

Abstract
Background:
Since the ancient times the skin aging application of honeybee venom (BV) is practiced and persisted until nowadays. The present study evaluated the effect of the honeybee venom (BV) on keratinocyte migration in wound healing model in vitro.
Objective:
To access BV further as a cosmetic ingredient and a potential external application for topical uses, we performed studies to investigate the biologic effect of BV treatment on keratinocyte proliferation and migration in vitro.
Material and Methods:
BV cytotoxicity was assessed by using a 3-[4,5-dimethyl-2-thiazolyl]-2,5-diphenyl tetrazolium bromide (MTT) assay over 24 h. To assess BV genotoxicity, damage to human epidermal keratinocyte (HEK) was evaluated using the Comet assay. HEK migration was evaluated using a commercial wound healing kit. The skin pro-inflammatory cytokines interleukin (IL)-8 and tumor necrosis factor (TNF)-α were examined to evaluate the pro-inflammatory response to BV.
Results:
It was found that BV (<100 μg/ml) was not cytotoxic and stimulated more HEK proliferation and migration compared to negative control, and did not induce DNA damage. There were also decreases in IL-8 and TNF-α expression levels in HEK at all time points.
Conclusion:
These findings highlight the potential of topical application of BV for promoting cell regeneration and wound treatment.
Keywords: Bee venom, Keratinocyte, migration, regeneration, skin
INTRODUCTION
Skin wound healing processes normally involves re-epithelization, contraction, formation of granulation tissue and collagen synthesis.[1,2] If any of the above phases are not balanced, the healing will be delayed and may leave scars after healing. Fibroblast proliferation affects the formation of granulation tissue, collagen synthesis and wound contraction, while keratinocyte migration affects re-epithelization.[3] Various growth factors or cytokines secreted by inflammatory cells, keratinocytes, fibroblasts, or the like are known to be involved in each of the phases of healing cuts.[4,5] That is, in the process of skin wound healing, macrophages are signalled to gather around the wound by carriers secreted by inflammatory cells. The macrophages begin to secrete cytokines and the fibroblasts move to the wound and proliferate therein forming a matrix in the dermis of the wound, thereby healing the wound. On the other hand, in wounds with an abnormal proliferation of inflammatory cells due to secondary infection, various inflammation-induced cytokines such as tumor necrosis factor (TNF)-α and interlukin (IL)-8 are secreted to suppress the proliferation of keratinocytes.[6,7] Thus, preventing scars as well as reconstructing skin is important. Because of its popularity in folk medicine, honeybee venom has become the subject of intense pharmacological and biological investigations. Numerous studies have proven its effectiveness in treating pathological conditions such as arthritis, rheumatism, pain, cancerous tumors, and skin diseases.[8] Recently bee venom also has been used as a cosmetic ingredient for antiaging, anti-inflammatory and antibacterial functions. Pure bee venom (BV) of honey bees (Apis mellifera) is generally obtained by collecting a large amount of BV by electric stunning using a BV collector without harming the honey bees, removing impurities from the collected BV, and lyophilizing the resultant. The obtained BV includes various components including peptides known to have anti-inflammatory activity, antibacterial activity, strong analgesic activity, and immune response enhancing effects.[8,9,10] For the purpose of accessing BV further as a cosmetic ingredient and a potential external application for topical uses, we performed studies to investigate the biological effect of BV treatment on keratinocyte proliferation and migration in vitro.
MATERIALS AND METHODS
BV of european honeybees, A. mellifera L.
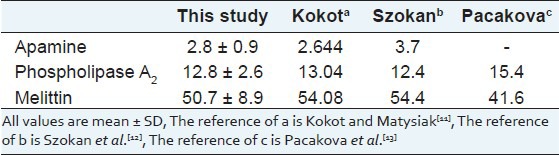
Colonies of natural honey bees used in this study were maintained at the National Academy of Agricultural Science, Suwon, South Korea. BV was collected by a bee venom collecting device (Chunggin, Korea) in a sterile manner under strict laboratory conditions. In brief, the bee venom collector was placed on the hive, and the bees were given enough electric shock to cause them to sting a glass plate from which dried BV was later scraped off. The collected venom was diluted in cold sterile water and then centrifuged at 10,000 g for 5 min at 4°C to discard residues from the supernatant. BV was lyophilized by freeze dryer and refrigerated at 4°C for later use. All the bioactive components of BV used in the experiment were confirmed with size exclusion gel chromatography (AKTAexplorer, Pharmacia, USA) by dissolving in 0.1 Mammonium formate adjusted to pH 4.5. A Sephadex TM75 column (Amersham Biosciences, USA) and then Source 15RPC ST eluent 0.1% trifluoracetic acid in 20% acetonitrile was used to confirm the presence of melittin, the major active ingredient of BV [Table 1]. Component of melittin, apamin, and phospholipase A2 was calculated with the following formulation. Standard of melittin, apamin, and phospholipase A2 were obtained commercially from Sigma (Sigma, USA).
Table 1.
The content (%) of major BV constituents (apamine, phospholipase A2 and melittin) and and concentration between this and three other studies
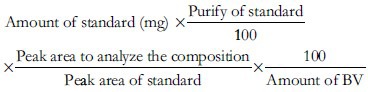
Culture of human epidermal keratinocytes
Human epidermal keratinocytes (HEK) isolated from human neonatal foreskin were purchased from Modern Tissue Technologies Inc. (Seoul, Korea). HEK were cultured on keratinocyte basal medium (Cambrex, East Rutherford, NJ) containing 10% fetal bovine serum, penicillin (100 IU/ml), and streptomycin (100 mg/ml) at 37°C in a humidified atmosphere containing 5% CO2.
Cell viability assay
HEK were assessed using a 3-4 5-dimethylthiazol-2-yl]-2, 5-diphenyltetrazolium bromide (MTT) assay. HEK were seeded in 96-well plates and treated with various concentrations of BV. The cells were cultured in a CO2 incubator for 48 h. After removing the media, MTT was added and the cells were incubated further at 37°C for 4 h. Supernatants were carefully discarded and DMSO was added. After incubating at 25°C for 30 min, the reactants were measured using a microplate reader (UV max, SunnyvalMolecular Devices, USA) at 540 nm.
Cell migration assay In Vitro
A HEK migration assay was performed using CytoSelectTM 24-Well Wound Healing Assay (Cell Biolabs, Inc., San Diego, CA). Inserts were placed in the plate wells with their wound field aligned in the same direction. Cells (5 × 105 cells/well) were added to each well by inserting the pipette tip through the opening end at the top of the insert. Cells were incubated for 24 h until a monolayer formed. Inserts were then removed from the well and the media were discarded. Various concentrations of BV and media were added to the wells. Cells were incubated for 48 h. The migration rate of the cells was measured in the wound field. The migration rate results were visualized with cell staining and calculated according to the following formula:
Migration rate = length of cell migration (nm)/migration time (h).
Comet assay for genotoxicity assessment
This experiment was conducted as described by Tice et al.[14] with some modifications using a CometAssayTM kit (Trevigen, Gaithersbug, MD). HEK (1 × 105 cells/ml) were combined with melted LMAgarose at a ratio of 1:10 (v/v), and 75 μl was immediately pipetted onto a CometSlide TM (Trevigen). The slides were placed flat at 4°C in the dark for 30 min in a high- humidity environment until the gel solidified. The slides were then immersed in pre-chilled lysis buffer comprising freshly prepared alkaline solution at pH > 13 for 60 min at room temperature in the dark. To perform the alkaline electrophoresis, the slides were transferred from the alkaline solution to a horizontal electrophoresis apparatus, placed flat onto a gel tray and centered between the electrodes. The voltage was set to approximately 1V/cm, and the electrophoresis was performed for 1 h. After electrophoresis, the slides were rinsed in distilled water and then immersed in 70% ethanol for 5min. The slides were air-dried and stained with the CometAssay silver staining solution (Trevigen). The comet tail length of HEK treatment was scored at 24 h and 72 h post-treatment. HEK treated with BV and organotin polyvinylchloride (PVC) served as positive controls and those without any treatment served as the negative control. The lengths of 100 randomly selected comet tails from each group were measured using a light microscope with image analysis software (Optimas, Media Cybernetics, Bethesda, MD).
Enzyme-linked immunosorbent assay (ELISA) for TNF- α and IL-8 expression
Supernatants from HEK treated with BV were obtained at 24, 48, and 72 h and the amounts of secreted human TNF-α and IL-8 induced by BV treatment were quantitatively measured using an ELISA Kit (SA Biosciences, Frederick, MD). Various samples (50 ml each) were added to a 96-well ELISA plate. The plate was incubated for 2 h at room temperature. Biotinylated antibody reagent was applied to each well and the plate was incubated at room temperature for 2 h. After washing the plate with PBS-Tween 20, diluted streptavidin-horseradish peroxidase was added, and the plate was incubated at room temperature for 30 min. After washing the plate, premixed tetramethylbenzemidine substrate solution was added. The plate was developed in a dark room for 30 min and read at 490 nm using a microplate reader (UV max). TNF-a and IL-8 concentrations were calculated using human recombinant TNF-a and human recombinant IL-8 as the standards.
Measurement of TNF-α and IL-8 mRNA with reverse-transcription–polymerase chain reaction (RT- PCR)
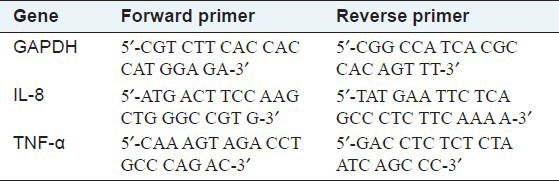
The RNA was extracted from the BV-treated HEK at 48 h using the RNAzol B (Tel-Test Inc, Friendwood, TX) reagent by the acid guanidinium thiocyanate-phenol-chloroform extraction procedure according to the manufacturer's instructions. RT-PCR was performed in a single reaction tube with an Access RT-PCR system (Promega, Madison, WI). Amplified PCR product was confirmed in a 1.5 % agarose gel and quantified with a gel calculation program (Optima 6.5, Baltimore, MD). The sequences of sense and anti-sense primers (Genotec, Korea) are shown in Table 2. The signal intensity of each band was quantified and normalized as GAPDH. Densitometric analysis was measured by using the Quantity One (Bio-Rad, Hercules, CA, USA) to scan the signals.
Table 2.
Primer sequences used in RT-PCR
Statistical analysis
Data are presented as mean ± standard error (SE). Experimental results were statistically analyzed using the Duncan's t-tests (SAS Enterprise Guide, SAS Institute Inc., Cary, NC).
RESULTS
Cytotoxicity based on MTT assay
To verify that BV was not toxic to the HEK cultures, an MTT assay was performed to determine cell viability of cells after treatment with various BV concentrations for 48 h. Results indicated that up to 10 mg/ml of BV treatment did not significantly affected cell viability [Figure 1]. One mg/ml BV treatment increased HEK proliferation compared with the control group. In the Figure 1 is described only until 10 mg/ml and not 100 mg/ml. Thus, in 72 h, 10 mg/ml induced significant decrease.
Figure 1.
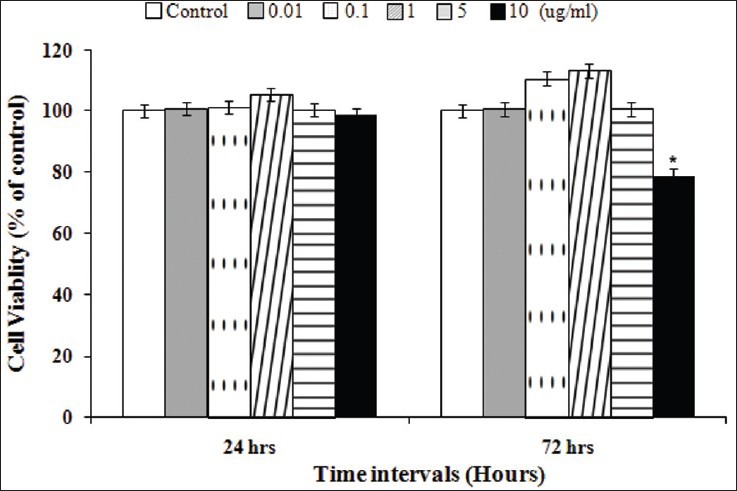
Cell viability assessed by MTT assay at 48 h compared with the negative control (cells only). BV (<100 mg/ml) induced no severe cytotoxicity in the keratinocytes. The concentration of 1 mg/ml BV slightly increased keratinocyte proliferation. Cell viability was reduced, however, in 100 mg/ml BV after 48 h. Values are expressed as mean ± SE. *P < 0.05 compared with the negative control
Effect of BV on cell migration
To evaluate migration, we determined the migration rate of cells in the defined wound area. The percentage of the migration rate was calculated by measuring the length of cell migration and expressed as percentage compared to the control group. The distance of cell migration in all groups was recorded after 48 h incubation. In the migration assay, the distance of cell migration was dramatically increased in the experimental groups exposed to BV compared to the control group [Figure 2]. In both 0.1 and 1mg/ml BV groups the distance of cell migration significantly increased compared to the control group (P < 0.05). These findings suggest that HEK migration occurred more rapidly in the BV-treated groups, indicating that BV stimulates keratinocyte migration.
Figure 2.
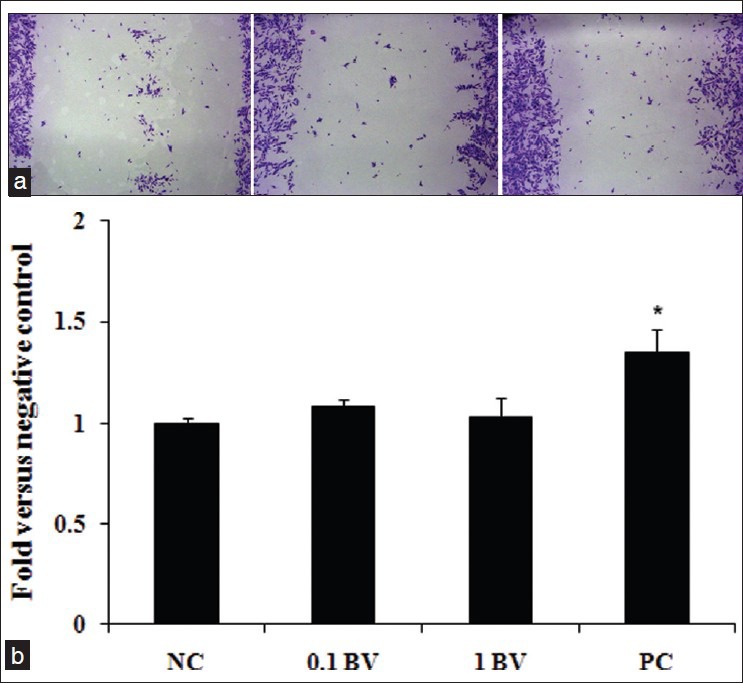
Effect of BV on keratinocyte migration in a migration assay. BV increases keratinocyte migration. Cells were treated in the presence or absence of BV and the incubated for 24 h. The migration rate of the cells into the wound field was measured. Cells were visualized with phase contrast cell staining. (a) Wounds in the control (cells only), (b) 0.1 mg/ml BV, C; 1 mg/ml BV. Values are expressed as mean ± SE. *P < 0.05 compared with the control
Genotoxicity of bv based on the comet assay
HEK cells treated with organotin-PVC (positive control) were longer tail length [Figure 3]. HEK cells treated with 1mg/ml BV had a tail length comparable to that of the negative control (cells only) at 24 and 72 h post-treatment [Figure 3]. There was no significant difference in the DNA tail length between BV-treated cells and the negative control (P>0.05). Genotoxicity induced by BV was not detected at 24 and 72 h post-treatment.
Figure 3.
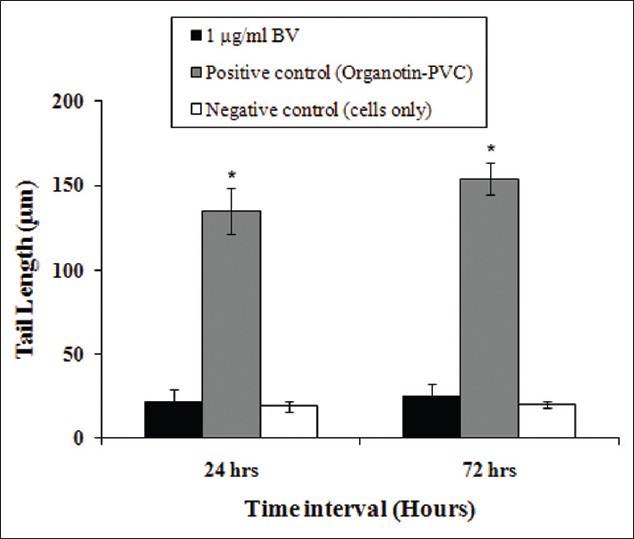
Genotoxicity of BV on keratinocytes based on comet tail length. BV did not increase the tail length of the cell compared with the negative control (cells only). Values are expressed as mean ± SE. *P < 0.05 compared with the negative control
Skin pro-inflammatory cytokines expressions
To determine the quantitative analysis of the pro-inflammatory cytokines, we measured the expression of IL-8 and TNF-α in HEK cells. In the positive control, the treatment of organotin-PVC significantly increased IL-8 [Figure 4a] and TNF-α [Figure 4b] secretion from 24 to 72 h. HEK cells treated with BV had the greatest decrease of IL-8 at 24 and 72 h. IL-8 expression was not significantly different between BV-treated HEK and the negative control [cells only, Figure 4a]. At 24 and 72 h after BV treatment, TNF-α expression was significantly decreased [Figure 4b]. TNF-α expression was not significantly different between BV-treated HEK and the negative control.
Figure 4.
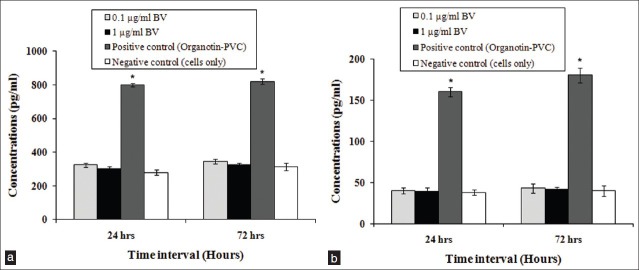
Effect of BV on IL-8. (a) and TNF-α. (b) production after 24 and 72 h of culture. Treatment with BV did not induce significant IL-8. (a) and TNF-α. (b) production compared with negative control (cells only). Values are expressed as mean ± SE. *P < 0.05 compared with the negative control
Gene expressions of skin pro-inflammatory cytokines
The specific expression of IL-8 and TNF-α mRNA was determined in HEK using RT-PCR amplification. RT-PCR using normalization to GAPDH showed the mean levels of gene expression for IL-8 in the treatment of 0.1 and 1 mg/ml BV were 1.18 ± 0.33 and 1.07 ± 0.21 fold, respectively, less than that for the positive control [Figure 5a]. As you can see Figure 5a, BV induced a significant decrease in IL-8 gene expression compared with the positive control (P < 0.05). Mean TNF-α gene expression levels in the positive control and in HEK treated with 0.1 and 1 mg/ml BV were, 1.1 ± 0.14 and 1.02 ± 0.34 fold, respectively less than that of the positive control [Figure 5b]. BV induced a significant decrease in TNF-α gene expression compared with the positive control (P < 0.05).
Figure 5.
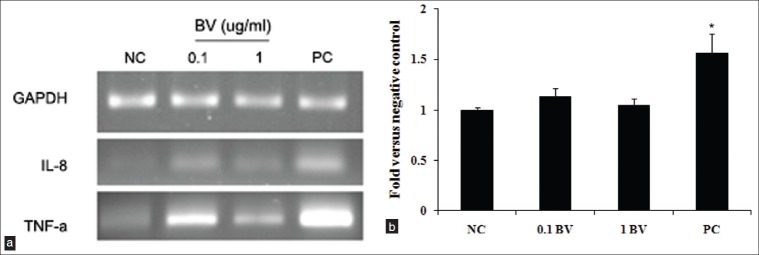
Effect of BV on IL-8. (a) and TNF-α. (b) mRNA expression in keratinocytes after 72 h culture. The relative intensity of IL-8 and TNF-α mRNA was normalized to the intensity of GAPDH mRNA. Treatment with BV did not induce significant levels of IL-8. (a) and TNF-α. (b) mRNA compared with negative control (cells only). Values are expressed as mean ± SE relative to the negative control (cells only). *P < 0.05 compared with the negative control
DISCUSSION
An essential feature of a healed wound is the restoration of an intact epidermal barrier through wound epithelialization of an intact epidermal barrier through wound epithelialization, also known as re-epithelialization.[15] The directed migration of keratinocytes is critical to wound epitheliazation and defects in this function are associated with the clinical phenotype of chronic non-healing wounds. Wound healing can be conceptually viewed as the result of three overlapping keratinocyte functions: migration, proliferation, and differentiation.[16] The sequence of events by which keratinocytes accomplish the task of re-epithelialization is generally believed to begin with dissolution of cell-cell and cell-substratum contacts. The majority of the review will focus on some of the mechanisms that regulate keratinocyte migration in the wound healing process.[16,17] Inflammatory cytokines are felt to have roles in wound healing including stimulation of keratinocytes and proliferation of fibroblasts. It has long been thought that proinflammatory cytokines, including TNF-α and IL-8, play an important role in wound healing.[16,17] They likely influence various processes at the wound site, including stimulation of keratinocyte proliferation, synthesis and breakdown of extracellular matrix proteins, fibroblast chemotaxis, and regulation of the immune response. In support of a role for proinflammatory cytokines in wound healing, TNF-α and IL-8 was shown to be strongly upregulated during the inflammatory phase of healing. Many types of wound dressing are commercially available and are promoted to reduce healing time. Several drawbacks, however, have been reported with some dressings during the last decade.[18,19] Wound-healing models are classified as either in vivo or in vitro.[20] Studying wound healing with the in vitro method is generally rapid and simple, and involves fewer ethical considerations than the vivo method. In the present study, we used a simple and basic in vitro wound-healing model to examine whether locally applied BV accelerates cell proliferation in wound-healing process.
BV from honeybee stings has been widely used in BV therapy to relive pain and treat inflammatory diseases. BV therapy is a treatment modality that may be thousands of years old and involves the application of live bee stings to patient skin or, in more recent years, the injection of BV into the skin with a hypodermic needle.[8,21] The chemistry and pharmacology of BV have been reported. BV contains several peptides including melittin, apamin, adolapin, mast cell degranulating peptide, enzymes, biologically activity amines, and non-peptide components.[8] Enzymes are composed of phospholipase A2, hyaluonidase, acid phosphomonesterase, α-D-glucosidase, and lysophospholipase.[8] Melittin is the major component of BV. It comprises some 50% of the weight of freeze-dried BV and account for a number of interesting properties of BV. In the present study, we confirmed the content 50.7 ± 8.9 % of BV by chromatography. BV also has been reported to be effective in treating allergies, scarring, burns, and skin diseases.[22] Because inhibition of the matrix metalloptoteinase (MMPs) production is useful for preventing collagen damage, BV can be used to inhibit MMP-1 and MMP-3 production as a skin disease remedy.[23] Primary normal keratinocyte culture models are used for cutaneous toxicity testing of new drugs, cosmetic products, and other chemicals and require phenotypically normal cell systems.[24] In addition, human keratinocytes cultures are far more sensitive than fibroblasts due to the fact that keratinocytes are cultured in a serum-free environment, whereas fibroblasts cultures require approximately 10% serum in the culture medium.[25]
In the present study, BV treatment enhanced keratinocyte migration. BV was cytocompatible at the cellular level, but the long-term use of BV as a wound-dressing agent remains unknown. Thus, in vitro screening at the molecular level was performed by single cell gel electrophoresis (comet assay). The comet assay is sensitive enough to assay DNA damage caused by genotoxic substances at the individual cell level.[26] In genotoxicity experiments, DNA damage in cells exposed to BV was comparable to that of the negative control at all time points. Cytokines, such as IL-8 and TNF-α, are involved in regulating cell proliferation. IL-8 is a powerful neutrophil attractant and is commonly produced by keratinocytes after external irritation.[27] Release of IL-8 is preceded by the release of TNF-α. Toxic substances may stimulate higher expression levels of both TNF-α and IL-8. Treatment with BV, however, decreased IL-8 and TNF-α gene expression levels compared to the negative control. BV facilitates keratinocyte migration in vitro and decreases the secretion of pro-inflammatory cytokines. BV also likely activates the DNA repair system or induces the death of severely damaged cells, which is compensated for by increased cell division. We hypothesized that BV might behave as a chemical stimulant that activates the BV subsequently engages the inhibition of TNF-α and IL-8 expression. Because BV used in the present study contains inhibiting compounds such as melittin and apamin, it is a major biological effects. Furthrmore, we assumed that the immune boosting effect of BV was from the melittin because it is a major bioactive component of BV as confirmed with a chromatography in the present study.
The present study using an in vitro wound healing model demonstrated that BV could be applied topically to accelerate wound healing by cell regeneration process. Further in vivo studies are needed to evaluate the effect of BV treatment in topical application.
ACKNOWLEDGMENTS
This work supported by a grant (Code #: PJ009534) from the BioGreen 21 program, Rural Development Administration, Korea.
Footnotes
Source of Support: Supported by a grant (Code #: PJ009534) from the BioGreen 21 program, Rural Development Administration, Korea
Conflict of Interest: None declared.
REFERENCES
- 1.Clark RA. Regulation of fibroplasia in cutaneous wound repair. Am J Med Sci. 1993;306:42–8. doi: 10.1097/00000441-199307000-00011. [DOI] [PubMed] [Google Scholar]
- 2.Martin P. Wound healing-aiming for perfect skin regeneration. Science. 1997;276:75–81. doi: 10.1126/science.276.5309.75. [DOI] [PubMed] [Google Scholar]
- 3.Lee SW, Kim SH, Kim JY, Lee Y. The effect of growth hormone on fibroblast proliferation and keratinocyte migration. J Plast Reconstr Surg. 2010;63:364–9. doi: 10.1016/j.bjps.2009.10.027. [DOI] [PubMed] [Google Scholar]
- 4.Lim CK, Yaacob NS, Ismail Z, Halim AS. In vitro biocompatibility of chitosan porous skin regenerating templates(PSRTs) using primary human skin keratinocytes. Toxicol In Vitro. 2010;24:721–7. doi: 10.1016/j.tiv.2010.01.006. [DOI] [PubMed] [Google Scholar]
- 5.Reiss MJ, Han YP, Garcia E, Goldberg M, Yu H, Garner WL. Matrix metalloproteinase-9 delays wound healing in a murine wound model. Surgery. 2010;147:295–302. doi: 10.1016/j.surg.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maas-Szabowski N, Shimotoyodome A, Fusenig NE. Keratinocyte growth regulation in fibroblast cocultures via double paracrine mechanism. J Cell Sci. 1999;112:1843–53. doi: 10.1242/jcs.112.12.1843. [DOI] [PubMed] [Google Scholar]
- 7.Han YP, Tuan TL, Wu H, Hughes M, Garner WL. TNF-α stimulates activation of pro-MMP2 in human skin through NF-κB mediated induction of MT-1 MMP. J Cell Sci. 2010;114:131–9. doi: 10.1242/jcs.114.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Piek T. New York: Academy Press; Venom of the Hymenoptera. [Google Scholar]
- 9.Franklin R, Baer H. Comparison of honeybee venoms and their components from various sources. J Allergy Clin Immunol. 1975;55:285–98. doi: 10.1016/0091-6749(75)90001-9. [DOI] [PubMed] [Google Scholar]
- 10.Somerfield SD, Stach JL, Mraz C, Gervais F, Skamene E. Bee venom inhibits superoxide production by human neutrophils. Inflammation. 1984;8:385–91. doi: 10.1007/BF00918214. [DOI] [PubMed] [Google Scholar]
- 11.Kokot ZJ, Matysiak J. Simultaneous determination of major constituents of honeybee venom by LC-DAD. Chromatographia. 2009;69:1–5. [Google Scholar]
- 12.Szokann G, Horvath J, Almas M, Saftics G, Palocz A. Liquid chromatographic analysis and separation of polipeptide components from honey bee venom. J Liq Chromatogr. 1994;17:3333–49. [Google Scholar]
- 13.Pacakova V, Stulik K, Pham TH, Jelinek I, Vins I, Sykora D. Comparison of high-performance liquid chromatography and capillary electrophoresis for the determination of some bee venom components. J Chromatogr A. 1995;700:187–93. [Google Scholar]
- 14.Tice RR, Agurell E, Anderson D, Burlinson B, Hartmann A, Kobayashi H, et al. Single cell gel/comet assay: Guidelines for in vitro and in vivo genetic toxicology testing. Environ Mol Mutagen. 2000;35:206–21. doi: 10.1002/(sici)1098-2280(2000)35:3<206::aid-em8>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 15.Storey A, McArdle F, Friedmann PS, Jackson MJ, Rhodes LE. Eicosapentaenoic acid and docosahexaenoic acid reduce UVB- and TNF- induces IL-8 secretion in keratinoctes and UVB-induced IL-8 in fibroblasts. J Invest Dermatol. 2004;124:248–55. doi: 10.1111/j.0022-202X.2004.23543.x. [DOI] [PubMed] [Google Scholar]
- 16.Sivamani RK, Garcia MS, Isseroff RR. Wound re-epithelialization: Modulating keratinocyte migration in wound healing. Front Biosci. 2007;12:2849–68. doi: 10.2741/2277. [DOI] [PubMed] [Google Scholar]
- 17.Werner S, Grose R. Regulation of wound healing by growth factors and cytokines. Physiol Rev. 2002;83:835–70. doi: 10.1152/physrev.2003.83.3.835. [DOI] [PubMed] [Google Scholar]
- 18.Young SR, Dyson M, Hickman R, Lang S, Osborn C. Comparison of the effects of semi-occlusive polyurethane dressings and hydrocolloid dressings on dermal repair: 1. Cellular changes. J Invest Dermatol. 1991;97:586–92. doi: 10.1111/1523-1747.ep12481927. [DOI] [PubMed] [Google Scholar]
- 19.Chakravarthy D, Rodway N, Schmidt S, Smith D, Evancho M, Sims R. Evaluation of three new hydrocolloid dressings: Retention of dressing integrity and biodegradability of absorbent components attenuate inflammation. J Biomed Mater Res. 1994;28:1165–73. doi: 10.1002/jbm.820281007. [DOI] [PubMed] [Google Scholar]
- 20.Amadeu TP, Coulomb B, Desmouliere A, Costa AM. Cutaneous wound healing: myofibroblastic differentiation and in vitro models. Int J Low Extrem Wounds. 2003;2:60–8. doi: 10.1177/1534734603256155. [DOI] [PubMed] [Google Scholar]
- 21.Castro HJ, Mendez-Lnocencio JI, Omidvar B, Omidvar J, Santilli J, Nielsen HS, et al. A Phase I Study of the safety of honeybee venom extract as a possible treatment for patients with progressive forms of multiple sclerosis. Allergy Asthma Proc. 2005;26:470–6. [PubMed] [Google Scholar]
- 22.Han SM, Lee KG, Yeo JH, Kweon HY, Woo SO, Baek HJ, et al. Inhibitory effect of bee venom against ultraviolet B induced MMP-1 and MMP-3 in human dermal fibroblasts. J Apic Res. 2007;46:94–8. [Google Scholar]
- 23.Fisher GJ, Kang S, Varani J, Bata-Csorgo Z, Wan Y, Datta S, et al. Mechanisms of photoaging and chronological skin aging. Arch Dermatol. 2002;138:1462–70. doi: 10.1001/archderm.138.11.1462. [DOI] [PubMed] [Google Scholar]
- 24.Flint OP. In vitro toxicity testing: purpose, validation and strategy. Lab Anim. 1990;18:11–8. [Google Scholar]
- 25.Sharpe GR, Fisher C. Time dependent inhibition of growth of human keratinocytes and fibroblasts by cyclosporine A: Effect on keratinocytes at therapeutic blood levels. Br J Dermatol. 1990;123:207–13. doi: 10.1111/j.1365-2133.1990.tb01848.x. [DOI] [PubMed] [Google Scholar]
- 26.Leroy T, Van Hummelen P, Anard D, Castelain P, Kirsch-Volders M, Lauwerys R, et al. Evaluation of three methods for the detection of DNA single-strand breaks in human lymphocytes: alkaline elution, nick translation, and single-cell gel electrophoresis. J Toxicol Environ Health. 1996;47:409–22. doi: 10.1080/009841096161573. [DOI] [PubMed] [Google Scholar]
- 27.Mohamadzadeh M, Müller M, Hultsch T, Enk A, Saloga J, Knop J. Enhanced expression of IL-8 in normal human keratinocytes and human keratinocyte cell line HaCaT in vitro after stimulation with contact sensitizers, tolerogens and irritants. Exp Dermatol. 1994;3:298–303. doi: 10.1111/j.1600-0625.1994.tb00292.x. [DOI] [PubMed] [Google Scholar]