

Abstract
Fanconi anemia (FA) is a genome-instability syndrome that has been associated with both cancer predisposition and bone-marrow failure. FA proteins are involved in cellular response to replication stress in which they coordinate DNA repair with DNA replication and cell cycle progression. One regulator of the replication stress response is the ATP-dependent DNA translocase FANCM which we have shown to be hyperphosphorylated in response to various genotoxic agents. However, the significance of this phosphorylation remained unclear. Here we show that genotoxic stress-induced FANCM phosphorylation is ATR dependent and that this modification is highly significant for the cellular response to replication stress. We identified serine (S1045) residue of FANCM that is phosphorylated in response to genotoxic stress and this effect is ATR dependent. We show that S1045 is required for FANCM functions including its role in FA pathway integrity, recruiting FANCM to the site of interstrand cross links, preventing the cells from entering mitosis prematurely, and efficient activation of the CHK1 and G2/M checkpoints. Overall, our data suggest that an ATR-FANCM feedback loop is present in the FA and replication-stress-response pathways, and that it is required for both efficient ATR/CHK1-checkpoint activation and FANCM function.
Keywords: DNA Repair, Phosphorylation, FANCM, Fanconi anemia, ATR
Introduction
Fanconi anemia (FA) is a genome-instability syndrome characterized by developmental abnormalities, bone-marrow failure, chromosome fragility, and a dramatically elevated cancer incidence (1). The FANCM protein is an ATP-dependent DNA translocase and the encoding gene is mutated in a single FA patient belonging to the complementation group FA-M (2, 3). FANCM along with FAAP24 and FANCM-interacting histone fold protein 1 (MHF1) and MHF2 physically and functionally interacts with additional proteins (including FANCA, FANCB, FANCC, FANCE, FANCF, FANCG, and FANCL, each representing an additional FA complementation group) and their associated factors (including FAAP100, FAAP20 and HES1) to form a larger complex known as the “FA nuclear core complex” (4-7). The FA nuclear core complex acts as an E3 ubiquitin ligase due to the E3 ubiquitin ligase domain in FANCL (8). In response to replication stress, FANCM recruits the core complex to chromatin, and this then monoubiquitinate FANCD2 and FANCI to activate the FA pathway (1). Downstream of this event, additional proteins (e.g., FANCD1/BRCA2, FANCJ/BACH1, FANCN/PALB2, FANCO/RAD51C, and FANCP/SLX4, each representing an additional FA complementation group) work together with FA-associated proteins (FAN1, RAD18, and RAD51) to effect DNA-repair and tolerance reactions. FANCM also plays other significant functions. For example, it provides resistance to ultraviolet (UV) rays and camptothecin (CPT), suppresses spontaneous sister-chromatid exchanges (SCE), and activates the synthesis (S)-phase checkpoint (via ATR) in response to replication stress (3, 9, 10). Despite this extensive catalog of FANCM functions and their clear importance for cellular response to replication stress, our understanding of the mechanisms whereby it acts remain unclear.
FA proteins are involved in cellular response to replication stress in which they coordinate DNA repair with DNA replication and cell cycle progression. In eukaryotes, DNA replication involves replication initiation, replication-fork progression, and replication termination (11, 12). Interruption during replication fork progression results in replication stress (11, 12). Negative influences include the presence of difficult-to-replicate DNA regions (e.g., rDNA, centromeres, telomeres and other repetitive sequences), DNA damage resulting from compounds generated by the cellular metabolism (e.g., reactive oxygen species) and genotoxic agents. The genotoxic agents include compounds that: cause interstrand crosslinks (ICLs), e.g., mitomycin C (MMC) and cisplatin; result in failure to remove covalently bound Topo I to DNA, e.g., camptothecin (CPT); and deplete the dNTP pool by inhibiting the enzyme ribonucleotide reductase, e.g. hydroxyurea (HU). The consequences of replication-fork stalling include the following. First, a long stretch of single-stranded DNA (ssDNA) is generated, and it is recognized as DNA damage by the cell-cycle-checkpoint machinery (13). Replication protein A (RPA), a single-strand binding protein, binds ssDNA and recruits the checkpoint kinase ATR (ATM- and Rad3-related) and its binding partner ATRIP (ATR-interacting protein) ATR then phosphorylates a large number of substrates to trigger the checkpoint response (11, 13). One such substrate is CHK1 kinase which phosphorylates CDC25A resulting in the inactivation of cyclin-dependent kinases (CDKs) to promote cell-cycle arrest (11, 13). Additionally, ATR/CHK1 signaling is important for activating the FA pathway (12, 14), including the core-complex proteins FANCA (S1449), FANCG (S7) and FANCE (T346 and S374), and this is necessary for cellular tolerance to MMC. Also, ATR/CHK1-mediated phosphorylation of FANCI and FANCD2 is essential for efficient monoubiquitination of these proteins (12, 14). Recently, FANCM was found to be required for the activation of ATR-mediated checkpoint signaling, and it was suggested to play an “upstream” role in ATR checkpoint activation (9, 15, 16). FANCM and its partner FAAP24 associate with checkpoint protein HCLK2 (9, 17). This activity of FANCM is independent of the FA core complex (9). Silencing of FANCM in mammalian cells confers a phenotype similar to that of cells lacking ATR, HCLK2, or CHK1: increased rates of spontaneous DNA damage, nuclear abnormalities and supernumerary centrosomes in unperturbed cells; and checkpoint defects in cells subjected to replication stress and DNA damage (17). The translocase activity of FANCM is essential for its role in checkpoint signaling (9, 15, 16). Notably, FANCM-deficient cells are sensitive to MMC and CPT, FANCM and FAAP24 possess intrinsic structure-based DNA-binding activity (preference for Y-shaped molecules), and both proteins interact constitutively with chromatin and with HCLK2 (9, 17). Based on these findings, it has been proposed that FANCM acts as a sensor of damage and/or mediator of ATR checkpoint signaling (9, 17).
FANCM is phosphorylated during the S and mitosis (M) phases of the cell cycle (2, 3, 18), and it becomes hyperphosphorylated in response to various forms of genotoxic stress (exposure to HU, MMC or CPT) (2, 3). Several lines of evidence suggest that multiple kinases are involved in FANCM phosphorylation, and the nature and extent of its phosphorylation have been proposed to play important roles in regulating its activity, both as a component of the FA core complex and otherwise (2, 3, 18). Although, the majority of FANCM phosphorylation during M-phase is mediated by Polo-like kinase 1 (PLK1) and results in FANCM degradation, it is not clear which kinases is relevant during S-phase and in the response to genotoxic stress (2, 3, 18). In this study, we have probed the role of ATR in FANCM phosphorylation, and found that it is relevant in the context of DNA damage. We also provide evidence for the importance of ATR-dependent phosphorylation of FANCM at serine 1045 (S1045) in the FA pathway and ATR signaling, and uncover a feed-back loop in the FA pathway. Our report provides the first evidence for a specific posttranslational modification of FANCM in response to genotoxic stress, and that this event is required for FANCM function.
Materials and Methods
Cells, cell culture, and chemicals
HeLa, U2OS and HEK293 cells were maintained using standard procedures (6). Parental HCT116 cells were purchased from ATCC (Manassas, VA) and HCT116 ATRflox/-cells were a kind gift from Dr. Stephen J. Elledge. Hydroxyurea (Sigma, St. Louis, MO) was resuspended in water to a stock concentration of 1 M. Mitomycin C (Sigma) was dissolved in 70% ethanol (Sigma) to a stock concentration of 500 ng/μL. Camptothecin (Sigma) was dissolved in DMSO (Sigma) to a stock concentration of 10 mM.
Cloning, constructs and retroviruses
The pMIEG3 bicistronic retroviral vector and pMIEG3-expressing FANCM were described previously (3). The FANCM S1045A mutant was introduced using the QuickChange Site-Directed Mutagenesis kit (Agilent Technologies, Santa Clara, CA). Retroviral packaging and generation of stable cell lines were as described earlier (3).
Antibodies
Antibodies against Actin, ATR, and CDC25A (F-6) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). CHK1 and pospho-CHK1 (S317) were purchased from Cell Signaling (Danvers, MA). Anti-FANCM, -FANCA, and -FANCD2 were described previously (2). Anti-FAAP24 was a kind gift from Dr. Stephen West (London Research Institute, Cancer Research UK). Anti-histone H2A antibody was purchased from Millipore (Billerica, MA). Custom-made phospho-specific antibody recognizing FANCM S1045 was generated by PhosphoSolutions (Aurora, CO).
Protein knockdown by siRNA/shRNA treatment
All siRNA oligos were purchased from Dharmacon (Lafayette, CO). For ATR knockdown, we used two siRNA oligos: ATR#1 targeting the coding region of ATR (CGAGACTTCTGCGGATTGC) and ATR#2 was purchased from Dharmacon (J-003202-19-00). A nonspecific control siRNA (D-001210-01) was used in all experiments. Cells were transfected as described previously (19). Endogenous FANCM was stably knocked down by retroviral-mediated expression of a shRNA targeting the 3’-UTR region of the FANCM mRNA (AAAGACCTCTCACAATATT). To generate retroviruses encoding shRNA’s specific to 3’-UTR region of the FANCM mRNA, we used a pSilencer 1.0-U6 vector (Ambion) containing the RNA PolIII-specific U6 gene promoter. Virus was generated as describe previously (20).
Purification and mass spectrometry analyses of FANCM complexes
HeLa cells were treated with 1.5 mM HU for 16 h and FANCM protein complex was purified from the nuclear extract using anti-FANCM antibody. The purified protein complex was resolved on an 8–16% SDS-PAGE gel and stained using either the SilverQuest silver-staining kit or colloidal blue staining kit according to the manufacturer’s instructions (Invitrogen, Carlsbad, CA). Phosphorylated FANCM was excised into three fragments for three protease digestions (Trypsin, Chymotrypsin and Glu-C), and phospho-peptides were identified by Ion-trap Mass Spectrometry analysis. All mass spectroscopy work was performed by the Mass Spectrometry Core at the University of Virginia (Charlottesville, VA).
Phosphatase treatment
HeLa cells expressing FLAGFANCM were treated with MMC for 16 h. Cells lysates were prepared using lysis buffer, and then FANCM was immunoprecipitated with anti-M2 agarose (Sigma). FLAG immunoprecipitates were mock treated or incubated at 30°C with 400 U of λ-protein phosphatase instructions (New England Biolabs, Ipswich, MA) for 60 min according to the manufacturer’s instructions prior to immunoblot analysis.
FANCD2 immunofluorescence
Cells were fixed in 2% paraformaldehyde in PBS for 20 minutes at room temperature (RT), and then deposited on 12-mm diameter glass coverslips coated with poly-d-lysine (centrifugation in a Thermo Shandon Cytospin apparatus at 163g for 2 minutes). After being washed with PBS, the cells were permeabilized with 0.2% Triton X-100 in PBS for 3 minutes, and washed again with PBS. Then anti-FANCD2 antibody E35 (1:200) in antibody buffer (PBS/3% BSA/0.05% Tween 20/0.04% sodium azide) was added for 1 hour at 37°C with. Cells were washed with PBS and incubated for 30 min at 37°C with rhodamine B-conjugated donkey anti-rabbit IgG antibody (1:200; Jackson ImmunoResearch Laboratories, West Grove, PA). Cells were washed with PBS, mounted in Vectashield with 4′,6-diamino-2-phenylindole (DAPI) (Vector Laboratories, Burlingame, CA) to stain DNA, and analyzed using a Zeiss Axiovert 200M microscope (Carl Zeiss, Thornwood, NY).
Drug sensitivity assay
Drug sensitivity was assessed using the chromogenic Cell Titer 96 Proliferation Assay (Promega Corporation, Madison, WI), with optical density recorded at 490 nm. Specifically, 3000 cells were grown in 96-well plates and treated with the indicated drugs for a week. The number of viable cells was then measured using the kit according to the manufacturer’s instructions. The data represent the percent survival compared to that in untreated cells. Each experiment was performed in triplicate, and mean values are shown with standard deviations.
Protein recruitment at laser-localized psorlaen ICLs
U2OS cells were transfected with FLAG-tagged FANCMWT or FANCMS1045A plasmid for 8 h at 37°C. Fresh medium was then added to the cells and they were grown for 48 h. Transfected cells were seeded in a 35-mm glass-bottom culture dish (MatTek Corporation, Ashland, MA). They were incubated with 6 μM trioxalen, at 37°C for 30 min prior to laser treatment, and processed further according to a previously described protocol (21). Briefly, localized irradiation was performed using a Nikon Eclipse TE2000 confocal microscope equipped with an SRS NL100 nitrogen laser-pumped dye laser (Photonics Instruments, St. Charles, IL) that fires 3-ns pulses at a repetition rate of 10 Hz at 365 nm, with a power of 0.7 nW measured at the back aperture of the 60x objective. The laser was directed to a specified rectangular region of interest (ROI) within the nucleus of a cell visualized with a Plan Fluor 60x/NA 1.25 oil objective. The laser beam was oriented by galvanometer-driven beam displacers and fired randomly throughout the ROI until the entire region was exposed. Throughout an experiment, cells were maintained at 37°C, 5% CO2, and 80% humidity using a Live Cell™ environmental chamber. After laser treatment, the cells were incubated at 37°C for 10 min and fixed immediately in freshly prepared 4% formaldehyde (in PBS) for 10 min at room temperature. Fixed cells were permeabilized with a PBS solution containing 0.5% Triton X-100, 1% BSA, 100 mM Glycine and 0.2 mg/mL EDTA on ice for 10 min, and subsequently digested with RNAse A at 37°C. For immunofluorescence staining, cells were incubated at 37°C for an hour with anti-γH2AX (Upstate, Millipore) and anti-FLAG (Sigma). Cells were incubated with corresponding secondary antibodies (Alexa Fluor goat anti-mouse or Alexa Fluor goat anti-rabbit, Molecular Probes, Invitrogen). After washing, they were mounted using ProLong Gold antifade reagent with DAPI (Molecular Probes, Invitrogen). The immunostained cells were visualized and imaged using a Hamamatsu EM-CCD digital camera attached to the Nikon Eclipse TE2000 confocal microscope.
UV-induced G2/M arrest analyzed by monitoring the mitotic index
Cells in PBS were UV-irradiated with 5 J/m2 UV and then cultured in complete medium for 16 h in the presence of 100 ng/mL nocodazole. The cells were then fixed in suspension (106 cells/mL) by the addition of 2 mL of 70% ethanol and incubation at −20°C for 24 h. After fixation, the cells were washed twice with PBS, suspended in 1 mL of PBS containing 0.25% Triton X-100, and incubated on ice for 15 min. After centrifugation, the cell pellet was suspended in 100 μL of PBS containing 1% bovine serum albumin (BSA) and anti-H3 (S10), a polyclonal antibody that specifically recognizes the phosphorylated form of histone H3 (Cell signaling), and incubated for 2 h at room temperature. The cells were then rinsed with PBS containing 1% BSA and incubated with fluoresceinisothiocyanate (FITC)-conjugated goat anti-rabbit immunoglobulin G antibody (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA), and diluted 1:200 in PBS containing 1% BSA. After a 1-h incubation at room temperature in the dark, the cells were washed again, resuspended in PBS containing 25 μg/mL of propidium iodide (PI) (Sigma, St. Louis, Mo.) and 0.1 mg/mL of RNase A (Sigma), and incubated at room temperature for 30 min. The percentage of positively staining cells was quantified by FACS analysis.
Preparation of Chromatin fractions
Chromatin fractions were prepared as described earlier (3).
Results
Kinetics of FANCM Phosphorylation and FANCD2 Monoubiquitination in Response to Genotoxic Stress are Similar
We previously reported that FANCM is phosphorylated in response to MMC- and HU-induced genotoxic stress (2). FANCD2, a key protein in the FA pathway, becomes monoubiquitinated under these conditions. A comparison of the time course of FANCM phosphorylation with that of FANCD2 monoubiquitination in response to both agents (Fig. 1A&S1A) revealed that both phenomena increase over time, reaching a peak at 24 h post treatment (Fig. 1A&S1A). The similarity in kinetics is consistent with phosphorylation of FANCM playing a role in the FA pathway.
Figure 1. DNA-damage-induced FANCM phosphorylation is ATR dependent.

(A) Time-dependent up-regulation of FANCM phosphorylation (pFANCM) and FANCD2 monoubiquitination (FANCD2-UB). HeLa cells were continually exposed to 1.5 mM HU for the indicated times were analyzed by immunoblotting using anti-FANCM and anti-FANCD2. (B) Inhibition of HU-induced FANCM hyperphosphorylation in ATR knockdown cells. Defective phosphorylation correlated with reduced FANCD2 monoubiquitination in U2OS cells. Cells treated with 1.5 mM HU for 16 h two days after being transfected with either siControl or siATR#1 were analyzed by immunoblotting with anti-ATR, anti-FANCM, anti-FANCD2, and anti-Actin. (C) Defective HU-induced FANCM hyperphosphorylation in ATR knockout cells correlates with reduced FANCD2 monoubiquitination. HCT116 ATRflox/- cells infected with either Ad-eGFP (lanes, 1, 2, 5) or Ad-eGFP-Cre (express Cre recombinase) (lanes, 3&4) were treated with 1.5 mM HU for 16 h two days post-infection, and were analyzed by immunoblotting with anti-ATR, anti-FANCM, anti-FANCD2, and anti-Actin. Please note that the sample from lane 2 and 5 are same and we have loaded it twice so that the comparison between lanes 4 and 5 is easier. Actin serves as loading control. Cell lysates were prepared by lysing cells directly in 2X Laemmli buffer and boiling for 10 min at 95°C.
FANCM Phosphorylation in Response to Genotoxic Stress is ATR Dependent
ATR plays an important role in the FA pathway, phosphorylating a number of the relevant proteins (12). We examined the ATR dependence of FANCM phosphorylation in response to genotoxic stress in two human cell lines (U2OS and HEK293), using siRNA oligos to knock down ATR and analyzing FANCM phosphorylation by immunoblotting (Fig. 1B&S1B, respectively). Genotoxic-stress-induced phosphorylation (+HU) of FANCM was strongly reduced in ATR knockdown cells of both types, and these effects correlated with a reduction of FANCD2 protein. Similar results were obtained using an siRNA oligo targeting a distinct site in the ATR coding region (Fig. S1C). These results are consistent with ATR playing a role in FANCM phosphorylation. We also tested the effects of ATR knockout, infecting cells of the HCT116 ATRflox/- (22) and isogenic HCT116 lines with Ad-eGFP-Cre. Assessment of the number of GFP-positive cells revealed the efficiency of infection to be greater than 90. After 48 h, the cells were either left untreated or were treated with HU for 16 h. Subsequent immunoblot analysis revealed a significant reduction of ATR from the ATRflox/- cells, accompanied by loss of FANCM phosphorylation (Fig. 1C). These results confirm that ATR is essential for FANCM phosphorylation in response to genotoxic stress.
Identification of FANCM Phosphopeptides and an ATR/ATM/DNA-PK consensus site at FANCM S1045
In order to purify phosphorylated FANCM and identify any phosphorylation sites, we immunoprecipitated FANCM from HU-treated HeLa cells (Fig. 2A). The band containing immunoprecipitated phospho-FANCM was excised from a colloidal Coomassie-stained gel and digested with three different proteases (Trypsin, Chymotrypsin and Glu-C), and the resulting peptides were analyzed by liquid chromatography (LC)–mass spectrometry (MS). The peptides analyzed covered ~85% of the FANCM protein (Fig. S2), and a total of 18 phosphorylation sites were identified. Given the role of ATR in FANCM phosphorylation, we inspected the phosphopeptides for the presence of the PIKK kinases (ATR, ATM, and DNA-PK) consensus site, S/TQ (13), and identified a peptide CTCLLSHSAVNS*QQNLELNSLK containing phospho-serine 1045 (pS1045) (Fig. 2B); the relative abundance of this peptide was higher than that of any other peptide in the spectrum (Fig. 2B).
Figure 2. FANCM S1045 is a novel ATR phosphorylation site.

(A) Hyperphosphorylation of FANCM in response to HU. HeLa cells were treated with 1.5 mM HU for 16h and FANCM was immunoprecipitated. A portion of the sample was immunoblotted using anti-FANCM. (B) LC-MS/MS spectra of the tryptic digests of the remainder of the FANCM protein samples. Relative peptide abundance is plotted against the mass to charge (m/z) ratio, revealing a novel phosphorylation site at S1045. Peak corresponding to phosphorylated tryptic peptide CTCLLSHSAVNS*QQNLELNSLK harbors phosphorylated S1045, as indicated by arrow. (C) Genotoxic stress induces phosphorylation at S1045. HeLa cells were treated continually with 1. 5 mM HU or 100 ng/mL MMC for 4 and 16h, and whole-cell lysates were analyzed by immunoblotting with an antibody that detects specifically FANCM phosphorylated at S1045 - pFANCM (S1045). (D) Loss of phosphorylation of endogenous FANCM at S1045 upon treatment with λ-phosphatase. FANCM from whole-cell lysates of HeLa cells that were treated with 100 ng/mL MMC were immunoprecipitated with anti-FANCM antiserum and the immunoprecipitates were either left untreated or treated with λ-phosphatase. (E). Loss of pS1045 in the absence of ATR. Isogenic HCT116 and HCT116-ATR-/flox cells were infected with Ad-eGFP-Cre, and input and IP samples were analyzed with the indicated antibodies. Actin served as loading control for Input samples and total FANCM as loading control for IP samples. (F) FANCM phosphorylation at S1045 following treatment with various inhibitors. HeLa cells expressing His6-FLAGMHF1 were pretreated with inhibitors of DNA-PK (NU7026), ATM (KU55933), or ATR (Caffeine), or with DMSO (control), for 1 h and then treated with 100 ng/mL MMC for 16 h to induce FANCM phosphorylation at S1045. Lysates were immunoprecipitated using anti-FLAG M2 agarose and immunoblotted with pFANCM (S1045) antibody. Total FANCM serves as loading control. The shift in the mobility of FANCM upon phosphorylation can be readily detected by running an 8% Tris-glycine gel for 3 h at 100V. Note that the apparent lack of mobility shift of phosphorylated FANCM upon treatment with genotoxic agents when compared to untreated, in figures C, D and F, is due to the fact that an 8-16% Tris-glycine gel was run only for 70 min to achieve a sharp band. For Fig. C&F Cell lysates were prepared by lysing cells directly in 2X Laemmli buffer and boiling for 10 min at 95°C.
FANCM S1045 Becomes Phosphorylated in Response to Genotoxic Stress
In order to detect and study the significance of phosphorylation at FANCM S1045, we raised an antiserum against a 15-amino acid long synthetic peptide that contains pS1045. The antiserum was affinity purified over a phosphorylated peptide column and cross-adsorbed to an unmodified peptide column. The anti-pS1045 thus purified detected FLAGFANCMWT but not FLAGFANCMS1045A by immunoblot analysis, in immunoprecipitates from HeLa cells stably expressing FLAGFANCMWT or FLAGFANCMS1045A, respectively (Fig. S3).
Because FANCM phosphorylation was induced in response to genotoxic stress (Fig. 2A), we tested pS1045 immunoreactivity following genotoxic stress. As shown in figures 2C and S4, the pS1045 signal increased over time after cells were treated with HU, MMC or UV. Thus, FANCM phosphorylation at S1045 is upregulated in response to genotoxic agents. Also, treatment of immunoprecipitated FLAGFANCMWT with lambda phosphatase eliminated signal detected with the anti-pS1045 (Fig. 2D), confirming the specificity of anti-pS1045.
FANCM S1045 Phosphorylation is ATR Dependent
Since S1045 is part of the ATR consensus motif S/TQ, we assessed whether its phosphorylation depends on ATR. To this end, we knocked out ATR in HCT116 ATRflox/- cells using Cre recombinase; the resulting reduction of ATR protein was significant (Fig. 2E). FANCM from lysates of ATR knockout cells were immunoprecipitated using anti-FANCM and immunoblotted with anti-pS1045. As shown in figure 2E, phosphorylation at S1045 was significantly diminished in these cells compared to controls, although anti-FANCM detected comparable levels of total FANCM protein in the two samples. These findings are consistent with the notion that phosphorylation at S1045 is ATR dependent (Fig. 2E). Moreover, of cells pre-treated with an inhibitor of DNA-PK (NU7026), ATM (KU55933) or ATR (Caffeine), the pS1045 signal was reduced only in the ATR inhibitor treated group (Fig. 2F). Thus, in human cells subjected to genotoxic stress, specifically ATR regulates FANCM phosphorylation at S1045.
Mutant FANCMS1045A Fails to Complement FANCM Deficiency
Cells from FA patients display a marked hypersensitivity to DNA crosslinking agents such as MMC, and this hypersensitivity can be complemented by ectopically expressing a wild-type cDNA of the defective gene (6). Among the core complex proteins, FANCM-deficient cells are uniquely sensitive to both CPT and MMC (3). If phosphorylation at S1045 is required for FA-pathway signaling and CPT resistance, a mutation affecting this residue should cause a phenotype similar to that observed in FANCM-depleted cells. We thus generated a FANCM-encoding construct in which S1045 is substituted with alanine (S1045A). Given that the S1045A form was not expressed in FANCM-deficient EUFA867 lymphoblast cells, which has served as a complementation system in previous analyses of FANCM (3), we established a complementation system using the U2OS cell line, in which endogenous FANCM was stably knocked down by retrovirus-mediated expression of an shRNA directed to the FANCM 3’-UTR (shFANCM-UTR) (Fig. 3A, lane 2). We then expressed either a wild-type (FANCMWT) or phosphomutant (FANCMS1045A) protein in these cells via retroviral gene transfer (Fig. 3A). Immunoprecipitation and immunoblot analysis revealed that FANCMS1045A interacted with proteins of the FA core complex in the same way as of FANCMWT (Fig. S5A). Nevertheless, the FANCMS1045A-expressing cells were not fully functional, as demonstrated by significant reductions in: FANCD2 monoubiquitination (Fig. S5B); FANCD2 focus formation (Fig. 3B); reconstitution of MMC and CPT killing (Fig. 3C&D); and MMC-induced chromatin association of the FA-core complex (Fig. S6). Thus, phosphorylation at S1045 is essential for the cells to respond appropriately to MMC- and CPT-induced genotoxic stress.
Figure 3. S1045 phosphorylation is essential for FANCM functions.

(A) Expression of FLAG tagged wild-type (FANCMWT) and S1045A mutant (FANCMS1045A) FANCM, in the context of knockdown of the endogenous protein. U2OS cells stably transduced with shRNA targeting the 3’UTR of FANCM (shFANCM-UTR) were further transduced with retroviral vector alone (Vector), or with retrovirus carrying FANCMWT or FAMCMS1045A. U2OS cells stably expressing a non-targeting shRNA (shControl) served as control. (B) Requirement of S1045 phosphorylation for assembly of FANCD2 foci. U2OS cells generated as described in (A) were either left untreated or exposed to 100 ng/mL MMC for 16 hours; the percentage of cells with 5 or more FANCD2 foci was assessed in at least 150 cells. The data represent the average of 3 independent experiments, with standard deviations. (C&D) MMC and CPT sensitivity of FANCM-depleted cells. Cells of indicated genotypes were continually exposed to indicated concentrations of MMC or CPT for 10 days and viable cells were subjected to the Cell Titer 96 Proliferation Assay. The data show the percentage growth compared with that of untreated cells; 1 representative result of 3 independent experiments is shown, with standard deviations. (E&F) Recruitment of FANCM forms to the ICL site. U2OS cells transfected with FLAG tagged FANCMWT and FANCMS1045A were tested for recruitment to the ICL sites were examined by immunofluorescence analysis. Representative images are shown in panel E, and quantitation is shown in F.
Phosphorylation of S1045 is Essential for Efficient Recruitment of FANCM to ICL Sites
FANCM is rapidly recruited at ICL sites generated by laser-activated psoralen conjugation (23). To investigate if FANCM recruitment to ICL sites is dependent on phosphorylation at S1045, we transfected U2OS cells with FLAGFANCMWT and FLAGFANCMS1045A and visualized protein recruitment at ICL sites by indirect immunofluorescence (anti-FLAG) as previously described (24). Analysis of eGFP staining revealed that more than 90% of the cells were transfected. As shown in figure 3E&F, recruitment of FLAGFANCMS1045A to ICL sites was greatly diminished relative to that of FLAGFANCMWT (occurred in only 11% vs. 35% of cells). These data suggest that the phosphorylation of FANCM at S1045 is essential for its efficient recruitment to ICL sites. Furthermore, we also observed a defective recruitment of endogenous FANCM to ICL sites in HeLa cells depleted of ATR (Fig. S7), thus providing strong evidence for a direct involvement of ATR in FANCM functions.
Phosphorylation of FANCM S1045 is Required for Efficient Blockage of Mitosis in Response to Genotoxic Stress
Previous studies suggested that FANCM is required to efficiently block the cell’s entry into mitosis following genotoxic stress and failure to block the cells entry into mitosis result in cells with abnormal nuclear morphology (9). This is consistent with FANCM-depleted cells frequently exhibiting abnormalities in nuclear morphology (Fig. 4A&B) (9). We assessed mitotic entry after ectopically expressing FANCMWT and FANCMS1045A in FANCM-depleted U2OS cells. Consistent with earlier observations,(9) the incidence of aberrant nuclear morphology increased 4–5 fold in the FANCM-depleted cells (Fig. 4A). Ectopic expression of FANCMWT, but not FANCMS1045A, corrected this phenotype (Fig. 4A), suggesting that phosphorylation at FANCM S1045 is required to prevent entry into mitosis in response to genotoxic stress.
Figure 4. FANCM S1045 phosphorylation is essential for the G2/M checkpoint.

(A) Frequency of nuclear abnormalities in U2OS cells described in Figure 3A. Data shown are mean from three independent experiments, with standard error. (B) Representative images of nuclear abnormalities (highlighted by red arrows) scored in A. (C) CDC25A degradation in cells of the indicated genotypes. Actin serves as loading control. Cells were treated with 3 mM hydroxyurea (HU) in the presence 25 mg/mL cyclohexamide for the indicated time and then lysed in 2X Laemmli buffer. (D) Mitotic index in cells of the indicated genotypes. Cells were treated with UV and tested 24 h later. Data shown are the mean of at least three separate experiments, with standard error.
The above-described block to mitosis is known to be mediated in part by the ATR/CHK1 signaling pathway, to ultimately invoke the G2/M checkpoint, and to be caused by the phosphorylation and degradation of CDC25A (9). Notably, CDC25A was rapidly degraded in HU-treated cells transduced with a control shRNA and, consistent with the earlier reports,(9) this effect was not as pronounced in cells depleted of FANCM (Fig. 4C). Although the effect was rescued by the expression of FANCMWT, this was not the case for expression of FANCMS1045A (Fig. 4C). These data suggest that phosphorylation at FANCM S1045 is required for efficient ATR/CHK1 signaling in response to replication stress.
To confirm that phosphorylation at FANCM S1045 is essential for efficient entry into mitosis following genotoxic stress, we assessed the mitotic index of the cells following exposure to UV-irradiation. ATR is activated following such treatment, and an important consequence is arrest at the G2/M checkpoint, as measured by a decrease in the percentage of mitotic (histone H3-p-S10-positive) cells in UV-treated versus untreated cells 24 h post treatment. As expected, the percentage of cells in mitosis was decreased among those expressing the control shRNA, but not among those expressing shFANCM- UTR (Fig. 4D). Ectopic expression of FANCMWT led to a reduction in the mitotic index similar to that observed in the control cells, whereas that of FANCMS1045A or an ATPase-dead mutant (FANCMK117R) did not (Fig. 4D). Although it is possible that the defect observed in FANCMS1045A expressing cells resulted from changes in distribution of the cells across the cell-cycle, an analysis of the FANCMWT-, FANCMS1045A- and FANCMK117R-expressing cells did not reveal differences in this regard (Fig. S8). These data indicate that the phosphorylation of FANCM on S1045 is required to prevent inappropriate entry into mitosis in the context of genotoxic stress.
Phosphorylation of FANCM S1045 is Essential for Efficient ATR/CHK1 Signaling in Response to Genotoxic Stress
Previous studies have shown that FANCM is essential for ATR/CHK1 signaling (9, 25), and the data presented above suggest that the phosphorylation of FANCM S1045 is ATR dependent. We next investigated the role of phosphorylation at S1045 in efficient ATR/CHK1 signaling. We hypothesized that the defect in genotoxic stress-induced CDC25A degradation observed in the context of FANCMS1045A may be due to defective CHK1 activation. We monitored CHK1 activation as it is ATR-mediated phosphorylation at S317. U2OS cells stably expressing shFANCM were transfected with the FANCMWT-, FANCMS1045A-, or FANCMK117R-encoding constructs, treated with 2 uM CPT for 30 min, and immunoblotted for phospho-CHK1 (S317). CPT treatment led to strong CHK1 phosphorylation in control cells, and this was diminished in FANCM knockdown cells (Fig. 5). CHK1 phosphorylation was rescued in FANCMWT-expressing cells but not in FANCMS1045A-, or FANCMK117R-expressing counterparts (Fig. 5). Thus, phosphorylation of FANCM at S1045 appears to be essential for ATR/CHK1 signaling in response to genotoxic stress. We also found an increase in spontaneous DNA damage in cells expressing mutant forms of the protein (i.e., an increase in the levels of γH2A.X phosphorylated at S139) (Fig. 5). Overall, these data suggest that the phosphorylation of FANCM at S1045 is essential for CHK1 activation, and that failure of this modification results in an increase in spontaneous DNA damage.
Figure 5. FANCM S1045 phosphorylation is essential for ATR/CHK1 signaling in response to DNA damage.
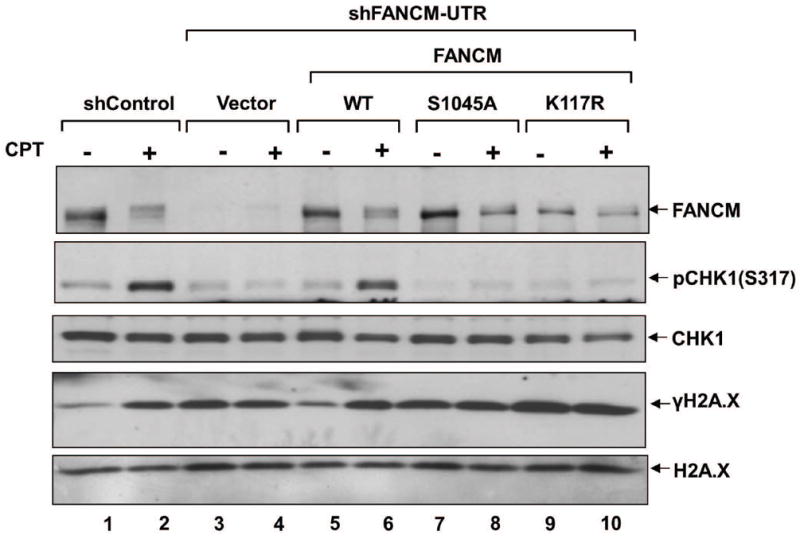
Cells of the indicated genotypes were exposed to CPT (2 μM) for 1 h and cell lysates that were prepared by lysing cells directly in 2X Laemmli buffer and boiling for 10 min at 95°C were analyzed by immunoblotting using the antibodies indicated to right. No significant cell death was observed at the 1 h time point after treatment with 2 μM CPT.
Discussion
Previously, we and others showed that FANCM is hyperphosphorylated in response to genotoxic stress, and that cells of a FANCM-deficient line (EUFA867) and others depleted of FANCM by either an siRNA or shRNA approach are sensitive to MMC as well as CPT (3, 26). Here we report that the genotoxic stress induced by FANCM phosphorylation is ATR dependent, and identify FANCM residue S1045 as a novel phosphorylation site. Using a phospho-specific antibody, we show for the first time that phosphorylation at this site is induced by genotoxic stress, and that ATR knockout or inhibition greatly reduces FANCM phosphorylation at S1045. Notably, phosphorylation at S1045 is essential for FANCM function in both the FA pathway and ATR/CHK1 signaling.
Kinetics of the genotoxic stress-induced phosphorylation of FANCM correlate with those of FANCD2 monoubiquitination under the same conditions, suggesting that the phosphorylation of FANCM plays an important role in the FA pathway—probably in signaling DNA damage. It is well established that genotoxic stress leads to the activation of three related protein kinases: ATR, ATM, and DNA-PK. Whereas the latter two are activated primarily by DNA DSBs, ATR responds principally to replication blockage or replication stress (27). ATR plays an important role in the FA pathway and phosphorylates several FA proteins, including FANCD2, FANCI, FANCA, FANCG and FANCE;(28-30) mutations at the relevant phosphorylation sites impair the monoubiquitination of FANCD2/FANCI. The genotoxic-stress-induced phosphorylation of FANCM is ATR dependent. Activated ATM, ATR and DNA-PK preferentially phosphorylate serine and threonine residues that are followed by a glutamine residue (S/TQ), and FANCM has 15 such sequences, 9 of which are conserved across FANCM orthologs. However, one of these (S1045) was abundantly represented in MS as being modified, in spite of the fact that peptides containing all 15 sites were recovered. The other 14 sites may not get phosphorylated at all or get phosphorylated at such a low degree that they were beyond the detection level of MS or may get phosphorylated in some other conditions. The significant increase in phosphorylation at S1045 following DNA damage suggests that this particular site is important for signal transduction. It is notable that although FANCMS1045 is proficient in binding to the FA core complex, nevertheless cells expressing it exhibit impaired FANCD2 monoubiquitination, complete loss of FANCM function with respect to MMC sensitivity, FANCD2 focus formation, and loading of the FA core complex onto chromatin in the context of genotoxic stress, and have unique phenotypes of CPT sensitivity and defective ATR/CHK1 signaling.
FANCM is known to stabilize replication forks, and perhaps its phosphorylation is required to for this response in the context of replication stress—to avoid fork collapse and the formation of DSBs. The fact that we also observed a large increased in spontaneous γ-H2A.X in the FANCM-depleted cells as well as their FANCMS1045A-reconstituted counterparts suggests that FANCM prevents stalled replication forks from developing into DSBs, and that FANCM phosphorylation is indispensable.
Although we have demonstrated that FANCM phosphorylation is dependent on ATR, it is formally possible that ATR does not act directly on FANCM. However, the finding that FANCM S1045 is phosphorylated normally after MMC treatment in the presence of a specific inhibitor of ATM or DNA-PK (Fig. 2F) indicates that these kinases are not required. Together with the fact that S1045 is at an ATR/ATM consensus site, this finding highly suggests that the role of ATR is direct. FANCM has been reported to play a role as a sensor of genotoxic stress (9). The findings that phosphorylation at S1045 1) is induced in the context of genotoxic stress, 2) is ATR dependent, and 3) is required for downstream ATR/CHK1 signaling suggest that the activities of two genotoxic stress sensors, ATR and FANCM, are coordinated via phosphorylation at FANCM S1045.
Previous reports placed FANCM upstream of ATR signaling (9, 25). However, our data indicate that following ATR phosphorylation of FANCM, the latter signals goes back to ATR. Such mutual control between DNA repair proteins has precedents. For example, studies of Xenopus FANCM (xFANCM) and FANCD2 (xFANCD2) revealed an interdependence: xFANCM is required for the monoubiquitination and recruitment of xFANCD2 to chromatin, but FANCD2 protein signals back to the xFA core complex by regulating FANCM phosphorylation (31). Similarly, the Mre11/Rad50/NBS1 complex (MRN) binds DNA breaks on chromatin independently of ATM but needs to recruit and activate ATM by phosphorylation before it can itself be phosphorylated and activated by ATM (32). We show here that FANCM and ATR affect one another in the context of genotoxic stress; further studies elaborating on how FANCM phosphorylation contributes to ATR function and vice versa will unravel their precise role in DNA damage signaling.
How FANCM phosphorylation contributes to DNA repair and the resolution of ICLs or CPT sensitivity is not clear yet. However, our favored model is that ATR phosphorylates FANCM at S1045 after detecting a block in replication, that phosphorylated FANCM is then translocated along the DNA to the repair site where it stabilizes the replication fork, activates the ATR/CHK1 checkpoint, and recruits the FA-core complex and other DNA-repair proteins. This model is consistent with the fact that FANCMS1045A, which cannot be phosphorylated on the relevant serine, is not recruited to ICL sites (Fig. 3E), and that when it is the only form of FANCM present, the FA-core complex is not recruited to the chromatin following MMC treatment (Fig. S6), ATR/CHK1 signaling is reduced, γ-H2A.X levels increase, and that the cells are sensitive to MMC and CPT. While definitive proof of the details of the proposed mechanism is not yet available, our results demonstrate that phosphorylation of FANCM at S1045 is essential for FANCM function, and show for the first time a link that co-ordinates the FA pathway and ATR checkpoint.
Supplementary Material
Acknowledgments
We thank Dr. Stephen J. Elledge for the HCT116 ATRflox/- cells.
Grant Support
This work was supported by National Institutes of Health research grants HL084082 and HL084082-03S1 and grants from the Ohio Cancer Research Associates and the Fanconi Anemia Research Fund to A.R.M. This work was also supported, in part, by the Intramural Research Program of the NIH, National Institute on Aging to M.M.S. T.R.S is supported by the Ramalingaswami re-entry fellowship, DBT-INDIA.
Footnotes
Conflict of Interest disclosure: The authors declare no competing financial interests.
References
- 1.Kim H, D’Andrea AD. Regulation of DNA cross-link repair by the Fanconi anemia/BRCA pathway. Genes Dev. 2012;26:1393–408. doi: 10.1101/gad.195248.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Meetei AR, Medhurst AL, Ling C, Xue Y, Singh TR, Bier P, et al. A human ortholog of archaeal DNA repair protein Hef is defective in Fanconi anemia complementation group M. Nat Genet. 2005;37:958–63. doi: 10.1038/ng1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Singh TR, Bakker ST, Agarwal S, Jansen M, Grassman E, Godthelp BC, et al. Impaired FANCD2 monoubiquitination and hypersensitivity to camptothecin uniquely characterize Fanconi anemia complementation group M. Blood. 2009;114:174–80. doi: 10.1182/blood-2009-02-207811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ling C, Ishiai M, Ali AM, Medhurst AL, Neveling K, Kalb R, et al. FAAP100 is essential for activation of the Fanconi anemia-associated DNA damage response pathway. EMBO J. 2007;26:2104–14. doi: 10.1038/sj.emboj.7601666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Singh TR, Saro D, Ali AM, Zheng XF, Du CH, Killen MW, et al. MHF1-MHF2, a histone-fold-containing protein complex, participates in the Fanconi anemia pathway via FANCM. Mol Cell. 2010;37:879–86. doi: 10.1016/j.molcel.2010.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ali AM, Pradhan A, Singh TR, Du C, Li J, Wahengbam K, et al. FAAP20: a novel ubiquitin-binding FA nuclear core complex protein required for functional integrity of the FABRCA DNA repair pathway. Blood. 2012;119:3285–94. doi: 10.1182/blood-2011-10-385963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yan Z, Delannoy M, Ling C, Daee D, Osman F, Muniandy PA, et al. A histone-fold complex and FANCM form a conserved DNA-remodeling complex to maintain genome stability. Mol Cell. 2010;37:865–78. doi: 10.1016/j.molcel.2010.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meetei AR, de Winter JP, Medhurst AL, Wallisch M, Waisfisz Q, van de Vrugt HJ, et al. A novel ubiquitin ligase is deficient in Fanconi anemia. Nat Genet. 2003;35:165–70. doi: 10.1038/ng1241. [DOI] [PubMed] [Google Scholar]
- 9.Collis SJ, Ciccia A, Deans AJ, Horejsi Z, Martin JS, Maslen SL, et al. FANCM and FAAP24 function in ATR-mediated checkpoint signaling independently of the fanconi anemia core complex. Mol Cell. 2008;32:313–24. doi: 10.1016/j.molcel.2008.10.014. [DOI] [PubMed] [Google Scholar]
- 10.Mosedale G, Niedzwiedz W, Alpi A, Perrina F, Pereira-Leal JB, Johnson M, et al. The vertebrate Hef ortholog is a component of the Fanconi anemia tumor-suppressor pathway. Nature structural & molecular biology. 2005;12:763–71. doi: 10.1038/nsmb981. [DOI] [PubMed] [Google Scholar]
- 11.Jones RM, Petermann E. Replication fork dynamics and the DNA damage response. Biochem J. 2012;443:13–26. doi: 10.1042/BJ20112100. [DOI] [PubMed] [Google Scholar]
- 12.Constantinou A. Rescue of replication failure by Fanconi anaemia proteins. Chromosoma. 2012;121:21–36. doi: 10.1007/s00412-011-0349-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nam EA, Cortez D. ATR signalling: more than meeting at the fork. Biochem J. 2011;436:527–36. doi: 10.1042/BJ20102162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Andreassen PR, D’Andrea AD, Taniguchi T. ATR couples FANCD2 monoubiquitination to the DNA-damage response. Genes Dev. 2004;18:1958–63. doi: 10.1101/gad.1196104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schwab RA, Blackford AN, Niedzwiedz W. ATR activation and replication fork restart are defective in FANCM-deficient cells. EMBO J. 2010;29:806–18. doi: 10.1038/emboj.2009.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luke-Glaser S, Luke B, Grossi S, Constantinou A. FANCM regulates DNA chain elongation and is stabilized by S-phase checkpoint signalling. EMBO J. 2010;29:795–805. doi: 10.1038/emboj.2009.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Collis SJ, Barber LJ, Clark AJ, Martin JS, Ward JD, Boulton SJ. HCLK2 is essential for the mammalian S-phase checkpoint and impacts on Chk1 stability. Nature cell biology. 2007;9:391–401. doi: 10.1038/ncb1555. [DOI] [PubMed] [Google Scholar]
- 18.Kee Y, Kim JM, D’Andrea AD. Regulated degradation of FANCM in the Fanconi anemia pathway during mitosis. Genes Dev. 2009;23:555–60. doi: 10.1101/gad.1761309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Singh TR, Ali AM, Busygina V, Raynard S, Fan Q, Du CH, et al. BLAP18/RMI2, a novel OB-fold-containing protein, is an essential component of the Bloom helicase-double Holliday junction dissolvasome. Genes Dev. 2008;22:2856–68. doi: 10.1101/gad.1725108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Singh TR, Saro D, Ali AM, Zheng XF, Du CH, Killen MW, et al. MHF1-MHF2, a histone-fold-containing protein complex, participates in the Fanconi anemia pathway via FANCM. Mol Cell. 2010;37:879–86. doi: 10.1016/j.molcel.2010.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Muniandy PA, Thapa D, Thazhathveetil AK, Liu ST, Seidman MM. Repair of laser-localized DNA interstrand cross-links in G1 phase mammalian cells. J Biol Chem. 2009;284:27908–17. doi: 10.1074/jbc.M109.029025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cortez D, Guntuku S, Qin J, Elledge SJ. ATR and ATRIP: partners in checkpoint signaling. Science. 2001;294:1713–6. doi: 10.1126/science.1065521. [DOI] [PubMed] [Google Scholar]
- 23.Yan Z, Delannoy M, Ling C, Daee D, Osman F, Muniandy PA, et al. A histone-fold complex and FANCM form a conserved DNA-remodeling complex to maintain genome stability. Mol Cell. 2010;37:865–78. doi: 10.1016/j.molcel.2010.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thazhathveetil AK, Liu ST, Indig FE, Seidman MM. Psoralen conjugates for visualization of genomic interstrand cross-links localized by laser photoactivation. Bioconjug Chem. 2007;18:431–7. doi: 10.1021/bc060309t. [DOI] [PubMed] [Google Scholar]
- 25.Horejsi Z, Collis SJ, Boulton SJ. FANCM-FAAP24 and HCLK2: roles in ATR signalling and the Fanconi anemia pathway. Cell cycle (Georgetown, Tex. 2009;8:1133–7. doi: 10.4161/cc.8.8.8204. [DOI] [PubMed] [Google Scholar]
- 26.Rosado IV, Niedzwiedz W, Alpi AF, Patel KJ. The Walker B motif in avian FANCM is required to limit sister chromatid exchanges but is dispensable for DNA crosslink repair. Nucleic Acids Res. 2009;37:4360–70. doi: 10.1093/nar/gkp365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang J, Yu Y, Hamrick HE, Duerksen-Hughes PJ. ATM, ATR and DNA-PK: initiators of the cellular genotoxic stress responses. Carcinogenesis. 2003;24:1571–80. doi: 10.1093/carcin/bgg137. [DOI] [PubMed] [Google Scholar]
- 28.Ishiai M, Kitao H, Smogorzewska A, Tomida J, Kinomura A, Uchida E, et al. FANCI phosphorylation functions as a molecular switch to turn on the Fanconi anemia pathway. Nature structural & molecular biology. 2008;15:1138–46. doi: 10.1038/nsmb.1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Collins NB, Wilson JB, Bush T, Thomashevski A, Roberts KJ, Jones NJ, et al. ATR-dependent phosphorylation of FANCA on serine 1449 after DNA damage is important for FA pathway function. Blood. 2009;113:2181–90. doi: 10.1182/blood-2008-05-154294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ho GP, Margossian S, Taniguchi T, D’Andrea AD. Phosphorylation of FANCD2 on two novel sites is required for mitomycin C resistance. Mol Cell Biol. 2006;26:7005–15. doi: 10.1128/MCB.02018-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sobeck A, Stone S, Landais I, de Graaf B, Hoatlin ME. The Fanconi anemia protein FANCM is controlled by FANCD2 and the ATR/ATM pathways. J Biol Chem. 2009;284:25560–8. doi: 10.1074/jbc.M109.007690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lavin MF. ATM and the Mre11 complex combine to recognize and signal DNA double-strand breaks. Oncogene. 2007;26:7749–58. doi: 10.1038/sj.onc.1210880. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.