

Abstract
Sleep disturbances are common in neurodegenerative diseases such as Alzheimer disease (AD). Unfortunately, how AD is mechanistically linked with interference of the body’s natural sleep rhythms remains unclear. Our recent findings provide insight into this question by demonstrating that sleep disruption associated with AD is driven by epigenetic changes mediated by the histone acetyltransferase (HAT) Tip60. In this study, we show that Tip60 functionally interacts with the AD associated amyloid precursor protein (APP) to regulate axonal growth of Drosophila small ventrolateral neuronal (sLNv) pacemaker cells, and their production of neuropeptide pigment dispersing factor (PDF) that stabilizes appropriate sleep-wake patterns in the fly. Loss of Tip60 HAT activity under APP neurodegenerative conditions causes decreased PDF production, retraction of the sLNv synaptic arbor required for PDF release and disruption of sleep-wake cycles in these flies. Remarkably, excess Tip60 in conjunction with APP fully rescues these sleep-wake disturbances by inducing overelaboration of the sLNv synaptic terminals and increasing PDF levels, supporting a neuroprotective role for Tip60 in these processes. Our studies highlight the importance of epigenetic based mechanisms underlying sleep disturbances in neurodegenerative diseases like AD.
Keywords: Epigenetics, histone acetyltransferases (HAT), Tip60, sleep, amyloid precursor protein (APP), axonal growth, neurodegeneration
Neurons, while being subjected to a variety of stimuli, are also able to convert such cues into higher order functions such as controlling behavior, storing memories and decision making. These unique properties are based on the highly flexible nature of neurons, a characteristic that is regulated by networks of extrinsic and intrinsic molecular pathways that together orchestrate precise gene expression profiles required for neuronal plasticity. Epigenetic control, which largely involves events of chromatin remodeling, appears to be one way in which transcriptional regulation of gene expression can be controlled in neurons.1 Of the epigenetic modifications identified so far in the nervous system, histone acetylation mediated by the antagonistic activities of histone acetyltransferase (HAT) and histone deacetylase (HDAC) enzymes,2 has been unequivocally shown to play a crucial role in regulating neuronal gene expression profiles critical for neuronal functions.3,4 HATs generally promote chromatin decondensation by catalyzing the transfer of an acetyl group from acetyl-CoA to the ε-amino group of specific lysine residues within the N-terminal tails of nucleosomal histones. This modification weakens histone-DNA as well as neighboring nucleosomal contacts to promote chromatin disruption that, in turn, facilitates factor binding and transcriptional activation. HATs also exhibit distinct substrate preference for specific histone, lysine and gene targets, and thereby generate different acetylation patterns within the genome.5,6 Such HAT generated acetylation patterns together with other DNA and histone modifications is thought to serve as a molecular bar code to recruit chromatin remodeling complexes and downstream regulatory factors that drive gene expression profiles required for particular cellular events, a paradigm referred to as the ‘histone-code hypothesis’7-9. In particular, loss of function of specific HATs with vital neuronal functions has been reported to impair neuronal acetylation status and contribute to degenerative effects in various cellular and animal models of neurodegenerative diseases.10-12
The HAT Tip60 (Tat interactive protein, 60 KDa) is a member of the MYST family of proteins that are related by a ~300 aminoacid domain containing a typical zinc finger and HAT domains.13 The HAT activity of Tip60 exerts pleiotropic cellular effects that include a variety of chromatin mediated processes such as transcription regulation, cell cycle check-point control, DNA damage repair and apoptosis to name a few (reviewed in ref. 14). In 2007, we first isolated the Drosophila homolog of Tip60 and further demonstrated an essential role for Tip60 during multicellular development.15 Subsequent work from our laboratory has demonstrated that Tip60 is robustly produced in the developing embryonic nervous system as well as in specific regions of the adult fly brain. Moreover, our studies further revealed that Tip60’s HAT activity is critical for nervous system development and function, an effect primarily mediated via transcriptional regulation of genes enriched for a variety of specific neuronal functions.16 Accordingly, we found that Tip60’s HAT activity controls synaptic plasticity17 and regulates apoptosis to prevent unwanted cell death in the developing Drosophila central nervous system (CNS).18 Consistent with our findings, Tip60 has been implicated in neurodegenerative diseases such as spinocerebellar ataxia (SCA1)19 and the age-related neurodegenerative Alzheimer disease (AD).20 Tip60’s role in the latter stems from observations that Tip60 forms a transcriptionally active complex with a cytosolic fragment derived from proteolytic processing of the AD-associated amyloid precursor protein (APP), termed the APP intracellular domain (AICD).20,21 The Tip60/AICD complex has been shown to increase histone acetylation22 and coactivate gene promoters which are linked to apoptosis and neurotoxicity associated with AD.23 Moreover, misregulation of certain putative target genes of the Tip60/AICD complex has been linked to AD related pathology.24,25 More recently, our laboratory has demonstrated that Tip60 and APP functionally interact to mediate lethality and apoptotic mediated neurodegeneration in the central nervous system (CNS) of an AD fly model, in vivo.18 Together, these studies support the concept that neuropathology associated with AD is due, at least in part, to epigenetic dysregulation, Tip60 being a likely candidate mediating such effects. However, little is known about how aberrant alterations of the neural epigenome by misregulation of Tip60 HAT activity in particular, affect specific neural circuits under AD linked neurodegenerative conditions.
Sleep abnormalities are a major and early feature of neurodegenerative diseases like AD that are also characterized by cognitive decline. While the causes of such sleep disturbances are unknown, they are thought to further exacerbate the effects of a fundamental process leading to neurodegeneration.26 Sleep dependent mechanisms of neural plasticity are believed to contribute to memory consolidation and thus are likely critical for learning and memory.27,28 Sleep disturbances in AD patients typically consists of sleep fragmentation with frequent awakenings in the night and an increment in the propensity to sleep during daytime.29 Transgenic mouse models for AD that overexpress human APP and exhibit plaque (via extracellular β amyloid deposits) and tangle pathologies have also been reported to exhibit decreased activity during the nocturnal (active) phase and increased activity during the day . Importantly, such changes in sleep-wake cycles were observed prior to when extracellular-Aβ deposition would be expected, suggesting that abnormalities in sleep-wake cycles may precede AD neuropathology.30 While the pathogenesis of sleep disturbances associated with AD and the precise mechanism by which APP overexpression contributes to such sleep abnormalities is unclear, neurodegeneration in brain regions that are involved in sleep regulation are thought to lead to sleep abnormalities.31 In addition to marked neuronal atrophy of the mammalian pacemaker region, the suprachiasmatic nucleus (SCN), dramatic decrease in circadian peptides like vasopressin and vasoactive intestinal peptide has been reported to underlie sleep disturbances in AD.32 Defects in cholinergic transmission observed in transgenic mouse models of AD have also been reported to contribute to AD associated sleep abnormalities.33,34 However, it has been difficult to unambiguously identify specific mechanisms and brain regions that play a causative role in mediating sleep abnormalities observed in AD patients. As such, analysis of sleep disturbances may offer important insights into the pathological mechanisms underlying AD.
Drosophila has become a well-accepted behavioral model for sleep research as it shares many features with mammalian sleep35,36 and is thus well suited to examine the fundamental functions of sleep, and the mechanisms that regulate it.37,38 In Drosophila, the small- and large- ventrolateral neurons (LNv) (henceforth referred to as sLNv and lLNv, respectively) are part of the well-characterized fly circadian circuitry39 as well as the “core” sleep circuitry in the fly.40,41 Both the circadian and sleep regulatory effects of the LNvs are mediated via the neuropeptide pigment dispersing factor (PDF) that serves as the main functional output from the LNvs to coordinate neural circuits that operate downstream of the LNvs42,43 (Fig. 1). A limited number of other fly brain regions have been proposed to contribute to sleep. These include the mushroom body and pars intercerebralis in the central brain and importantly, both regions are thought to receive rhythmic signal from the sLNv axon terminals.44 These features bear resemblance to the regulatory effects that the mammalian pacemaker, the suprachiasmatic nucleus (SCN) has on controlling sleep-wake cycles as well as coordinating this with other brain areas to enhance behavioral adaptation.45 All of these features make the Drosophila LNv sleep circuit a powerful model to study the mechanisms underlying sleep regulation.
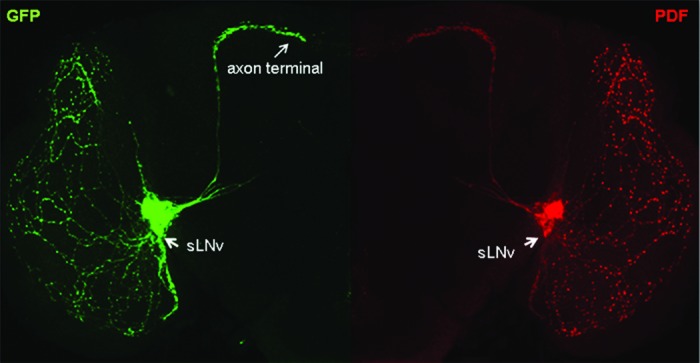
Figure 1.Drosophila ventrolateral neurons expressing GFP shown in green and the circadian neuropeptide pigment dispersing factor (PDF) shown in red. GFP and PDF Immunostaining label the circadian pacemaker cells, the small ventrolateral neurons (sLNv) including their dorsally projecting axons and terminal arbors in the central region of the adult fly brain. Axon growth and PDF expression in sLNv is epigenetically regulated by Tip60 HAT activity.
In the study by,46 we set out to test the hypothesis that APP and Tip60 are both required to mediate selective neuronal processes such as sLNv morphology and function that when misregulated, are linked to AD pathology. We found that both sLNv and lLNv cell types in the adult flies endogenously express Tip60. Furthermore, expression of a dominant negative mutant Tip60 that is defective in its epigenetic HAT function (Tip60HAT mut) in the LNvs using the LNv specific PDF-Gal4 driver causes sleep disturbances consisting of fragmented night sleep and daytime sleepiness, reminiscent of those observed in AD. Our analysis also revealed that the sLNvs are particularly susceptible to loss of Tip60’s HAT activity and exhibit diminished expression of PDF as well as retraction of the sLNv axon terminals that are required for pre-synaptic release of PDF in the dorsal protocerebrum (Table 1). Importantly, these effects mediated by loss of Tip60 HAT activity were confined to the sLNvs in the adult flies and there no marked effect on these neurons during early larval development. These neuroanatomical defects likely contributed to the sleep disturbances by disrupting PDF-mediated interaction of the sLNvs with downstream circuits. Overexpression of APP within the LNvs also resulted in similar reduction in sLNv PDF expression and led to disruption of night sleep and increased daytime sleepiness, an effect that was dependent on the presence of the C-terminus of APP that is required for generation of the AICD fragment that interacts with Tip60. Intriguingly, disruption of Tip60 HAT activity under APP induced neurodegenerative conditions (APP; Tip60HAT mut) was found to exacerbate retraction of the sLNv axonal terminals and caused complete loss of PDF, although the sleep disturbances were same as in flies exhibiting only loss of Tip60 HAT activity in their sLNvs (Table 1). Importantly, the anatomical defects we observed were dependent on the presence of the C-terminus of APP. Together, the degenerative effects we observe specifically in the sLNvs suggest that Tip60 mediated epigenetic dysregulation can render selective neuronal populations more vulnerable to APP induced neurodegeneration with detrimental consequences on associated behavioral outputs. A functional interaction between Tip60 and the AICD fragment of APP has been shown by us and others to epigenetically regulate genes essential for neurogenesis.18,23,24 Such an effect is thought to be mediated by recruitment of the Tip60/AICD-containing complex to certain gene promoters in the nervous system that are then epigenetically modified by Tip60 via site-specific acetylation of specific histones and accordingly activated or repressed. While the mutation in our dominant negative HAT-defective version of Tip60 (dTip60HAT mut) reduces Tip60 HAT activity, it does not interfere with its ability to assemble into a protein complex.16,47 Thus, dTip60HAT mut likely exerts its dominant negative action over endogenous wild-type Tip60 via competition with the endogenous wild-type Tip60 protein for access to the Tip60/AICD complex and/or additional native Tip60 complexes, with subsequent negative consequences on chromatin histone acetylation and gene regulation critical for promoting sLNv axon growth. A number of recent studies indicate that Tip60 not only functions as a transcriptional co-activator but also as a co-repressor. Although the exact mechanism for how Tip60 represses genes remains unclear, it is thought to possibly occur through direct recruitment and interaction of Tip60 with transcriptional silencers and/or histone deacetylases.47,48 As such, the presence of the HAT mutant Tip60 in the Tip60/AICD complex or additional native Tip60 complexes could mediate gene expression changes that lead to activation or de-repression of factors that promote axonal degeneration thereby causing the observed detrimental effects on sLNv axon growth.
Table 1. Tip60 induced defects on axonal growth and/or PDF expression in Drosophila sLNv affects sleep.
| Mutant Phenotype | |||
|---|---|---|---|
| Genotype |
Axon growth |
Effect on PDF |
Sleep Defects |
| Tip60HAT mut |
Retracted |
Partial loss |
Yes |
| APP; Tip60HAT mut |
Severely retracted |
Complete loss |
Yes |
| Tip60OE |
No effect |
Elevated |
Yes |
| APP; Tip60OE | Overelaborated | Elevated | No |
sLNv directed expression of HAT defective mutant Tip60 (Tip60HAT mut) causes retraction of axons and moderate decrease in PDF expression. These defects are exacerbated in flies that co-express the Tip60 HAT mutant with APP (APP; Tip60HAT mut) resulting in much shorter axons and complete loss of Pdf in sLNv. Sleep defects were observed under both these conditions and consisted of disrupted night sleep and daytime sleepiness. Night sleep was disrupted in flies overexpressing wild type Tip60 (Tip60OE) that also exhibited elevated levels of PDF in sLNv. Overexpression of Tip60 with APP (APP; Tip60OE) also induces sLNv PDF levels, however, without any marked effect on sleep, suggesting the induction of compensatory sleep inducing neural signals in these flies. The overelaborated sLNv axon terminals in the protocerebrum in the APP, Tip60OE flies could play a role in transducing such sleep promoting signals.
In light of these observations, we then hypothesized that overexpression of HAT competent Tip60 under APP overexpressing conditions would override APP mediated neurodegenerative effects and alleviate the observed sleep disturbances. LNv directed overexpression of Tip60 (Tip60OE) enhanced PDF expression in the sLNv with no marked effect on the sLNv axon growth. These flies also exhibit impaired ability to maintain sleep at night (Table 1), an effect we speculate could be mediated through untimely activation of downstream arousal promoting neural circuits by the excess PDF. Overexpression of wild type Tip60 in the LNvs in conjunction with APP containing its C-terminus (APP; Tip60OE) also increased sLNv PDF expression. Additionally, these flies also exhibited extensive arborization of the sLNv axon terminals in the dorsal protocerebrum. However, despite these anatomical changes, co-expression of Tip60 along with APP that contained its C-terminus restored the normal sleep-wake cycle (Table 1), consistent with our hypothesis.
So how can Tip60 overexpression in conjunction with APP rescue the night time sleep disruption and day time sleepiness we observe in APP overexpressing flies or when Tip60 itself is misregulated? The absence of any observable effect on sleep in the APP; Tip60OE flies despite the increase in sLNv PDF suggests the presence of additional sleep promoting compensatory mechanisms that help override the sleep defects induced by PDF overexpression. A clue to pinpointing a possible rescue mechanism comes from our observation that significant exacerbation of axonal arborization was only observed as a result of co-expression of wild type Tip60 and APP, and not when Tip60 was overexpressed alone; this may account for the difference in sleep phenotype between these two genotypes. Based on these findings, we propose a model by which such APP/Tip60 induced overelaboration of the sLNv axon terminals into the dorsal protocerebrum could play a role in restoring the sleep-wake cycles as these additional synaptic terminals may provide additional neural input sites for signals from sleep promoting neurons that are located in the vicinity (Fig. 2). This model garners support from two major observations: the sLNv axon terminals have been reported to express postsynaptic GABAB receptors and GABAergic sleep promoting neurons have also been observed in the vicinity of the sLNv axon terminals in the adult CNS,49 suggesting that the sLNvs might receive slow inhibitory GABAergic input from such neurons in the vicinity through the dorsal terminals. Furthermore, recent electron microscopy studies also indicate the presence of sparsely distributed input synapses at the sLNv axon terminals34 that could also play a role in transducing sleep promoting neural signals.

Figure 2. Model for cellular events that potentially contribute to rescue of sleep-wake cycle by Tip60 under APP overexpressing conditions. (A) Loss of PDF and severe shortening of sLNv axons contributes to sleep defects in flies co-expressing the Tip60 HAT mutant with APP (APP; Tip60HAT mut) likely by interfering with PDF dependent signaling to neural circuits operating downstream of sLNv. (B) Overelaborated sLNv synaptic arbors in flies overexpressing wild type Tip60 in conjunction with APP (APP; Tip60OE) may provide additional input sites for signals from sleep-promoting neurons in the vicinity that counteract the arousing effect of PDF overexpression on nocturnal sleep
Accumulating evidence indicates that axonal dysfunction and degeneration in AD may persist long before the disease related neuropathologies are detectable, and it is believed that these early axonal dystrophies in the affected neurons may significantly contribute to disease symptoms.50 In this regard, our observation that loss of Tip60 HAT activity causes retraction and loss thereof of sLNv axonal synaptic terminals and/or PDF signaling suggests that disruption of neuronal connectivity within particular neuronal circuits involved in sleep regulation may be an early event in the AD process, and may account for the sleep abnormalities that persist in AD patients long before pathophysiological manifestation of the disease sets in. Moreover, the neurodegenerative process in AD disseminates across different brain regions and types of neurons making it difficult to discern which of these susceptibility regions contribute to the behavioral defects. In this regard, while the LNv encompassing neural circuit is a much simpler and circumscribed version of the complex and widely distributed mammalian sleep circuitry, it lends itself as a tractable system to understand aspects of APP mediated circuit level control of sleep that is not well understood to date. Additionally, the dependence of the observed sleep defects in the APP overexpressing flies on the presence of the C-terminus of APP suggests that AICD mediated intracellular changes could lead to behavioral dysfunction prior to overt neuropathology. Furthermore, our data demonstrating the modulatory effects that Tip60 HAT activity or lack thereof have on the sLNvs under APP overexpressing conditions, potentially by interacting with the AICD fragment provides novel mechanistic insights into epigenetic regulation of neural circuits that underlie the sleep abnormalities persistent in AD patients. Although a number of HATs with vital neuronal functions including Tip60 have been implicated in neurodegenerative diseases, it remains to be determined whether the function of such HATs is altered due to aging, a major risk factor known to date for many neurodegenerative diseases including AD. Future investigations into identifying specific gene targets of the Tip60/AICD complex within the sLNv neurons and the downstream mechanism by which Tip60 regulates axonal growth as well as its apparent neuroprotective role in maintaining normal sleep-wake cycles under APP induced neurondegenerative conditions (Fig. 2) should serve as the groundwork when exploring the utility of specific HAT activators as early intervention therapeutic strategies to prevent or delay the progression of age-linked neurodegenerative disorders.
Acknowledgments
Research in our laboratory is supported by NIH grant R01HD057939 to F.E.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/fly/article/24141
References
- 1.Borrelli E, Nestler EJ, Allis CD, Sassone-Corsi P. Decoding the epigenetic language of neuronal plasticity. Neuron. 2008;60:961–74. doi: 10.1016/j.neuron.2008.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Legube G, Trouche D. Regulating histone acetyltransferases and deacetylases. EMBO Rep. 2003;4:944–7. doi: 10.1038/sj.embor.embor941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Feng J, Fouse S, Fan G. Epigenetic regulation of neural gene expression and neuronal function. Pediatr Res. 2007;61:58R–63R. doi: 10.1203/pdr.0b013e3180457635. [DOI] [PubMed] [Google Scholar]
- 4.Peleg S, Sananbenesi F, Zovoilis A, Burkhardt S, Bahari-Javan S, Agis-Balboa RC, et al. Altered histone acetylation is associated with age-dependent memory impairment in mice. Science. 2010;328:753–6. doi: 10.1126/science.1186088. [DOI] [PubMed] [Google Scholar]
- 5.Sterner DE, Berger SL. Acetylation of histones and transcription-related factors. Microbiol Mol Biol Rev. 2000;64:435–59. doi: 10.1128/MMBR.64.2.435-459.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roth SY, Denu JM, Allis CD. Histone acetyltransferases. Annu Rev Biochem. 2001;70:81–120. doi: 10.1146/annurev.biochem.70.1.81. [DOI] [PubMed] [Google Scholar]
- 7.Nowak SJ, Corces VG. Phosphorylation of histone H3 correlates with transcriptionally active loci. Genes Dev. 2000;14:3003–13. doi: 10.1101/gad.848800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rice JC, Allis CD. Histone methylation versus histone acetylation: new insights into epigenetic regulation. Curr Opin Cell Biol. 2001;13:263–73. doi: 10.1016/S0955-0674(00)00208-8. [DOI] [PubMed] [Google Scholar]
- 9.Fischle W, Wang Y, Allis CD. Histone and chromatin cross-talk. Curr Opin Cell Biol. 2003;15:172–83. doi: 10.1016/S0955-0674(03)00013-9. [DOI] [PubMed] [Google Scholar]
- 10.Stilling RM, Fischer A. The role of histone acetylation in age-associated memory impairment and Alzheimer’s disease. Neurobiol Learn Mem. 2011;96:19–26. doi: 10.1016/j.nlm.2011.04.002. [DOI] [PubMed] [Google Scholar]
- 11.Selvi BR, Cassel JC, Kundu TK, Boutillier AL. Tuning acetylation levels with HAT activators: therapeutic strategy in neurodegenerative diseases. Biochim Biophys Acta. 2010;1799:840–53. doi: 10.1016/j.bbagrm.2010.08.012. [DOI] [PubMed] [Google Scholar]
- 12.Konsoula Z, Barile FA. Epigenetic histone acetylation and deacetylation mechanisms in experimental models of neurodegenerative disorders. J Pharmacol Toxicol Methods. 2012;66:215–20. doi: 10.1016/j.vascn.2012.08.001. [DOI] [PubMed] [Google Scholar]
- 13.Miyagi S, Mishima Y, Iwama A. [Structure and function of MYST family histone acetyltransferases] Rinsho Ketsueki. 2011;52:490–6. [PubMed] [Google Scholar]
- 14.Sapountzi V, Logan IR, Robson CN. Cellular functions of TIP60. Int J Biochem Cell Biol. 2006;38:1496–509. doi: 10.1016/j.biocel.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 15.Zhu X, Singh N, Donnelly C, Boimel P, Elefant F. The cloning and characterization of the histone acetyltransferase human homolog Dmelin Drosophila melanogaster: Dmelis essential for multicellular development. Genetics. 2007;175:1229–40. doi: 10.1534/genetics.106.063685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lorbeck M, Pirooznia K, Sarthi J, Zhu X, Elefant F. Microarray analysis uncovers a role for Tip60 in nervous system function and general metabolism. PLoS One. 2011;6:e18412. doi: 10.1371/journal.pone.0018412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sarthi J, Elefant F. dTip60 HAT activity controls synaptic bouton expansion at the Drosophila neuromuscular junction. PLoS One. 2011;6:e26202. doi: 10.1371/journal.pone.0026202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pirooznia SK, Sarthi J, Johnson AA, Toth MS, Chiu K, Koduri S, et al. Tip60 HAT activity mediates APP induced lethality and apoptotic cell death in the CNS of a Drosophila Alzheimer’s disease model. PLoS One. 2012;7:e41776. doi: 10.1371/journal.pone.0041776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gehrking KM, Andresen JM, Duvick L, Lough J, Zoghbi HY, Orr HT. Partial loss of Tip60 slows mid-stage neurodegeneration in a spinocerebellar ataxia type 1 (SCA1) mouse model. Hum Mol Genet. 2011;20:2204–12. doi: 10.1093/hmg/ddr108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cao X, Südhof TC. A transcriptionally [correction of transcriptively] active complex of APP with Fe65 and histone acetyltransferase Tip60. Science. 2001;293:115–20. doi: 10.1126/science.1058783. [DOI] [PubMed] [Google Scholar]
- 21.Słomnicki LP, Leśniak W. A putative role of the Amyloid Precursor Protein Intracellular Domain (AICD) in transcription. Acta Neurobiol Exp (Wars) 2008;68:219–28. doi: 10.55782/ane-2008-1691. [DOI] [PubMed] [Google Scholar]
- 22.Kim HS, Kim EM, Kim NJ, Chang KA, Choi Y, Ahn KW, et al. Inhibition of histone deacetylation enhances the neurotoxicity induced by the C-terminal fragments of amyloid precursor protein. J Neurosci Res. 2004;75:117–24. doi: 10.1002/jnr.10845. [DOI] [PubMed] [Google Scholar]
- 23.Kinoshita A, Whelan CM, Berezovska O, Hyman BT. The gamma secretase-generated carboxyl-terminal domain of the amyloid precursor protein induces apoptosis via Tip60 in H4 cells. J Biol Chem. 2002;277:28530–6. doi: 10.1074/jbc.M203372200. [DOI] [PubMed] [Google Scholar]
- 24.Baek SH, Ohgi KA, Rose DW, Koo EH, Glass CK, Rosenfeld MG. Exchange of N-CoR corepressor and Tip60 coactivator complexes links gene expression by NF-kappaB and beta-amyloid precursor protein. Cell. 2002;110:55–67. doi: 10.1016/S0092-8674(02)00809-7. [DOI] [PubMed] [Google Scholar]
- 25.Hernández F, Nido JD, Avila J, Villanueva N. GSK3 inhibitors and disease. Mini Rev Med Chem. 2009;9:1024–9. doi: 10.2174/138955709788922647. [DOI] [PubMed] [Google Scholar]
- 26.Kang JE, Lim MM, Bateman RJ, Lee JJ, Smyth LP, Cirrito JR, et al. Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science. 2009;326:1005–7. doi: 10.1126/science.1180962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Walker MP, Stickgold R. Sleep, memory, and plasticity. Annu Rev Psychol. 2006;57:139–66. doi: 10.1146/annurev.psych.56.091103.070307. [DOI] [PubMed] [Google Scholar]
- 28.Stickgold R, Walker MP. Sleep-dependent memory consolidation and reconsolidation. Sleep Med. 2007;8:331–43. doi: 10.1016/j.sleep.2007.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ancoli-Israel S, Parker L, Sinaee R, Fell RL, Kripke DF. Sleep fragmentation in patients from a nursing home. J Gerontol. 1989;44:M18–21. doi: 10.1093/geronj/44.1.M18. [DOI] [PubMed] [Google Scholar]
- 30.Sterniczuk R, Dyck RH, Laferla FM, Antle MC. Characterization of the 3xTg-AD mouse model of Alzheimer’s disease: part 1. Circadian changes. Brain Res. 2010;1348:139–48. doi: 10.1016/j.brainres.2010.05.013. [DOI] [PubMed] [Google Scholar]
- 31.Schwartz JR, Roth T. Neurophysiology of sleep and wakefulness: basic science and clinical implications. Curr Neuropharmacol. 2008;6:367–78. doi: 10.2174/157015908787386050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Swaab DF, Fliers E, Partiman TS. The suprachiasmatic nucleus of the human brain in relation to sex, age and senile dementia. Brain Res. 1985;342:37–44. doi: 10.1016/0006-8993(85)91350-2. [DOI] [PubMed] [Google Scholar]
- 33.Wisor JP, Edgar DM, Yesavage J, Ryan HS, McCormick CM, Lapustea N, et al. Sleep and circadian abnormalities in a transgenic mouse model of Alzheimer’s disease: a role for cholinergic transmission. Neuroscience. 2005;131:375–85. doi: 10.1016/j.neuroscience.2004.11.018. [DOI] [PubMed] [Google Scholar]
- 34.Yasuyama K, Meinertzhagen IA. Synaptic connections of PDF-immunoreactive lateral neurons projecting to the dorsal protocerebrum of Drosophila melanogaster. J Comp Neurol. 2010;518:292–304. doi: 10.1002/cne.22210. [DOI] [PubMed] [Google Scholar]
- 35.Hendricks JC, Sehgal A. Why a fly? Using Drosophila to understand the genetics of circadian rhythms and sleep. Sleep. 2004;27:334–42. doi: 10.1093/sleep/27.2.334. [DOI] [PubMed] [Google Scholar]
- 36.Shaw P. Awakening to the behavioral analysis of sleep in Drosophila. J Biol Rhythms. 2003;18:4–11. doi: 10.1177/0748730402239672. [DOI] [PubMed] [Google Scholar]
- 37.Ho KS, Sehgal A. Drosophila melanogaster: an insect model for fundamental studies of sleep. Methods Enzymol. 2005;393:772–93. doi: 10.1016/S0076-6879(05)93041-3. [DOI] [PubMed] [Google Scholar]
- 38.Cirelli C. The genetic and molecular regulation of sleep: from fruit flies to humans. Nat Rev Neurosci. 2009;10:549–60. doi: 10.1038/nrn2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nitabach MN, Taghert PH. Organization of the Drosophila circadian control circuit. Curr Biol. 2008;18:R84–93. doi: 10.1016/j.cub.2007.11.061. [DOI] [PubMed] [Google Scholar]
- 40.Parisky KM, Agosto J, Pulver SR, Shang Y, Kuklin E, Hodge JJ, et al. PDF cells are a GABA-responsive wake-promoting component of the Drosophila sleep circuit. Neuron. 2008;60:672–82. doi: 10.1016/j.neuron.2008.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sheeba V, Fogle KJ, Kaneko M, Rashid S, Chou YT, Sharma VK, et al. Large ventral lateral neurons modulate arousal and sleep in Drosophila. Curr Biol. 2008;18:1537–45. doi: 10.1016/j.cub.2008.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lin Y, Stormo GD, Taghert PH. The neuropeptide pigment-dispersing factor coordinates pacemaker interactions in the Drosophila circadian system. J Neurosci. 2004;24:7951–7. doi: 10.1523/JNEUROSCI.2370-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lear BC, Zhang L, Allada R. The neuropeptide PDF acts directly on evening pacemaker neurons to regulate multiple features of circadian behavior. PLoS Biol. 2009;7:e1000154. doi: 10.1371/journal.pbio.1000154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Helfrich-Förster C, Wulf J, de Belle JS. Mushroom body influence on locomotor activity and circadian rhythms in Drosophila melanogaster. J Neurogenet. 2002;16:73–109. doi: 10.1080/01677060213158. [DOI] [PubMed] [Google Scholar]
- 45.Moore RY. Suprachiasmatic nucleus in sleep-wake regulation. Sleep Med. 2007;8(Suppl 3):27–33. doi: 10.1016/j.sleep.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 46.Pirooznia SK, Chiu K, Chan MT, Zimmerman JE, Elefant F. Epigenetic regulation of axonal growth of Drosophila pacemaker cells by histone acetyltransferase tip60 controls sleep. Genetics. 2012;192:1327–45. doi: 10.1534/genetics.112.144667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yan Y, Barlev NA, Haley RH, Berger SL, Marmorstein R. Crystal structure of yeast Esa1 suggests a unified mechanism for catalysis and substrate binding by histone acetyltransferases. Mol Cell. 2000;6:1195–205. doi: 10.1016/S1097-2765(00)00116-7. [DOI] [PubMed] [Google Scholar]
- 48.Li B, Samanta A, Song X, Iacono KT, Bembas K, Tao R, et al. FOXP3 interactions with histone acetyltransferase and class II histone deacetylases are required for repression. Proc Natl Acad Sci U S A. 2007;104:4571–6. doi: 10.1073/pnas.0700298104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hamasaka Y, Wegener C, Nässel DR. GABA modulates Drosophila circadian clock neurons via GABAB receptors and decreases in calcium. J Neurobiol. 2005;65:225–40. doi: 10.1002/neu.20184. [DOI] [PubMed] [Google Scholar]
- 50.Luo L, O’Leary DD. Axon retraction and degeneration in development and disease. Annu Rev Neurosci. 2005;28:127–56. doi: 10.1146/annurev.neuro.28.061604.135632. [DOI] [PubMed] [Google Scholar]