

Abstract
Oncogene-induced senescence (OIS) is crucial for tumour suppression. Senescent cells implement a complex pro-inflammatory response termed the senescence-associated secretory phenotype (SASP). The SASP reinforces senescence, activates immune surveillance and paradoxically also has pro-tumourigenic properties. Here, we present evidence that the SASP can also induce “paracrine senescence” in normal cells both in culture and in human and mouse models of OIS in vivo. Coupling quantitative proteomics with small molecule screens, we identified multiple SASP components mediating paracrine senescence, including TGFβ family ligands, VEGF, CCL2 and CCL20. Amongst them, TGFβ ligands play a major role by regulating p15INK4b and p21CIP1. Expression of the SASP is controlled by inflammasome-mediated IL-1 signalling. The inflammasome and IL-1 signalling are activated in senescent cells and IL-1α expression can reproduce SASP activation, resulting in senescence. Our results demonstrate that the SASP can cause paracrine senescence and impact on tumour suppression and senescence in vivo.
Keywords: Senescence, SASP, secretome, IL-1α, paracrine, TGFβ, inflammasome
INTRODUCTION
Senescence protects damaged cells from neoplastic transformation by inducing a stable growth arrest 1,2. Concomitant with this arrest, cells secrete a complex mixture of factors referred to as the senescence-associated secretory phenotype (SASP) or senescence-messaging secretome (SMS) 3,4. The SASP can exert opposing and contradictory effects 4. Initial studies focused on the pro-tumourigenic properties of the SASP 5-7 but the SASP also mediates important tumour suppressive effects 3. Specific components of the SASP such as IGFBP-7, PAI-1, IL-6 and CXCR2-binding chemokines (such as IL-8 or GROα) can reinforce senescence 8-11. The SASP also contributes to the surveillance and elimination of senescent cells by the immune system 12-14.
It is unclear whether pro-senescence effects can be exerted in non-cell-autonomous fashion (paracrine) in addition to cell-autonomous fashion (autocrine) and whether senescence can be transmitted to normal cells. Early experiments where ‘young’ and ‘old’ fibroblasts were mixed suggested that senescence was exclusively cell intrinsic 15,16 although more recently a ‘bystander senescence’ response has been suggested 17. However, although some factors secreted by senescent cells, such as IL-6, reinforce senescence in an intracrine fashion 9, others like IGFBP-7 can display paracrine effects 11. In this investigation, we present unequivocal evidence supporting that senescence can be transmitted in a paracrine fashion, and provide insights into the pathways regulating and mediating paracrine senescence.
RESULTS
Paracrine transmission of senescence by cells undergoing OIS
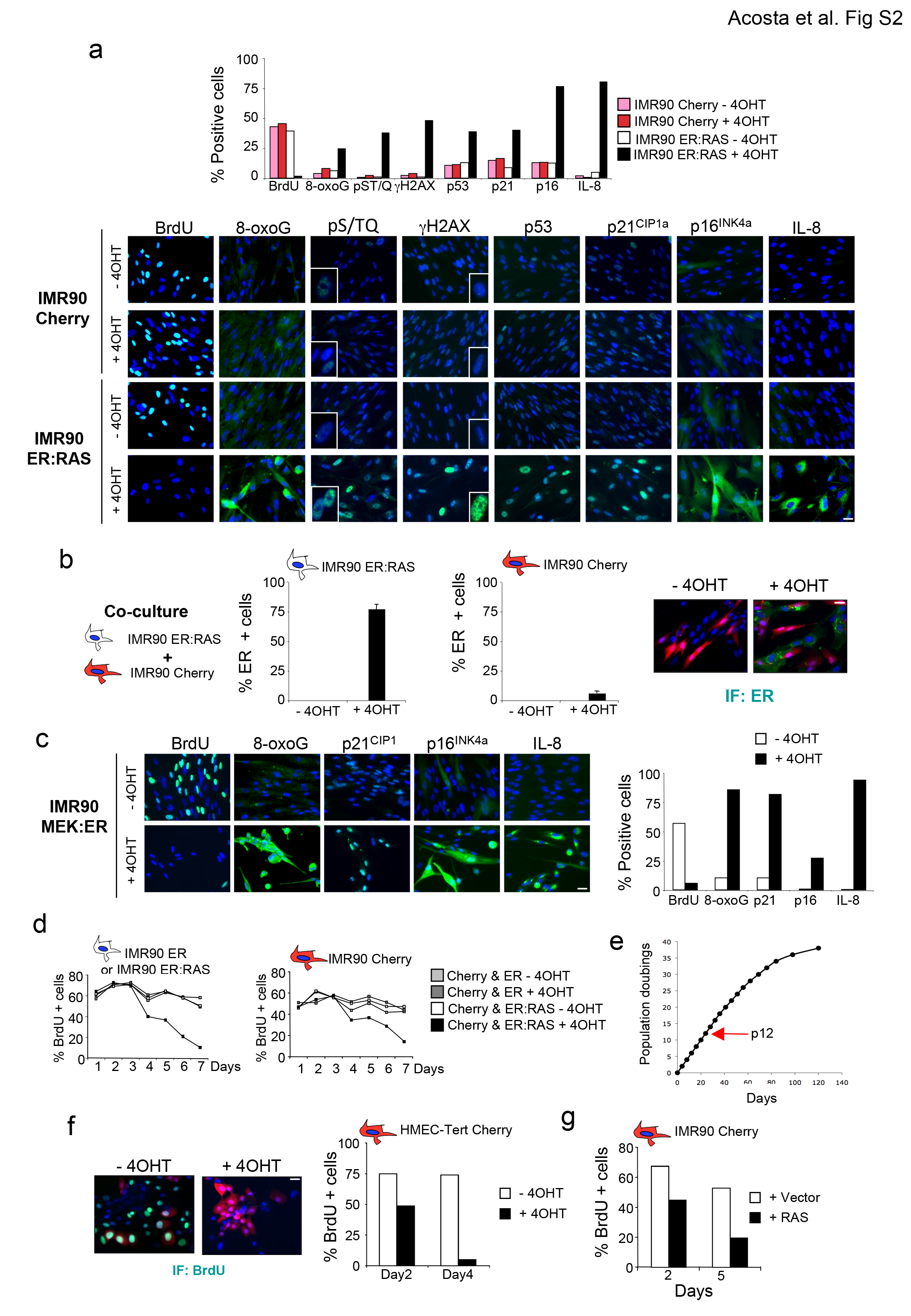
To understand whether cells undergoing OIS can transmit senescence in a non-cell-autonomous manner, we established co-cultures of senescent and normal human IMR90 fibroblasts. Cells were distinguished by expressing an mCherry fluorescent marker in the normal IMR90 cells. The mCherry positive and negative populations were monitored using high content analysis (HCA) microscopy (Fig 1a, Fig S1a, b). We used IMR90-ER:RAS cells expressing a chimeric fusion protein that activates upon treatment with 4-hydroxytamoxifen (4OHT). RAS activation triggers growth arrest, induces senescence effectors and the SASP (Fig S2a). IMR90-ER:RAS cells co-cultured with normal IMR90-mCherry cells also undergo arrest upon 4OHT treatment (Fig 1a, middle). Importantly, normal IMR90-mCherry cells also stopped proliferating when co-cultured with cells undergoing OIS, which suggested a non-cell-autonomous (paracrine) transmission of senescence (Fig 1a, right panel). Controls confirmed that mCherry positive cells did not express ER:RAS (Fig S2b). Normal IMR90-Cherry cells also underwent arrest when co-cultured with IMR90 MEK:ER cells (Fig 1b), an alternative model of OIS (Fig S2c). Expression of the ER binding domain alone, or treatment with 4OHT did not affect the growth of IMR90 ER cells or IMR90 mCherry cells co-cultured with them (Fig 1c, S2d). These experiments were performed using early passage IMR90 cells (Fig S2e) to exclude confounding effects of replicative senescence. Normal human mammary epithelial cells (HMECs) also underwent arrest upon co-culture with HMECs undergoing OIS (HMEC-ER:RAS, Fig 1d centre and S2f). Moreover, HMEC-ER:RAS cells induced the arrest of normal IMR90 fibroblasts (Fig 1d right, S2g), suggesting that paracrine senescence can be transmitted between different cell types. These results clearly show that senescence can be transmitted.
Figure 1. Cells undergoing oncogene-induced senescence (OIS) can induce paracrine arrest of normal cells.

(a) Co-culture with cells undergoing OIS induces the arrest of normal IMR90 cells. Normal IMR90 human fibroblasts expressing the fluorescent marker mCherry (IMR90 Cherry), were mixed with IMR90 ER:RAS cells. When indicated 200 nM 4OHT was added to activate ER:RAS. Growth curves represent the number of IMR90 ER:RAS (left) or IMR90 Cherry cells (right) present in the co-cultures. Data is a representative experiment of n=2 independent experiments for days 0-7 and mean +/− s.d. of n= 3 independent experiments for day 8. The source data for 2 independent experiments and statistics for day 8 (Student’s t-test) are provided in Supplementary Table S8. (b) Co-culture with IMR90 MEK:ER cells induces the arrest of normal IMR90 cells. The percentage of positive BrdU cells for each population in the cocultures 4 days after 4OHT induction is shown. Data is a representative experiment. The source data for 2 independent experiments are provided in Supplementary Table S8. (c) IMR90 ER:RAS or IMR90 ER cells were co-cultured with normal IMR90 Cherry fibroblasts. BrdU incorporation at day 7 shows that co-culture with IMR90 ER:RAS but not with IMR90 ER induces the arrest of normal IMR90 Cherry cells (centre). Data is a representative experiment. The source data for 2 independent experiments are provided in Supplementary Table S8. Representative images are shown (right). Scale bar, 30 μm. (d) Normal HMEC or IMR90 cells suffer growth arrest when co-cultured with HMEC cells undergoing OIS. HMEC-hTERT (centre) or IMR90 (right) cells expressing Cherry as a fluorescent marker were co-cultured with HMEC-TERT ER:RAS or HMEC-TERT vector cells in the presence of 100 nM 4OHT. Growth curves showing growth arrest of HMEC-TERT Cherry (centre) or IMR90-Cherry cells (right) are shown. Data is a representative experiment.
Paracrine senescence is a stable arrest mediated by secreted factors
We noted that normal cells arrested with a slight delay compared to cells undergoing OIS in co-cultures (Fig 2a). We hypothesized that this delay could be attributed to a paracrine response, as the induction of SASP components (CXCL1, IL-8, CCL-20, ActivinA or VEGFc) occurred as early as 2-3 days after RAS activation (Fig 2b, S3a).
Figure 2. Paracrine senescence is a stable arrest mediated by soluble factors.

(a) Quantification of the percentage of IMR90 ER:RAS (centre) and IMR90 Cherry cells (right) incorporating BrdU during the course of 7 days after activation with 4OHT in co-cultures. Data is a representative experiment. The source data for 2 independent experiments are provided in Supplementary Table S8. (b) Kinetics of the production of secreted factors during OIS. Conditioned media from IMR90 ER:RAS cells was collected and used to detect CXCL1 (left) or IL-8 (right) levels by ELISA. Data is a representative experiment. The source data for 2 independent experiments are provided in Supplementary Table S8. (c) Paracrine senescence is a stable arrest. Cells were co-cultured separated by transwells. After a week IMR90 cells in the bottom were split, seeded and cultured alone for 14 additional days. CV (centre) and SA-β-Gal staining (right) is shown. Scale bar, 50 μm. (d) Conditioned media from senescent cells induces paracrine senescence. Diagram explaining the CM experiments (left). CM was collected from IMR90 vector or IMR90 ER:RAS cells grown 7 days in the presence 200 nM 4OHT. The effect of CM on IMR90 cells was evaluated by BrdU staining (right). Data is a representative experiment of n=2 independent experiments for days 1-3 and mean +/− s.d. of n= 3 independent experiments for day 4. The source data for 2 independent experiments and statistics for day 4 (Student’s t-test) are provided in Supplementary Table S8. (e) Fibroblast undergoing OIS induce growth arrest on normal neighboring fibroblasts. IMR90 ER:RAS cells were seeded in ‘clusters’ surrounded of normal IMR90-Cherry cells with the help of cloning cylinders (left). Cells were grown in the presence or absence of 200 nM 4OHT for 7 days. Percentage of BrdU positive cells was calculated in consecutive fields in the external (IMR90-Cherry) and internal region (IMR90-ER:RAS). Data is mean +/− s.d. of n= 3 optical fields. (f) An experiment testing serial transfer of senescence by conditioned media showed a limited arrest with absence of SA-β-Gal positive cells in ‘tertiary’ cells. Data is representative of n=2 independent experiments. Scale bar, 50 μm.
To test whether soluble factors mediate paracrine senescence, we used transwell inserts that ensure physical separation of the cells (Fig 2c, S3b). IMR90 cells at the bottom displayed a senescent morphology and became arrested when co-cultured in the presence of senescent cells in the top chamber (Fig S3b). Next, we co-cultured normal cells and IMR90-ER:RAS for 7 days using transwells. At that point we split the IMR90 cells, and cultured them alone for 14 additional days (Fig 2c). Cells that have undergone paracrine senescence continued displaying features of senescence, suggesting that the transmitted phenotype is stable (Fig 2c). RT-PCR analysis discarded cross-contamination between cells or transmission of the RAS oncogene in the transwell experiments (Fig S3c).
To confirm that factors secreted by senescent cells were sufficient to induce paracrine senescence, we exposed normal IMR90 cells to conditioned media (CM) from IMR90 cells expressing active forms of RAS, RAF or MEK. Whereas cells exposed to CM from control cells grew normally, those treated with CM from senescent cultures showed reduced BrdU incorporation with a higher percentage staining positive for SA-β-Gal (Fig 2d, S3d). The cell arrest persisted after withdrawal of the CM (Fig S3e). Similar results were observed on mouse embryo fibroblasts (MEFs, Fig S3f).
As paracrine senescence seemed dependent on soluble factors, we reasoned that its effects should be spatially restricted. To test this, we seeded a ‘cluster’ of IMR90-ER:RAS cells surrounded by normal IMR90-mCherry cells (Fig 2e). Normal IMR90-mCherry cells in close proximity to the IMR90-ER:RAS cluster (in 3 optical fields, equivalent to up to 1 mm) showed reduced incorporation of BrdU following induction of OIS (Fig 2e). In contrast, normal IMR90-mCherry cells located further (>1mm) from the cluster were unaffected by RAS activation. Similarly, while CM from cells undergoing OIS (‘primary’ senescence) triggered paracrine senescence (‘secondary’), CM derived from those cells just slowed down the proliferation of normal cells (‘tertiary’) without inducing SA-β-galactosidase positive cells (Fig 2f and S3d). These data do not exclude that either cell-to-cell contact or the extracellular matrix contribute to paracrine senescence but establish a restricted transmissibility of senescence by soluble factors.
Paracrine senescence resembles a full senescence response
Next we co-cultured IMR90-mCherry and IMR90-ER:RAS cells and monitored the expression of senescence markers and effectors by HCA using thoroughly validated antibodies (Fig S1c-e). Upon activation of RAS, IMR90-ER:RAS cells in the co-cultures displayed high DNA and oxidative damage and upregulated expression of the CDKIs, p16INK4a and p21CIP1, and of IL-8, a component of the SASP (Fig 3a, top centre). Normal cells (IMR90-mCherry) in the co-cultures also showed increased levels of oxidative and DNA damage and activation of p16INK4a, p21CIP1 and IL-8 expression, suggesting a full transmission of senescence (Fig 3a, top right). A similar induction of senescent features was observed in normal cells co-cultured with IMR90 MEK:ER cells undergoing OIS (Fig S4a).
Figure 3. Paracrine senescence depends on the p16INK4a/Rb and p53/p21CIP1 tumour suppressor networks.

(a) IMR90-Cherry cells were co-cultured with IMR90 ER:RAS cells in the presence or absence of 200 nM 4OHT. 7 days after 4OHT treatment, cells were subjected to IF. The percentage of IMR90 ER:RAS (top, centre) or IMR90 Cherry cells (top, right) positive for each of the markers is shown. Data is a representative experiment. The source data for 2 independent experiments are provided in Supplementary Table S8. Representative pictures are shown in the bottom. Scale bar, 30 μm. (b) mRNA expression profiling of IMR90, IMR90 ER:RAS or IMR90 cells cultured in Transwells together with IMR90 vector or IMR90 ER:RAS cells during 7 days in the presence of 200 nM 4OHT and 0.5 % FBS. The plot shows the correlation between cells undergoing OIS and paracrine senescence. (c) Hierarchical clustering of genes changing more than 2-fold, centred in a cluster that defines the equivalence between OIS and paracrine senescence. (d) GSEA of a signature associated with senescence 18 in IMR90 cells undergoing paracrine senescence. (e) A signature derived from IMR90 cells undergoing paracrine senescence is found enriched in mouse PANIN 44 and human serrated sessile adenomas (SSA) 36. NES, normalized enrichment score; FDR, false discovery rate. (f) Paracrine senescence is dependent on the p53/p21CIP1a and p16INK4a pathways. IMR90 cells were transfected with the indicated siRNAs. The next day, CM from IMR90 ER:RAS or IMR90 vector cells was added. The proliferation of IMR90 ER:RAS cells was assessed by BrdU incorporation 2 days after CM addition (right).). Data are mean ± s.d., n = 3 independent experiments. The source data and statistics (Student’s t-test) are provided in Supplementary Table S8. IMR90 cells transfected with the different siRNAs were subjected to IF as a control for knockdown efficiency (centre).
Global gene expression exposed a high correlation between IMR90 cells undergoing OIS and paracrine senescence (Fig 3b and S4b). Unsupervised hierarchical clustering grouped OIS and paracrine senescence (Fig 3c) and a transcriptional signature associated with senescence 18 was significantly upregulated during paracrine senescence (Fig 3d). In addition, qRT-PCR confirmed that CDK inhibitors and the SASP were induced during paracrine senescence (Fig S4c and Table S1).
To understand whether paracrine senescence is commonly associated with senescence, we compared paracrine senescence and OIS induced by MEK activation 19 observing a significant overlap of upregulated genes (Fig S4d). Moreover, we derived a ‘paracrine senescence’ signature and used gene set enrichment analysis (GSEA) to interrogate its association with senescence transcriptomes. Different human and mouse cell types undergoing replicative, oncogene or stress-induced senescence displayed an enrichment of the ‘paracrine senescence’ signature (Fig 3e and S4e, f). Amongst them HMEC cells undergoing OIS expressed key SASP components suggesting a similar implementation of paracrine senescence (Fig S4g). The ‘paracrine senescence’ signature was also associated with murine pancreatic intraepithelial neoplasias (PanIN) and human serrated sessile adenomas (SSAs, Fig 3e, S4f), lesions that are both enriched in senescent cells. To examine whether paracrine senescence depends upon the same genetic networks as OIS, we knocked down key effectors of senescence in IMR90 cells and either exposed them to conditioned media of senescent cells or co-cultured them with cells undergoing OIS. These experiments revealed that the paracrine senescence arrest depends on the activation of p16INK4a, p21CIP1 and p53 (Fig 3f, S4h).
Multiple components of the SASP mediate paracrine senescence
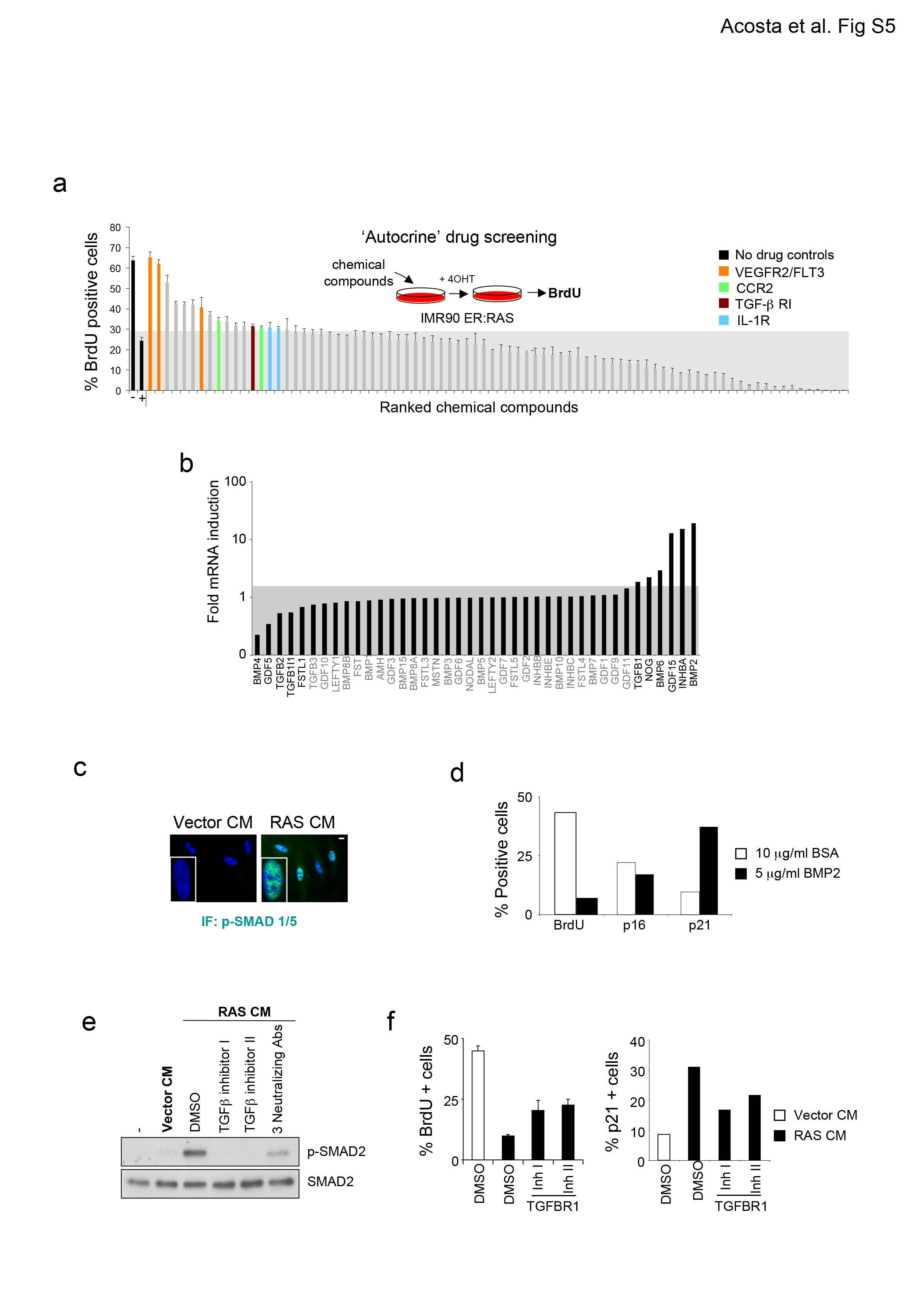
We next catalogued the secretome of cells undergoing OIS using stable isotope labelling of amino acids in culture (SILAC, Fig 4a). Unbiased quantitative proteomics offered several advantages for breadth of coverage and its ability to detect changes on protein expression not apparent from gene expression profiling (Fig 4b). Amongst the top hits identified, were chemokines, TGFβ family ligands or VEGF (Fig 4c and Table S2.). To identify which factors mediate paracrine senescence, we compiled a collection of 78 chemical compounds targeting their receptors or key downstream pathways activated by the SASP (Table S3). Normal IMR90 cells treated with the drug library were exposed to CM from cells undergoing OIS and BrdU incorporation was assessed 48 h later. Out of the compounds partially impairing the arrest, several targeted the VEGFR2/FLT3, TGFBR1 and CCR2 receptors (Fig 4d and Table S3). These compounds were confirmed to inhibit paracrine senescence in a dose-dependent manner, except the CCR2 inhibitor that exhibited a biphasic effect (Fig 4e). By using RNAi to knock down the expression of CCR2 or the TGFβ receptors ALK4, ALK5 (also known as TGFBR1) and ALK7, we confirmed their role (Fig 4f, g). These results suggest that multiple factors secreted by senescent cells mediate paracrine senescence.
Figure 4. Multiple components of the SASP are involved on paracrine senescence.

(a) Diagram summarizing the proteomics approach. (b) Comparison between mRNA and protein expression for the secretome of cells undergoing OIS. Overall Pearson correlation is 0.64. A lower correlation was observed for proteins induced more than 4-fold (red line, Pearson correlation=0.15). This suggests post-trancriptional upregulation of SASP components (e.g. MMP7, IGFBP5, IGFBP6, THBS1, THBS2 and IL6ST). (c) Plot of 2 forward and reverse SILAC experiments. Significant changes in at least 2/3 experiments are coloured. (d) Screening for compounds inhibiting paracrine senescence. IMR90 fibroblasts were grown in the indicated CM in the presence of a collection of 78 drugs. 2 days later, BrdU incorporation was measured. −, IMR90 treated with DMSO and grown in CM of IMR90 ER:RAS − 4OHT; +, IMR90 treated with DMSO and grown in CM of IMR90 ER:RAS +4OHT. A gray area represents an arbitrary cut-off of 120 % the value of BrdU in the no drug control (+). Inhibitors over the cut-off targeting VEGFR2 and/or FLT3 (orange), CCR2 (green) or TGFBR1 (brown) are marked. Data are mean ± s.d., n = 3 independent screen plates. (e) IMR90 cells cultured 2 days with the indicated CM and drugs (concentrations 10 μM to 10 nM). Proliferation was evaluated by BrdU incorporation. Graph shows one representative experiment out of two independent experiments. (f) Infected IMR90 cells were treated with CM and growth evaluated by CV (top). Data are one representative experiment out of two independent experiments. IF against CCR2 is shown as a control for the efficiency of the shRNAs used (bottom). Scale bar, 10 μm. (g) Knockdown of receptors of the TGFβ family partially rescue paracrine senescence. IMR90 cells infected with the indicated vectors were treated with CM of senescent or control cells and senescence evaluated 10-14 days after by CV (top left) and SA-β-Gal staining (bottom left). Knock down efficiency was measured by qRT-PCR (right). Data are one representative experiment out of 2 independent experiments. The source data for 2 independent experiments are provided in Supplementary Table S8. Scale bar, 50 μm.
The TGFβ pathway mediates paracrine senescence
We also interrogated the chemical compounds library for their ability to influence RAS-induced senescence (Fig S5a, Table S3). In addition to the compounds identified as affecting paracrine senescence the ‘autocrine senescence screen’ showed that inhibition of IL-1R signalling also prevented OIS (Fig 5a and S5a). The comparison between both screens suggested that TGFBR1 inhibitors had a more pronounced effect on ‘paracrine’ rather than on ‘autocrine’ senescence (Fig 5a). In fact, although GSEA unveiled an association of TGFβ1 signalling with both OIS and paracrine senescence (Fig 5b), most TGFβ-dependent genes were more prominently upregulated during paracrine senescence than OIS (Fig 5c).
Figure 5. A role for TGFβ signalling on mediating paracrine senescence.

(a) Comparing the paracrine and autocrine screens. Autocrine screen is in Sup Fig S5A. (b) GSEA of TGFβ target genes during OIS and paracrine senescence.. (c) qRT-PCR for a set of TGFβ target genes in OIS and paracrine senescence. Data are mean ± s.d., n = 3 independent experiments (d) Induction of TGFβ ligands during OIS by qRT-PCR (bars). Induction in the SILAC experiments is in blue. Data are mean ± s.d., n = 3 independent experiments (e) IMR90 cells were treated with CM and 10 μg/ml of antibodies for 2 days. Growth was evaluated by BrdU incorporation (right). Percentage of cells positive for p-SMAD2/3 is presented (left). Data are mean ± s.d., n = 4 independent experiments. p was calculated using Student’s t-Test. pSMAD 2/3 statistics: ActivinA p=0.0037; TGFβ p =0.91; BMP2 p =0.52; ActivinA/TGFβ p =0.0029; ActivinA/TGFβ/BMP2 p =0.040; TGFβ inhibitor II p value 0.0007. For BrdU, statistics: ActivinA p=0.009905573; TGFβ p =0.282677043; BMP2 p =0.233112557; ActivinA/TGFβ p =0.000723521; ActivinA/TGFβ/BMP2 p =0.002428509; TGFβ inhibitor II p =4.44×10−5. Source data are in Supplementary Table S8. (f) TGFBR1 inhibitors partially prevent paracrine senescence. IF against phosphorylated SMAD2/3. Scale bar, 10 μm. Representative pictures (left). IMR90 cells were treated as indicated. INK4b and CDKN1a expression measured by qRT-PCR (right). Data are mean ± s.d., (n = 3 independent experiments, p, Student’s t-Test). pSMAS2/3 statistics: Inhibitor I p= 4.49×10−5; Inhibitor II 4.53×10−5. INK4B statistics: Inhibitor I p =0.00083; Inhibitor II p= 0.010. CDKN1A statistics: Inhibitor I p = 0.00054; Inhibitor II p = 0.00014. Source data are provided in Supplementary Table S8. (g) Alk5 deletion suppresses KrasG12D-driven OIS in PANIN. Diagram showing mice strains used (top left). GSEA shows TGFβ activation in PANIN (bottom left). Ki67 and SA-β-Gal staining (centre) and quantification (right) showing decreased senescence in pancreatic lesions of Pdx1-cre KrasG12D/+ TGFβR1fl/fl mice. Scale bar, 100 μm. Boxplot represent first and third quartiles (n=5 mice per condition). Inside lines shows median. Whiskers extend to highest or lowest observation. p= 0.0184 for both experiments calculated using Mann-Whitney.
TGFBR1-type receptors bind multiple TGFβ family ligands 20. Although TGFβ1 was also induced, other ligands of the TGFβ and BMP branches, including BMP6, BMP2, InhibinA and GDF15, were more acutely upregulated during senescence (Fig 5d, S5b). BMP-like ligands and TGFβ-like ligands signal through activation of different SMAD family members. The phosphorylation of both SMAD2/3 and SMAD1/5 was upregulated in cells undergoing paracrine senescence (Fig 5e, S5c), corroborating the involvement of both branches of TGFβ signalling on senescence. The effect of BMP2 on senescence has been reported 21 and further confirmed by us (Fig S5d). Moreover, combination of blocking antibodies targeting either TGFβ1, Activin A (a homodimer of Inhibin A) and BMP2, partially rescue the arrest observed during paracrine senescence (Fig 5e). TGFBR1 inhibitors prevented the phosphorylation of SMAD2/3 (Fig 5f and S5e) and blunted the paracrine senescence arrest (Fig 5f). These effects correlated with impaired p15INK4b and p21CIP1 induction (Fig 5f, S5g) consistent with previous observations 22.
We next investigated whether TGFβ signalling influence senescence in vivo. We used mouse bearing a conditional Pdx1-driven activated Kras allele (KRasG12D) 23. KRasG12D is a potent oncogene in pancreas, but its tumourigenic properties are restrained by its ability to cause OIS, observed in premalignant PanIN lesions 24. GSEA showed that TGFβ signalling was associated with these PanIN lesions (Fig 5g, bottom left). Pdx1-cre KrasG12D mice were crossed with a conditional allele lacking TGFβR1 (TGFβR1fl/fl) 25(Fig 5g, top left). Lesions observed in Pdx1-cre KrasG12D/+ mice had characteristics of OIS, with low proliferation and stained positive for SA-β-Gal (Fig 5g). The OIS was attenuated in Pdx1-cre KrasG12D/+ TGFβR1fl/fl lesions (Fig 5g). Importantly Pdx1-cre KrasG12D/+ TGFβR1fl/fl mice succumbed to a mixture of pancreatic and skin cancer in less than 3 months, while only a subset of Pdx1-cre KrasG12D/+ animals progress to pancreatic cancer, and with latency of over a year 26,27.
Activation of the inflammasome controls SASP production
As multiple components of the SASP execute paracrine senescence, we searched for factors co-ordinating their expression. We screened factors for their ability to induce IL-6 and IL-8, identifying IL-1α as one of the most robust inducers (Fig S6a). IL-1α signalling has been implicated in regulating IL-6 and IL-8 on senescence 28. A more thorough analysis identified IL-1α as a potent inducer of multiple SASP components (Fig 6a, b). Moreover expression of IL-1α caused a SASP-like response phenocopying cells undergoing OIS (Fig 6c, left). Although cells expressing Inhibin A or TGFβ induced some SASP components such as IL-8 or CCL2 (Fig S6b), they did not mimick the SASP (Fig 6c, centre). Inhibiting TGFBR1 did not affect the secretome induced by IL-1α (Fig 6c, right). In addition, while IL-1α inhibition partially prevented induction of IL-8 or CCL2 by TGFβ, the converse was not true (Fig S6b), suggesting that IL-1 has a more prominent role than TGFβ signalling in controlling the SASP.
Figure 6. The inflammasome regulates the senescence secretome.

(a-b) IMR90 cells were infected with a vector that expresses IL-1α or a control and IF of the indicated SASP components performed. Scale bar, 30 μm. (b) Quantification of (a). (c) IL-1α activates a SASP-like response. IMR90 cells were infected with retroviruses expressing RASG12V, IL-1α or Inhibin A. When indicated 4 μM TGFBR1 inhibitor II was used. CM was used to probe chemokine and cytokine antibody arrays. (d) Gene set enrichment analysis (GSEA) of IL1R pathway in the gene expression profile of IMR90 cells undergoing OIS (left) and mouse PANIN (right). FDR, false discovery rate; NES, normalized enrichment score. (e) qRT-PCR analysis showing the expression of a set of genes involved on IL1R signalling. Data are one representative experiment out of 2 independent experiments. (f) IB with antibodies against IL-1α and IL-1β in CM collected from IMR90 ER:RAS (RAS) or IMR90 vector cells (Vector) incubated during 7 days with 200 nM 4OHT and 0.5 % FBS. Pro, precursor form; mat, mature form. (g) Activation of the inflammasome during OIS. IMR90 ER:RAS cells (left) and murine models of SSA (centre) and PANIN (right) display increased Caspase 1 activity. Data are mean ± s.d., n = 4, 3, 6 and 8 different samples for control (GI tract), SSA, control (pancreas), and PanIN respectively. (h) IMR90 ER:RAS cells cultured in the presence or absence of 200 nM 4OHT were subjected to IF with antibodies recognizing inflammasome components. A control of IMR90 ER:RAS cells + 4OHT without primary ab (No ab) is shown in the lower row. Scale bar, 10 μm. (i) IMR90 ER:RAS or IMR90 vector cells were cultured in the presence of 200 nM 4OHT and 0.5 % FBS during 7 days with 10 μM Caspase-1 inhibitor or 20 μM IL1R antagonist. After that time, cells were processed and qRT-PCR against different SASP components performed. Data are mean ± s.d., n = 3 independent experiments. Data from this experiment is also presented in Fig S6j.
GSEA showed that IL-1 signalling was associated with paracrine senescence and OIS both in culture and in vivo (Fig 6d and S6c-d). Molecules involved in IL-1 signalling (such as IRAK family kinases) were also induced during OIS (Fig 6e, S6e). IL-1α and IL-1β are synthesized as precursors. In particular, pro-IL-1β is inactive until processed by the inflammasome, a multiprotein complex comprising of Caspase1 and several adapter molecules 29,30. Cells undergoing OIS secreted the processed, mature forms of both IL-1α and IL-1β, suggesting inflammasome activation (Fig 6f). Indeed, IMR90 cells undergoing OIS displayed Caspase1 activity (Fig 6g, left). The inflammasome was also activated during OIS in vivo: in BrafV600E-driven murine SSAs 31 and in KrasG12D-driven PanIN lesions (Fig 6g, centre and right). Protein levels of inflammasome components such as Caspase 1, ASC (also known as PYCARD), and NLPR3 (but not NLPR1) were up-regulated during OIS (Fig 6h, S6f) without noticeable changes to their mRNA levels (Fig S6g), suggesting protein stabilization. Cells undergoing OIS secreted NLPR3 and Caspase1, a characteristic of Caspase1-dependent unconventional secretion (Fig S6h) 32. Finally, treatment with Caspase 1 or IL-1R inhibitors (Fig 6i, S6i) but not TGFBRI inhibitors (Fig S6j), blunted the expression of SASP components during OIS, suggesting that activation of the inflammasome was a cause rather than an effect of the SASP.
The Inflammasome and IL-1 signalling reinforce senescence
Ectopic IL-1α expression arrested IMR90 cells (Fig 7a) causing senescence (Fig 7b) accompanied by increased oxidative and DNA damage, and induction of p53 and p21CIP1 (Fig 7c, S7a). Conversely, knock down of the IL-1 receptor or inflammasome components partially prevented OIS (Fig 7d). These results were extended using siRNAs targeting downstream adapter molecules of the IRAK family (Fig S7b-d). Treatment with Caspase 1 inhibitors partially prevented the induction of p21CIP1 and the cell cycle arrest observed during OIS (Fig 7e), while inhibition of IL-1R signalling either using shRNAs (Fig 7f, S7e) or blocking antibodies targeting IL-1α and IL-1β (Fig S7f) also affected paracrine senescence.
Figure 7. IL-1 signalling regulates senescence.

(a) IMR90 were infected as indicated and proliferation measured by CV. (b) SA-β-Gal staining. Scale bar, 50 μm. (c) IF using the indicated antibodies. Data is a representative experiment. Source data for 2 independent experiments are provided in Supplementary Table S8. (d) IMR90 ER:RAS were infected as indicated and cell growth analysed by CV (right). qRT-PCR showing knockdown efficiency (left). Data are one representative experiment out of 2 independent experiments. (e) IMR90 ER:RAS cells were tretaed as indicated. BrdU incorporation and p21CIP1 expression were measured. Data are mean ± s.d., n = 3 independent experiments. p was calculated using Student’s t-Test. For BrdU, IL1R inh p = 5.30×10−5; Casp inh I p = 0.013; Casp inh II p = 0.013. For p21, IL1R inh p = 6.73×10−6; Casp inh I p = 1.42×10−5; Casp inh II p = 0.00047. Source data are provided in Supplementary Table S8. (f) Knockdown of IL1R prevents paracrine senescence. IMR90 cells infected and treated as indicated. After 10-14 days, cells were stained for SA-β-Gal. Data is representative experiment. Source data for 2 independent experiments are provided in Supplementary Table S8. (g-i) Inhibition of IL-1R reduces senescence and immune clearance in vivo. (g) NrasG12V or NrasG12V/D38A transposons and a transposase were co-delivered into mouse livers via hydrodynamic injection. Mice were treated with carrier or drugs for 12 days. (h) Quantification of Nras- (top), p21Cip1- (centre) and p16Ink4a- (bottom) positive cells on liver sections. Student’s t-test (n=5 mice per condition): carrier/ NrasG12V vs. IL1R/ NrasG12V, *P< 0.0150, Carrier/ NrasG12V vs. Inhibitor combination/ NrasG12V, **P< 0.002, p21: Carrier/ NrasG12V vs. IL1R/ NrasG12V, *P< 0.0341, Carrier/ NrasG12V vs. Inhibitor combination/ NrasG12V, *P< 0.0116, p16: Carrier/ NrasG12V vs. Inhibitor combination/ NrasG12V, *P< 0.01. 1, delivery of NrasG12V/D38A and treatment with vehicle. 2-7 delivery of NrasG12V. Treatments: 2, vehicle; 3, IL1R inhibitor; 4, CCR2 inhibitor; 5, VEGFR2 inhibitor; 6, TGFBR1 inhibitor; 7, combination of the 4 inhibitors. Data are mean ± s.e.m. (i) Representative pictures of H&E and IHC for Nras, p2Cip1 and p16Ink4a. Scale bar, 100 μm.
We next took advantage of a model where OIS is induced in mouse livers through stable, transposon-mediated transfer of oncogenic Nras (NrasG12V). This model was used to show that senescent hepatocytes undergo immune-mediated clearance (designated “senescence surveillance”), important for tumour suppression 12. Using this model, we tested whether blockade of IL-1 and other SASP components affected hepatocyte senescence. After injection with the NrasG12V transposons, mice were treated daily with the indicated compounds for 12 days (Fig 7g). While the percentage of cells positive for Nras expression was similar 6 days after transduction (Fig S7g), 12 days after injection the percentage was higher in mice treated with either IL-1R inhibitor or a combination of drugs (targeting IL-1R, VEGFR2, CCR2 and TGFBR1), reflecting reduced clearance of senescent hepatocytes by the immune system and/or senescence inhibition. To analyze the effect on senescence we measured p16Ink4a and p21Cip1 levels, observing that treatment with IL-1R inhibitor or the drug combination reduced the percentage of senescent hepatocytes (Fig 7h, i and S7h). Treatment with IL-1R inhibitor also resulted in a significant percentage of NRas-positive cells proliferating (Fig S7i). The effect of inhibiting IL-1 was further confirmed using IL-1α neutralizing antibodies (Fig S7j). Overall, the above results highlight the relevance of IL-1 signalling and SASP regulation for senescence in vivo.
Paracrine senescence is observed in mouse and human models of OIS in vivo
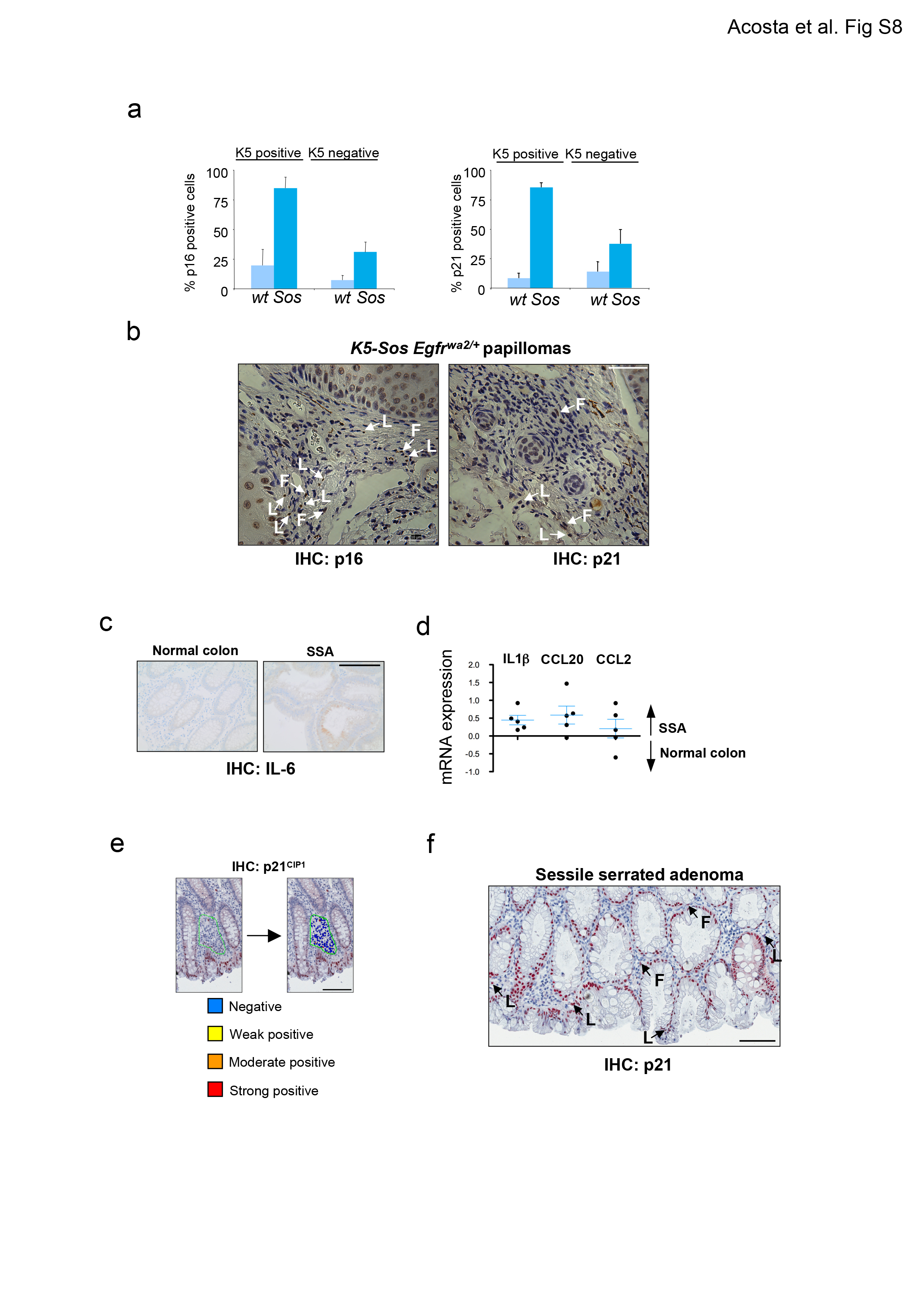
To investigate if paracrine senescence occurs in pathophysiologically relevant conditions in vivo, mouse and human models of OIS were analyzed. First we revisited the model where OIS is induced in mouse hepatocytes by NrasG12V 12. The senescent hepatocytes are found surrounded by clusters of immune cells 12 (Fig 8a). We observed that many cells in these clusters stained positive for senescence markers (Fig 8a). To confirm these findings, we used Keratin5-Sos Egfrwa2/+ transgenic mice 33. These mice develop papillomas with characteristics of OIS as confirmed by staining for p16Ink4a and p21Cip1 within the basal and suprabasal layers of the papilloma (Fig 8b). Whilst there were no senescent cells in the tissue close to normal skin, we observed senescent cells present in the Keratin 5-negative tissue adjacent to the senescent papillomas (Fig 8b, S8a). Examination of their morphological features identified fibroblast, lymphocytes and plasma cells, but not cells with epithelial characteristics amongst the senescent cells in the vicinity of papillomas (Fig S8b).
Figure 8. Paracrine senescence is observed in mouse and human models of OIS in vivo.

(a) Paracrine senescence in the liver. Immunohistochemistry images showing senescent hepatocytes surrounded by clusters of immune cells. Immune cells positive for p21Cip (left) and p16Ink4a (right) are marked by asterisks. Scale bar, 50 μm. (b) Stroma cells in the vicinity to papillomas in the skin of K5-Sos Egfrwa2/+ mice show the presence of cells positive for p21Cip1 and p16Ink4a. Arrows show senescent cells in the Keratin 5 negative population. Staining for p16Ink4a (pictures, top) and p21Cip1 (pictures, bottom) is shown in red. Keratin 5 is shown in green. Nuclei are shown in blue. Merged pictures are presented. A close up is shown to the right, and a wider view in the left. Bars are 10 μm. Graph showing the quantification of p16Ink4a (top, centre) and p21Cip1 (top right) nuclear levels in cells in the epidermis (Keratin 5 positive) or dermis (Keratin 5 negative) are shown (left).
For p21: wt K5+ n=102; wt K5− n=108; Sos K5+ n=460; Sos K5− n=536. For p16: wt K5+ n=100; wt K5− n=105; Sos K5+ n=597; Sos K5− n=939. n= number of cells. Scale bar, 10 μm. (c-e) Stromal cells in the vicinity to human SSA present high levels of p21CIP1 expression. (c) Epithelial cells in the crypts from serrated sessile adenomas (SSA) are positive for activated BRAF and p21CIP1a, but negative for KI-67. Close up shown here pictures correspond to images shown at the bottom of the panel. Scale bar, 25 μm in top panel and 100 μm in bottom panel. (d) IHC showing representative staining of CCL2 in normal colon and SSA. 6/11 samples showed higher CCL2 expression in SSA than normal colon, while 2/11 have a higher pattern of expression in normal colon than SSA. Scale bar, 100 μm. (e) The percentage of stromal p21CIP1 positive cells close to the normal colon or SSA is significantly different Box represents the first and third quartiles and the line inside shows the median. Whiskers extend upward and downward to the highest or lowest observation. Unpaired t-test (p=0.03, n=10 samples/condition).
Finally, we looked for evidence of paracrine senescence during human tumourigenesis studying SSA. Colon SSAs are mainly driven by activating BRAF mutations that trigger OIS31,34,35. Epithelial tissue from human SSA but not normal colonic crypts, was positive for senescence markers like p21CIP1a and negative for proliferation markers such as KI-67 (Fig 8c). SASP components such as CCL2 and IL-6 were induced in SSAs (Fig 8d and S8c). Analysis of expression data 36 also showed the upregulation of IL-1β and other SASP components in SSA (Fig S8d). Using automated imaging analysis (Fig S8e) we measured a significant increase in p21CIP1a positive cells (Fig 8c, e, p=0.03) or p21CIP1 positive/Ki67 negative stromal cells (p=4.6 10−5) close to SSA compared to tissue close to normal colonic crypts. These cells had immune or fibroblast morphology (Fig S8f). These data suggests that senescence can be transmitted in a non-cell-autonomous fashion in both mouse and human models of OIS in vivo.
DISCUSSION
The SASP can potentiate the tumourigenic properties of cancer cells, recruit the immune system to eliminate premalignant cells 5,12,13 or reinforce senescence 3. Here we add paracrine senescence to the repertoire of functions regulated by the SASP. Paracrine senescence could be relevant in different scenarios in vivo. As well as reinforcing the arrest of a cell undergoing OIS, its secretome could induce arrest on the surrounding epithelium, expanding the senescent “footprint” of the preneoplastic lesion and promoting their immune clearance. Alternatively, cells undergoing OIS could propagate senescence to the surrounding tissue, as suggested here (Fig 8). Other scenarios are possible; therapy-induced senescence (TIS) influences chemotherapy 37. Radiation can induce a ‘bystander response’ 38 which could be partially explained by paracrine senescence. Clearance of senescent cells is also beneficial for age-associated disorders 39. Therefore, it is worthy investigating whether paracrine senescence mediates the deleterious effects of senescent cells on tissue homeostasis.
Not all cells surrounding preneoplastic lesions undergo paracrine senescence. Levels of soluble factors, gradients of their concentration, the susceptibility of different cell types and even whether the cells are dividing or arrested are possible factors influencing which cells undergo paracrine senescence in vivo. The nature of these cells and the functional implications of paracrine senescence remain open questions for future studies. Intriguingly recent work suggest that tumours have the ability to induce p16INK4a expression in their surrounding stroma and infiltrated immune cells 40, similar as we observed here.
In this study, we identified multiple SASP factors with previous unrecognized roles in controlling senescence e.g. TGFβ family members (Activin A and GDF15), VEGF, and the chemokines CCL2 and CCL20. Although multiple components of the SASP amplify the response, at least in part by activating NF-κB 41,42, IL-1α is a key regulator not linked previously with senescence induction 28. There is a wealth of information on the activation of the inflammasome by infectious agents, but less is known about links with other cellular stresses. The ability of the inflammasome to regulate senescence adds to the pro- and anti-tumour effects of the inflammasome 43.
In conclusion cells undergoing OIS can transmit paracrine senescence to their neighbours. Here, we identified that paracrine senescence is a complex response regulated by the inflammasome and IL-1 signalling. Understanding the significance and regulation of paracrine senescence can be the first step towards manipulating it for therapeutic benefit.
METHODS
Chemical compounds and neutralizing antibodies
4-hydroxytamoxifen (4OHT) was from Sigma. Ac-YVAD-CMK and z-YVAD-FMK (Caspase-1 inhibitor I and VI) were from Calbiochem. Chemical compounds and concentrations used are summarized in Table S4. Neutralizing antibodies targeting BMP2, TGFβ1, ActivinA, IL-1α and IL-1β (R&D) were used at 10 μg/mL.
Cell culture
HEK293T and IMR90 cells were obtained from the ATCC. Cells were maintained as described 45. HMEC-hTert cells were cultured in mammary epithelial cell growth media (PromoCell). Co-culture of IMR90 and HMEC was carried out in mammary epithelial cell growth media (PromoCell) supplemented with 10% FBS. We used 0.02 μm Anopore cell culture inserts (Nunc-Thermo) for Trans-well co-culture experiments. Cell numbers and cell viability were determined using Guava Viacount reagent (Millipore) and a Guava Cytometer (Millipore).
Retroviral and lentiviral infection
Retroviral and lentiviral infection were performed as previously described 45,46.
Plasmids
pRetroSUPER (pRS) plasmids expressing shRNAs targeting p16INK4a, p53 or p21CIP1 and pBABE-RasV12 pLNC-ER:RAS and pLNC-MEK:ER have been described previously 8. MSCV puro based retroviral plasmid encoding IL-1α was generated by cloning its cDNA from pCMV6 IL-1α (Origene). pGIPZ-based shRNA vectors targeting ALK4, ALK5, ALK7, IL1R1, CASP1, ASC and TP53 were from SIGMA.
BrdU incorporation, growth curves, colony formation assays and Senescence-associated β-Galactosidase staining
Conditioned media
2×106 of the indicated cells were seeded in a 10 cm dish and incubated during 7 days with 200 nM 4OHT in DMEM 0.5 % FBS. After incubation, conditioned media (CM) was collected, centrifuged at 5000g and filtered through a 0.2 μm pore filter. CM was mixed with DMEM 40 % FBS in a proportion of 3 to 1 to generate CM containing 10% FBS.
Transfection of siRNAs
IMR90 fibroblasts were reverse-transfected with 30 nM siRNA using a 3.5% solution of HiPerFect transfection reagent (QIAGEN). AllStars scrambled siRNA served as negative controls. For a list of siRNAs, see Table S5.
Gene expression analysis
RT-qPCR was performed as described 47. Primer sets and TaqMan Gene Expression Assays (Applied Biosystems) used are listed in Table S6.
Microarray analysis
For global gene expression studies, cRNA was hybridized to Human Gene 1.0 ST arrays (Affymetrix). Microarray data processing and analysis was carried out at EMBL. Data were normalized using Robust Multichip Average (RMA) and differentially expressed genes were identified using Linear Models for MicroArrays (LIMMA). A cut-off of a Benjamini-Hochberg false discovery rate (BH) <0.05 was used to identify significant genes. All analyses were carried out in R (v2.13.0).
Gene set enrichment analysis (GSEA)
We used GSEA (v2.07) to examine the association between gene sets and gene expression. We ranked the genes by log2 fold-change obtained from LIMMA contrasts. Pre-ranked GSEA was performed using curated Molecular Signatures Database (v3.0) with a size of 5-1200 genes (http://www.broadinstitute.org/gsea/msigdb/index.jsp). To define the ‘paracrine senescence’ signature, genes upregulated >2 fold were chosen.
Immunofluorescence and high content analysis
IF was performed as previously described 47 using the antibodies listed in Table S7. Image acquisition was performed using an automated high throughput microscope (IN Cell Analyzer 1000 or 2000, GE Healthcare). High content analysis (HCA) was performed, as described elsewhere 45,47,48. Briefly, two or three fluorescence images corresponding to DAPI, primary antibody/Alexa Fluor 488-secondary antibody and Cherry were acquired for each field using the IN Cell Analyzer 1000 or IN Cell Analyzer 2000 automated epifluorescence (GE Healthcare). No less than 1000 cells were acquired. HCA was performed using the IN Cell Investigator software (v1.7; GE Healthcare). DAPI staining was used to identify cells. The nuclei were then segmented using top-hat segmentation. A collar segmentation routine was set to define cell segments. To determine the cellular expression of the analyzed protein or detect the Cherry labelled cells, the mean intensity of pixels in the reference channel (Alexa Fluor 488 or Cherry) within the specified nuclear region (Object Nuclear Intensity) was measured. Each cell was assigned a nuclear intensity value used to set up a threshold filter. The threshold filter used a histogram for data visualization. To set the filter cut-off, expression in control cells was measured to define the negative population, followed by the analysis of the positive control (Fig S1a). As a result, the software classified each cell as either positive or negative for the expression of the analyzed protein or signal (Fig S1b). The mean of the nuclear intensity was routinely analyzed with equivalent results. Antibodies used for the analysis were tested (shRNAs or siRNAs) to assess their specificity (Fig S1c-e).
Mass spectrometry
This was carried out as described in 49
Identification of secreted proteins
To identify secreted proteins, a filtering step was applied using the Secreted Protein Database’s Ranks 0 to 3 and Gonzalez and co-workers “Secreted” or “SPTM” classifications 50,51. This criteria required proteins to be annotated as either ranks 0 to 2, “Secreted”, or as both “SPTM” and rank 0 or 1.
Caspase-1 assay
50 μg of cell extracts or 200 μg of pancreatic or colonic tissue protein extracts were incubated with WEHD-AFC substrate for 2 hours (cells) or 6 hours (tissue) and measured in a microplate reader, excited at 400 nm, fluorescence emission at 505 nm.
Antibody arrays
Human chemokine or human cytokine V arrays (Ray Biotech, Inc) were processed and quantified using ImageJ software (NIH).
Western Blot analysis
Protein extracts were processed and analyzed as previously described 45.
Experiments with Pdx1-Cre KrasG12D/+ mice
Experiments were performed according to UK Home office regulations. Mice carrying a conditional Pdx1-Cre KrasG12D/+ allele 23,52 were intercrossed with mice carrying a conditional knockout ALK5/TGBR1 allele 25. Sections of formalin fixed paraffin embedded mouse pancreas from 6 weeks old mice were stained with antibody against Ki67. The total number of Ki67-positive cells per PanIN, and the total cells per PanIN were counted, and the percentage of Ki67-positive cells per PanIN calculated. The mean score for each mouse was calculated and these scores were plotted on a box plot. SA β-gal staining was carried out on cryosections of pancreas harvested from 6 week old mice and assessed using a histoscore method taking account of both the intensity of staining and the proportion of each PanIN staining. This was calculated using the formula Σ(0 × no staining)+(1 × low intensity)+(2 × high intensity). The mean score for each mouse was calculated and these scores were plotted on a box plot. 5 mice of each genotype (3 male and 2 female) were assessed. Statistical significance was assessed using a Mann-Whitney test to compare genotypes.
In vivo hepatocyte senescence experiments
The VEGFR2/Flt3/c-Kit inhibitor (Calbiochem 676500) was dissolved in 2% Ethanol, 5% Tween 80, 20% PEG 400, 73% isotonic NaCl solution and orally applied twice daily at 4 mg/kg body weight. The IL1-R antagonist (Calbiochem 407616) was dissolved in isotonic NaCl solution and applied via intraperitoneal injection every 2nd day at 200mg/kg body weight. RS102895 (Sigma R1903) a CCR2 antagonist was applied via the drinking water at a dose of 10mg/kg/day per mouse. From the TGF-βRI kinase inhibitor (Calbiochem 616452) a 3.4mM stock solution in DMSO was prepared. Twice daily 100 μl of a 1 to 10 dilution in PBS was injected subcutaneous. For each inhibitor four C57bl6 mice and four control animals (age 4-6 weeks, all males) were used. Treatment started at day −2 and continued until day 12. At day 0 hydrodynamic tail vein injection of a transposon-based Nras expression plasmid together with an expression plasmid for the sleeping beauty 13 transposase 12 was performed. At day 12 the animals were sacrificed and livers collected. Samples were fixed and subjected to IHC analysis. Microscopic analyses were performed using Axio Imager M2 (Zeiss). Five high power fields were counted on two liver sections from each mouse liver (200×, > 200 counted cells per field).
IHC of mouse skin samples
4 weeks old wild type or K5-Sos Egfrwa2/+ mice (heterozygous for a hypomorphic form of Egfr 33) were used for the experiments (equal ratios of male and female). Normal skin or papilloma samples were isolated from the tail, fixed over night in 4% PFA and then embedded in paraffin for IHC analysis.
IHC of human colon samples
Pseudo-anonymized human FFPE tissue samples from 9 patients with sessile serrated adenomas (SSA) that were resected endoscopically were provided by the Tissue Bank of the National Center for Tumor Diseases Heidelberg (project no. 841) after approval by the ethics committee (no. 206/2005, Medical Faculty, Heidelberg, Germany). IHC was carried out on 3-μm sections. BRAF V600E specific IHC (clone VE1) was performed on an automated immunostainer (Ventana BenchMark XT, Ventana Medical Systems, Tucson, Arizona, USA) as previously described53. The settings included pretreatment with cell conditioner 1 for 60 min, incubation with undiluted VE1 hybridoma supernatant at 37°C for 32 min and signal enhancement with the Ventana amplification kit (catalogue number 760-080). For Ki-67 (clone MIB-1, Dako, 1:400) and p21WAF1/Cip1 (clone SX118, DAKO, 1:25) antigens were retrieved using alkaline buffer (pH 9, Dako, Glostrup, Denmark). The latter stainings were performed using the Techmate™ 500+ automated staining system (Dako) with the Avidin–Biotin Complex method. p21 and Ki-67 positive nuclei in the tumor stroma were counted per area using virtual microscopy (Spectrum™ Version 11.0.0.725, Image scope v11.0.2.725, Aperio Technologies, Vista, CA, USA). For statistical evaluation the p21 to KI-67 ratio was determined and compared using the non-parametrical Wilcoxon rank sum test.
Statistical data analysis
Significance levels were denoted as: *P≤ 0.05, **P≤ 0.01 and ***P≤ 0.001. Sources for statistical data are provided in Table S8.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
ACKNOWLEDGEMENTS
We are grateful to M. Stampfer, G. Núñez and D. Escors for reagents and to T. Bird, S. Forbes, V. Episkopou, T. Rodríguez, P. Schirmacher, S. Parrinello, M. Narita, G. Peters and D. Beach for advice and critical reading of the manuscript. We also thank the tissue bank of the National Center for Tumour Diseases Heidelberg for providing colon tissues. Core support from MRC and grants from MRCT, CRUK and the AICR funded the research in J. Gil’s laboratory. J. Gil is also supported by the EMBO Young Investigator Programme.
Footnotes
Accession numbers. Microarray data has been deposited at the Gene Expression Omnibus (GEO) under the accession number GSE41318.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
REFERENCES
- 1.Kuilman T, Michaloglou C, Mooi WJ, Peeper DS. The essence of senescence. Genes Dev. 2010;24:2463–79. doi: 10.1101/gad.1971610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729–40. doi: 10.1038/nrm2233. [DOI] [PubMed] [Google Scholar]
- 3.Kuilman T, Peeper DS. Senescence-messaging secretome: SMS-ing cellular stress. Nat Rev Cancer. 2009;9:81–94. doi: 10.1038/nrc2560. [DOI] [PubMed] [Google Scholar]
- 4.Coppe JP, et al. A human-like senescence-associated secretory phenotype is conserved in mouse cells dependent on physiological oxygen. PLoS One. 2010;5:e9188. doi: 10.1371/journal.pone.0009188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Krtolica A, Parrinello S, Lockett S, Desprez PY, Campisi J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc Natl Acad Sci U S A. 2001;98:12072–7. doi: 10.1073/pnas.211053698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coppe JP, Kauser K, Campisi J, Beausejour CM. Secretion of vascular endothelial growth factor by primary human fibroblasts at senescence. J Biol Chem. 2006;281:29568–74. doi: 10.1074/jbc.M603307200. [DOI] [PubMed] [Google Scholar]
- 7.Coppe JP, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–68. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Acosta JC, et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell. 2008;133:1006–18. doi: 10.1016/j.cell.2008.03.038. [DOI] [PubMed] [Google Scholar]
- 9.Kuilman T, et al. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008;133:1019–31. doi: 10.1016/j.cell.2008.03.039. [DOI] [PubMed] [Google Scholar]
- 10.Kortlever RM, Higgins PJ, Bernards R. Plasminogen activator inhibitor-1 is a critical downstream target of p53 in the induction of replicative senescence. Nat Cell Biol. 2006;8:877–84. doi: 10.1038/ncb1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wajapeyee N, Serra RW, Zhu X, Mahalingam M, Green MR. Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7. Cell. 2008;132:363–74. doi: 10.1016/j.cell.2007.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kang TW, et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature. 2011 doi: 10.1038/nature10599. [DOI] [PubMed] [Google Scholar]
- 13.Xue W, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656–60. doi: 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krizhanovsky V, et al. Senescence of activated stellate cells limits liver fibrosis. Cell. 2008;134:657–67. doi: 10.1016/j.cell.2008.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hayflick L. The Limited in Vitro Lifetime of Human Diploid Cell Strains. Exp Cell Res. 1965;37:614–36. doi: 10.1016/0014-4827(65)90211-9. [DOI] [PubMed] [Google Scholar]
- 16.Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25:585–621. doi: 10.1016/0014-4827(61)90192-6. [DOI] [PubMed] [Google Scholar]
- 17.Nelson G, et al. A senescent cell bystander effect: senescence-induced senescence. Aging Cell. 2012;11:345–9. doi: 10.1111/j.1474-9726.2012.00795.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fridman AL, Tainsky MA. Critical pathways in cellular senescence and immortalization revealed by gene expression profiling. Oncogene. 2008;27:5975–87. doi: 10.1038/onc.2008.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Collado M, et al. Tumour biology: senescence in premalignant tumours. Nature. 2005;436:642. doi: 10.1038/436642a. [DOI] [PubMed] [Google Scholar]
- 20.Massague J. TGFbeta in Cancer. Cell. 2008;134:215–30. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaneda A, et al. Activation of Bmp2-Smad1 signal and its regulation by coordinated alteration of H3K27 trimethylation in Ras-induced senescence. PLoS Genetics. 2011;7:e1002359. doi: 10.1371/journal.pgen.1002359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reynisdottir I, Polyak K, Iavarone A, Massague J. Kip/Cip and Ink4 Cdk inhibitors cooperate to induce cell cycle arrest in response to TGF-beta. Genes Dev. 1995;9:1831–45. doi: 10.1101/gad.9.15.1831. [DOI] [PubMed] [Google Scholar]
- 23.Hingorani SR, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–50. doi: 10.1016/s1535-6108(03)00309-x. [DOI] [PubMed] [Google Scholar]
- 24.Morton JP, et al. Mutant p53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. Proc Natl Acad Sci U S A. 2010;107:246–51. doi: 10.1073/pnas.0908428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Larsson J, et al. Abnormal angiogenesis but intact hematopoietic potential in TGF-beta type I receptor-deficient mice. EMBO J. 2001;20:1663–73. doi: 10.1093/emboj/20.7.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morton JP, et al. LKB1 haploinsufficiency cooperates with Kras to promote pancreatic cancer through suppression of p21-dependent growth arrest. Gastroenterology. 2010;139:586–97. 597, e1–6. doi: 10.1053/j.gastro.2010.04.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morton JP, et al. Dasatinib inhibits the development of metastases in a mouse model of pancreatic ductal adenocarcinoma. Gastroenterology. 2010;139:292–303. doi: 10.1053/j.gastro.2010.03.034. [DOI] [PubMed] [Google Scholar]
- 28.Orjalo AV, Bhaumik D, Gengler BK, Scott GK, Campisi J. Cell surface-bound IL-1alpha is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc Natl Acad Sci U S A. 2009;106:17031–6. doi: 10.1073/pnas.0905299106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–32. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 30.Strowig T, Henao-Mejia J, Elinav E, Flavell R. Inflammasomes in health and disease. Nature. 2012;481:278–86. doi: 10.1038/nature10759. [DOI] [PubMed] [Google Scholar]
- 31.Carragher LA, et al. V600EBraf induces gastrointestinal crypt senescence and promotes tumour progression through enhanced CpG methylation of p16INK4a. EMBO Mol Med. 2010;2:458–71. doi: 10.1002/emmm.201000099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Keller M, Ruegg A, Werner S, Beer HD. Active caspase-1 is a regulator of unconventional protein secretion. Cell. 2008;132:818–31. doi: 10.1016/j.cell.2007.12.040. [DOI] [PubMed] [Google Scholar]
- 33.Sibilia M, et al. The EGF receptor provides an essential survival signal for SOS-dependent skin tumor development. Cell. 2000;102:211–20. doi: 10.1016/s0092-8674(00)00026-x. [DOI] [PubMed] [Google Scholar]
- 34.Kriegl L, et al. Up and downregulation of p16(Ink4a) expression in BRAF-mutated polyps/adenomas indicates a senescence barrier in the serrated route to colon cancer. Modern Pathology. 2011;24:1015–22. doi: 10.1038/modpathol.2011.43. [DOI] [PubMed] [Google Scholar]
- 35.Bennecke M, et al. Ink4a/Arf and oncogene-induced senescence prevent tumor progression during alternative colorectal tumorigenesis. Cancer Cell. 2010;18:135–46. doi: 10.1016/j.ccr.2010.06.013. [DOI] [PubMed] [Google Scholar]
- 36.Caruso M, et al. Over-expression of cathepsin E and trefoil factor 1 in sessile serrated adenomas of the colorectum identified by gene expression analysis. Virchows Archiv. 2009;454:291–302. doi: 10.1007/s00428-009-0731-0. [DOI] [PubMed] [Google Scholar]
- 37.Schmitt CA. Cellular senescence and cancer treatment. Biochimica et Biophysica Acta. 2007;1775:5–20. doi: 10.1016/j.bbcan.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 38.Prise KM, O’Sullivan JM. Radiation-induced bystander signalling in cancer therapy. Nature Reviews Cancer. 2009;9:351–60. doi: 10.1038/nrc2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baker DJ, et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011;479:232–6. doi: 10.1038/nature10600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Burd CE, et al. Monitoring tumorigenesis and senescence in vivo with a p16(INK4a)-luciferase model. Cell. 2013;152:340–51. doi: 10.1016/j.cell.2012.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chien Y, et al. Control of the senescence-associated secretory phenotype by NF-{kappa}B promotes senescence and enhances chemosensitivity. Genes Dev. 2011;25:2125–36. doi: 10.1101/gad.17276711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jing H, et al. Opposing roles of NF-{kappa}B in anti-cancer treatment outcome unveiled by cross-species investigations. Genes Dev. 2011;25:2137–46. doi: 10.1101/gad.17620611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zitvogel L, Kepp O, Galluzzi L, Kroemer G. Inflammasomes in carcinogenesis and anticancer immune responses. Nature Immunology. 2012;13:343–51. doi: 10.1038/ni.2224. [DOI] [PubMed] [Google Scholar]
- 44.Ling J, et al. KrasG12D-induced IKK2/beta/NF-kappaB activation by IL-1alpha and p62 feedforward loops is required for development of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21:105–20. doi: 10.1016/j.ccr.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barradas M, et al. Histone demethylase JMJD3 contributes to epigenetic control of INK4a/ARF by oncogenic RAS. Genes Dev. 2009;23:1177–82. doi: 10.1101/gad.511109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Banito A, Gil J. Induced pluripotent stem cells and senescence: learning the biology to improve the technology. EMBO Rep. 2010;11:353–9. doi: 10.1038/embor.2010.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Banito A, et al. Senescence impairs successful reprogramming to pluripotent stem cells. Genes Dev. 2009;23:2134–9. doi: 10.1101/gad.1811609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bishop CL, et al. Primary cilium-dependent and -independent Hedgehog signaling inhibits p16(INK4A) Mol Cell. 2010;40:533–47. doi: 10.1016/j.molcel.2010.10.027. [DOI] [PubMed] [Google Scholar]
- 49.Acosta JC, Snijders AP, Gil J. Unbiased characterization of the senescence-associated secretome using SILAC-based quantitative proteomics. Methods in Molecular Biology. 2013;965:175–84. doi: 10.1007/978-1-62703-239-1_11. [DOI] [PubMed] [Google Scholar]
- 50.Chen Y, et al. SPD--a web-based secreted protein database. Nucleic Acids Research. 2005;33:D169–73. doi: 10.1093/nar/gki093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gonzalez R, et al. Screening the mammalian extracellular proteome for regulators of embryonic human stem cell pluripotency. Proc Natl Acad Sci U S A. 2010;107:3552–7. doi: 10.1073/pnas.0914019107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jackson EL, et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001;15:3243–8. doi: 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Capper D, et al. Assessment of BRAF V600E mutation status by immunohistochemistry with a mutation-specific monoclonal antibody. Acta Neuropathologica. 2011;122:11–9. doi: 10.1007/s00401-011-0841-z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.