 2H, TAM Bn CH2), 5.08 (s, 2H, TAM Bn CH2), 5.27 (s, 4H, HOPO Bn CH2), 6.76 (d, 2H, HOPO Ar H, J = 7.0), 6.88 (s, 1H, ME Ar H), 6.93 (s, 2H, ME Ar H), 7.10 (d, 2H, HOPO Ar H, J = 7.0), 7.12–7.42 (m, 20H, Bn Ar H), 7.86 (d, 1H, TAM Ar H, J = 8.0), 7.89–7.96 (m, 2H, TAM Ar H, TAM NHCO), 8.06 (t, 1H, TAM NHCO, J = 5.5), 8.25 (t, 2H, HOPO NHCO, J = 5.8); 13C NMR (125 MHz): δ 14.05, 22.56, 28.27, 37.62, 39.99, 43.26, 43.34, 53.38, 74.67, 77.15, 104.68, 126.11, 126.14, 126.30, 126.55, 128.44, 128.61, 128.66, 128.70, 128.72, 128.84, 128.98, 130.10, 132.12, 135.26, 135.50, 135.78, 138.79, 139.02, 146.41, 150.35, 156.12, 159.39, 163.00, 163.85, 164.91; LRFAB-MS(+): m/z 1151 (M+H+).
ME-bis-(1-Me-3,2-HOPO)-TAM-N1 (24)
To a solution of ME-bis-(1-Me-3,2-HOPO-Bn)-TAM-Bn2-N1-BOC (23) (0.030 g, 0.026 mmol) in glacial acetic acid (3 mL) was added concentrated HCl (3 mL). The solution was stirred under N2 in the dark for 30 hours. After TLC indicated the absence of any starting material, the solvent was evaporated under high vacuum with heating to 40°C. The residue was suspended in MeOH and the solvent removed (×5) to facilitate removal of HOAc. The residue was suspended in MeOH (4 mL) and added dropwise to a rapidly stirring solution of Et2O (300 mL) and the solution was stirred overnight in the dark. The purple product was filtered and immediately transferred to a vacuum oven and left under high vacuum at 40°C for 2 days (0.015, 79.5%). 1H NMR (MeOH, 500 MHz): δ 3.25 (t, 2H, H2N-CH2), 3.57 (s, 6H, HOPO CH3), 3.73 (t, 2H, N1 CONH-CH2-CH2), 4.58 (br. s, 6H, ME CH2), 6.59 (d, 2H, HOPO Ar-H), 7.08 (d, 2H, HOPO Ar-H), 6.90–7.20 (m, 5H, TAM Ar-H, ME Ar-H); LRFAB-MS(+): m/z 690 (M+H+).
Relaxivity Measurements
1H NMR
The water proton longitudinal relaxation rates of 1 mM aqueous solutions of complexes 6, 7, and 8 were measured by using a Stelar Spinmaster spectrometer (Mede, Italy) operating at 0.5 T and 25 °C. In the case of visible precipitation of the complex, dilutions were made to ensure complete dissolution. The standard inversion-recovery method was employed (16 experiments, 4 scans) with a typical 90° pulse width of 3.5 ms, and the reproducibility of the T1 data was ± 0.5%. The temperature was controlled with a Stellar VTC-91 air-flow heater equipped with a copper-constantan thermocouple (uncertainty of ± 0.1 °C). The proton 1/T1 NMRD profiles were measured on a fast field-cycling Stelar Spinmaster FFC relaxometer over a continuum of magnetic field strengths from 0.00024 to 0.5 T (corresponding to 0.01–20 MHz proton Larmor frequencies). The relaxometer operates under computer control with an absolute uncertainty in 1/T1 of ± 1%.
Dynamic Light Scattering measurements
DLS experiments were performed using a Zetasizer, Nano ZS instrument (Malvern Instruments) with a He-Ne laser operating at 633 nm. The machine operates under computer control, and the intensity-size distributions and mean hydrodynamic diameters were determined from measured correlation curves by the instrument software package. Sample solutions of 21 and 24 were prepared by dissolving a known amount of ligand in Millipore H2O, and titrating into each solution enough standardized Gd stock solution to result in a slight excess (~5%) of ligand. The solutions were brought to pH = 7.4 by the addition of HCl/NaOH, and filtered through a 0.20 μm syringe filter immediately before measurements.
Supplementary Material
Acknowledgments
We acknowledge financial support from the NIH (Berkeley, grant HL69832), NATO (Torino, travel grant PST.CLG.980380) and MIUR (Torino, PRIN2005: 2005039914). We thank Dr. Stefano Avedano for help with the relaxometric measurements and Dr. Ankona Datta for help with the manuscript.
References
- 1.Paper no. 27 in the series “High Relaxivity Gadolinium MRI Agents”; for the previous paper in the series see Werner EJ, Kozhukh J, Botta M, Moore EG, Avedano S, Aime S, Raymond KN. 1,2-Hydroxypyridonate/Terephthalamide Complexes of Gadolinium(III): Synthesis, Stability, Relaxivity, and Water Exchange Properties. Inorg Chem. 2009;48:277–286. doi: 10.1021/ic801730u.
- 2.Aime S, Botta M, Terreno E. Gd(III)-based Contrast Agents for MRI. In: van Eldik R, Bertini I, editors. Advances in Inorganic Chemistry. Vol. 57. Elsevier; San Diego: 2005. pp. 173–237. [Google Scholar]
- 3.Caravan P. Strategies for increasing the sensitivity of gadolinium based MRI contrast agents. Chem Soc Rev. 2006;35:512–523. doi: 10.1039/b510982p. [DOI] [PubMed] [Google Scholar]
- 4.Merbach AE, Tóth E, editors. The Chemistry of Contrast Agents in Medical Magnetic Resonance Imaging. Wiley; Chichester: 2001. [Google Scholar]
- 5.Caravan P, Ellison J, McMurry T, Lauffer R. Gadolinium(III) chelates as MRI contrast agents: Structure, dynamics, and applications. Chem Rev. 1999;99:2293–2352. doi: 10.1021/cr980440x. [DOI] [PubMed] [Google Scholar]
- 6.Raymond KN, Pierre VC. Next Generation, High Relaxivity Gadolinium MRI Agents. Bioconjugate Chem. 2005;16:3–8. doi: 10.1021/bc049817y. [DOI] [PubMed] [Google Scholar]
- 7.Werner EJ, Datta A, Jocher CJ, Raymond KN. High-Relaxivity MRI Contrast Agents: Where Coordination Chemistry Meets Medical Imaging. Angew Chem Int Ed. 2008;47:8568–8580. doi: 10.1002/anie.200800212. [DOI] [PubMed] [Google Scholar]
- 8.Hajela SP, Johnson AR, Xu J, Sunderland CJ, Cohen SM, Caulder DL, Raymond KN. Synthesis of Homochiral Tris(2-alkyl-2-aminoethyl)amine Derivatives from Chiral α-Amino Aldehydes and Their Application in the Synthesis of Water Soluble Chelators. Inorg Chem. 2001;40:3208–3216. doi: 10.1021/ic001021x. [DOI] [PubMed] [Google Scholar]
- 9.O’Sullivan B, Doble DMJ, Thompson MK, Siering C, Xu J, Botta M, Aime S, Raymond KN. The Effect of Ligand Scaffold Size on the Stability of Tripodal Hydroxypyridonate Gadolinium Complexes. Inorg Chem. 2003;42:2577–2583. doi: 10.1021/ic0261575. [DOI] [PubMed] [Google Scholar]
- 10.Werner EJ, Avedano S, Botta M, Hay BP, Moore EG, Aime S, Raymond KN. Highly Soluble Tris-hydroxypyridonate Gd(III) Complexes with Increased Hydration Number, Fast Water Exchange, Slow Electronic Relaxation, and High Relaxivity. J Am Chem Soc. 2007;129:1870–1871. doi: 10.1021/ja068026z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harris WR, Raymond KN. Ferric ion sequestering agents. 3. The spectrophotometric and potentiometric evaluation of two new enterobactin analogs: 1,5,9-N,N′,N″-tris(2,3-dihydroxybenzoyl) cyclotriazatridecane and 1,3,5-N,N′,N″-tris(2,3-dihydroxybenzoyl)triaminomethylbenzene. J Am Chem Soc. 1979;101:6534–6541. [Google Scholar]
- 12.Harris WR, Weitl FL, Raymond KN. Synthesis and evaluation of an enterobactin model compound. 1,3,5-Tris-(2,3-dihydroxybenzoylaminomethyl)benzene and its iron(III) complex. J Chem Soc, Chem Commun. 1979:177–178. [Google Scholar]
- 13.Hay BP, Dixon DA, Vargas R, Garza J, Raymond KN. Structural Criteria for the Rational Design of Selective Ligands. 3. Quantitative Structure-Stability Relationship for Iron(III) Complexation by Tris-Catecholamide Siderophores. Inorg Chem. 2001;40:3922–3935. doi: 10.1021/ic001380s. [DOI] [PubMed] [Google Scholar]
- 14.Loomis LD, Raymond KN. Solution equilibria of enterobactin and metal-enterobactin complexes. Inorg Chem. 1991;30:906–911. [Google Scholar]
- 15.Hou Z, Stack TDP, Sunderland CJ, Raymond KN. Enhanced iron(III) chelation through ligand predisposition: syntheses, structures and stability of tris-catecholate enterobactin analogs. Inorg Chim Acta. 1997;263:341–355. [Google Scholar]
- 16.Piyamongkol S, Zhou T, Liu ZD, Khodr HH, Hider RC. Design and characterization of novel hexadentate 3-hydroxypyridin-4-one ligands. Tetrahedron Lett. 2005;46:1333–1336. [Google Scholar]
- 17.Pierre VC, Botta M, Aime S, Raymond KN. Substituent Effects on Gd(III)-Based MRI Contrast Agents: Optimizing the Stability and Selectivity of the Complex and the Number of Coordinated Water Molecules. Inorg Chem. 2006;45:8355–8364. doi: 10.1021/ic061262q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gorden AEV, Xu J, Raymond KN, Durbin P. Rational Design of Sequestering Agents for Plutonium and Other Actinides. Chem Rev. 2003;103:4207–4282. doi: 10.1021/cr990114x. [DOI] [PubMed] [Google Scholar]
- 19.Doble DMJ, Melchior M, O’Sullivan B, Siering C, Xu J, Pierre VC, Raymond KN. Toward Optimized High-Relaxivity MRI Agents: The Effect of Ligand Basicity on the Thermodynamic Stability of Hexadentate Hydroxypyridonate/Catecholate Gadolinium(III) Complexes. Inorg Chem. 2003;42:4930–4937. doi: 10.1021/ic026240s. [DOI] [PubMed] [Google Scholar]
- 20.Doble DMJ, Botta M, Wang J, Aime S, Barge A, Raymond KN. Optimization of the Relaxivity of MRI Contrast Agents: Effect of Poly(ethylene glycol) Chains on the Water-Exchange Rates of GdIII Complexes. J Am Chem Soc. 2001;123:10758–10759. doi: 10.1021/ja011085m. [DOI] [PubMed] [Google Scholar]
- 21.Thompson MK, Doble DMJ, Tso LS, Barra S, Botta M, Aime S, Raymond KN. Hetero-tripodal hydroxypyridonate gadolinium complexes: Syntheses, relaxometric properties, water exchange dynamics, and human serum albumin binding. Inorg Chem. 2004;43:8577–8586. doi: 10.1021/ic048607u. [DOI] [PubMed] [Google Scholar]
- 22.Pierre VC, Botta M, Aime S, Raymond KN. Tuning the Coordination Number of Hydroxypyridonate-Based Gadolinium Complexes: Implications for MRI Contrast Agents. J Am Chem Soc. 2006;128:5344–5345. doi: 10.1021/ja057805x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pierre VC. Ph D Thesis. University of California; Berkeley: 2005. [Google Scholar]
- 24.Accardo A, Tesauro D, Roscigno P, Gianolio E, Paduano L, D’Errico G, Pedone C, Morelli G. Physicochemical properties of mixed micellar aggregates containing CCK peptides and Gd complexes designed as tumor specific contrast agents in MRI. J Am Chem Soc. 2004;126:3097–3107. doi: 10.1021/ja039195b. [DOI] [PubMed] [Google Scholar]
- 25.Fatin-Rouge N, Tóth E, Meuli R, Bünzli J-CG. Enhanced imaging properties of a GdIII complex with unusually large relaxivity. J Alloys Compd. 2004;374:298–302. [Google Scholar]
- 26.Tóth É, Bolskar RD, Borel A, Gonzalez G, Helm L, Merbach AE, Sitharaman B, Wilson LJ. Water-soluble gadofullerenes: Toward high-relaxivity, pH-responsive MRI contrast agents. J Am Chem Soc. 2005;127:799–805. doi: 10.1021/ja044688h. [DOI] [PubMed] [Google Scholar]
- 27.Sitharaman B, Bolskar RD, Rusakova I, Wilson LJ. Gd@C60[C(COOH)2]10 and Gd@C60(OH)x: Nanoscale Aggregation Studies of Two Metallofullerene MRI Contrast Agents in Aqueous Solution. Nano Lett. 2004;4:2373–2378. [Google Scholar]
- 28.Stubbs M, McSheehy PMJ, Griffiths JR, Bashford CL. Causes and consequences of tumour acidity and implications for treatment. Mol Med Today. 2000;6:15–19. doi: 10.1016/s1357-4310(99)01615-9. [DOI] [PubMed] [Google Scholar]
- 29.Tannock IF, Rotin D. Acid pH in tumors and its potential for therapeutic exploitation. Cancer Res. 1989;49:4373–4384. [PubMed] [Google Scholar]
- 30.Laus S, Sitharaman B, Tóth É, Bolskar RD, Helm L, Asokan S, Wong MS, Wilson LJ, Merbach AE. Destroying Gadofullerene Aggregates by Salt Addition in Aqueous Solution of Gd@C60(OH)x and Gd@C60[C(COOH2)]10. J Am Chem Soc. 2005;127:9368–9369. doi: 10.1021/ja052388+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fatin-Rouge N, Tóth E, Perret D, Backer RH, Merbach AE, Bünzli J-CG. Lanthanide Podates with Programmed Intermolecular Interactions: Luminescence Enhancement through Association with Cyclodextrins and Unusually Large Relaxivity of the Gadolinium Self-Aggregates. J Am Chem Soc. 2000;122:10810–10820. [Google Scholar]
- 32.Xu J, Churchill DG, Botta M, Raymond KN. Gadolinium(III) 1,2-hydroxypyridonate-based complexes: Toward MRI contrast agents of high relaxivity. Inorg Chem. 2004;43:5492–5494. doi: 10.1021/ic049028s. [DOI] [PubMed] [Google Scholar]
- 33.Xu J, Franklin SJ, Whisenhunt DW, Raymond KN. Gadolinium complex of tris[(3-hydroxy-1-methyl- 2-oxo-1,2-didehydropyridine-4-carboxamido)ethyl]-amine: A New Class of gadolinium magnetic resonance relaxation agents. J Am Chem Soc. 1995;117:7245–7246. [Google Scholar]
- 34.Venuti MC, Rastetter WH, Neilands JB. 1,3,5-Tris(N,N′,N″-2,3-dihydroxybenzoyl)aminomethylbenzene, a synthetic iron chelator related to enterobactin. J Med Chem. 1979;22:123–124. doi: 10.1021/jm00188a002. [DOI] [PubMed] [Google Scholar]
- 35.Weitl FL, Raymond KN. Ferric ion sequestering agents. 1. Hexadentate O-bonding N,N′,N″-tris(2,3-dihydroxybenzoyl) derivatives of 1,5,9-triazacyclotridecane and 1,3,5-triaminomethylbenzene. J Am Chem Soc. 1979;101:2728–2731. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.