

Abstract
Block copolymers of poly(ethylene glycol) (PEG) and poly(ε-caprolactone) (PCL) with chemically addressable functional groups were synthesized and characterized. Ring opening polymerization of ε-caprolactone (CL) and 1,4,8-trioxaspiro-[4,6]-9-undecanone (TSU) using α-methoxy, ω-hydroxyl poly(ethylene glycol) (mPEG) as the initiator afforded a copolymer with cyclic ketals being randomly distributed in the hydrophobic PCL block. At an initiator/catalyst molar ratio of 10/1 and a TSU/CL weight ratio of 1/4, a ketal-carrying copolymer (ECT2-CK) with Mn of 52 kDa and a ketal content of 15 mol% was obtained. Quantitative side chain deacetalization revealed the reactive ketones without noticeable polymer degradation. In our study, 10 mol% of cyclic ketals were deprotected and the ketone-containing copolymer was designated as ECT2-CO. Reaction of ECT2-CO with 2-(2-(aminooxy)acetoxy)-ethyl acrylate gave rise to an acrylated product (ECT2-AC) containing an estimated 3–5 acrylate groups per chain. UV-initiated radical polymerization of ECT2-AC in dichloromethane resulted in a crosslinked network (xECT2-AC). Thermal and morphological analyses employing Differential Scanning Calorimetry (DSC) and Atomic Force Microscopy (AFM) operated in PeakForce Tapping mode revealed the semicrystalline nature of the network, containing stiff crystalline lamellae dispersed in a softer amorphous interstitial. Macroscopic and nanoscale mechanical characterizations showed that ECT2-CK exhibited a significantly lower modulus than PCL of a similar molecular weight. While ECT2-CK undergoes a plastic deformation with a distinct yield point and a cold drawing region, xECT2-AC exhibited a compliant, elastomeric deformation with a Young’s modulus of 0.5 ± 0.1 MPa at 37 °C. When properly processed, the crosslinked network exhibited shape memory behaviors, with shape fixity and shape recovery values close to 1 and a shape recovery time of less than 4 s at 37 °C. In vitro studies showed that xECT2-AC films did not induce any cytotoxic effects to the cultured mesenchymal stem cells. The crosslinkable polyester copolymers can be potentially used as tissue engineering scaffolds and minimally invasive medical devices.
Keywords: Poly(ε-caprolactone), Poly(ethylene glycol), Copolymers, Functional Groups, Photocrosslinking, Elastomeric, Shape Memory, Tissue Engineering
Introduction
Biodegradable polymers have been widely explored for use as tissue engineering scaffolds or as intelligent medical devices [1–3]. Many tissues/organs in the human body are mechanically active and medical devices are frequently mechanically deployed using minimally invasive surgical techniques. In some cases, the engineered scaffolds and the medical devices are implanted in mechanically stressed tissues. Therefore, polymeric materials targeted for aforementioned biomedical applications should not only be mechanically robust but also exhibit mechanical properties comparable to the target tissues [4, 5]. Elastomeric materials that have the ability to recover from large and repetitive deformations can effectively transmit the external mechanical stimulations to the cultured cells in vitro [6–8]. These materials can also fulfill the mechanical functions of the target tissues in vivo without inducing mechanical irritation to the host tissue [9, 10].
Poly(ε-caprolactone) (PCL) is one of the most extensively investigated degradable polymers because it can be readily synthesized at low cost and possesses rheological and viscoelastic properties superior to many of its resorbable-counterparts[11–14]. However, many physical, chemical and biological barriers associated with PCL homopolymers have to date limited their biomedical applications. For instance, due to its high crystallinity, PCL is relatively stiff compared to many soft tissues; it undergoes a plastic deformation when stretched beyond the linear viscoelastic region [12]. Therefore, materials based solely on PCL are not conducive substrates for the engineering of mechanically active soft tissues. Furthermore, PCL’s inherent hydrophobicity has resulted in poor cell-scaffold interaction, and the segregated crystalline domains prevent homogeneous distribution of therapeutic molecules in the scaffolds [15]. The combined crystallinity and hydrophobicity also give rise to slow degradation [16, 17]. Finally and most importantly, the lack of reactive groups along the PCL backbone has prohibited the facile adjustment of its physical and biological properties.
Methods to functionalize PCL include blending of PCL with other polymers bearing functional groups [18, 19], polymerization of CL in the presence of comonomers containing reactive moieties [20–23] and the termination of living PCL chains with selected molecules with appropriate functional groups [24]. Commonly used methods to tailor the mechanical properties of PCL include hybridization of PCL and organic or inorganic particles into composites [25–28], copolymerization of CL with lactide [29, 30], glycolide [31], 1,3-trimethylene carbonate [32], 2-oxepane-1,5-dione [33, 34] or blending of PCL with starch [35] or poly(D,L-lactic acid) [36]. Covalent crosslinking of functional PCL copolymers allows for their mechanical properties to be readily tuned. To this end, photocrosslinkable PCL macromers have been synthesized by the reaction of PCL diol with acryloyl chloride [37]. Similarly, poly(caprolactone fumarate), synthesized by condensation polymerization of PCL diols and fumaryl chloride [38], was crosslinked photochemically. Materials with the most desirable rheological and mechanical properties at body temperature were obtained by combining chemical crosslinks and physical associations between crystalline domains [39].
The semicrystalline nature of PCL-based materials, suggests that, when chemically crosslinked and properly processed, these materials can exhibit shape memory effects, with the shape recovery being triggered by external stimuli [40]. Shape memory materials have been designed as inflatable stents to treat the clotted artery and intelligent suture for wound closure [41]. Strategic placement of reactive handles along the PCL backbone will enable the development of implantable devices or biocompatible scaffolds with desirable biological and mechanical properties.
Herein, we report the synthesis of functional PCL copolymers with readily addressable functional groups that allow for facile modification of their morphological and mechanical properties. First, a PCL based copolymer (ECT2-CK, 4, Scheme 1) was prepared via mPEG-initiated ring opening copolymerization of ε-caprolactone (CL) and 1,4,8-trioxaspiro-[4,6]-9-undecanone (TSU) using Sn(Oct)2 as the catalyst. The inclusion of the PEG block offers the opportunity to fine-tune the solubility, crystallinity and flexibility of the base material, as well as protein-polymer interactions [42–46]. The resulting copolymer was subjected to further modification for selective deprotection of the cyclic ketal groups to reveal the reactive ketone groups, which were further converted to acrylates via a hydroxylamine-ketone coupling reaction. Photocrosslinking of the copolymers carrying pendent acrylates (ECT2-AC, 7, Scheme 1) resulted in a soft and elastomeric network with shape memory properties. The cytocompatible nature of the crosslinked networks was confirmed by culturing mesenchymal stem cells in the presence of crosslinked films.
Scheme 1.

Synthetic pathways for the preparation of crosslinked polyester networks. (A): Synthesis of ECT2-CK (4) by mPEG (1)-initiated ring opening polymerization (ROP) of CL (2) and TSU (3). The TSU content in feed was 20wt%. (B): Synthesis of acrylated ECT2 (ECT2-AC, 7) via a ketone containing intermediate (ECT2-CO, 5). (C): UV-initiated radical polymerization of ECT2-AC in DCM afforded a crosslinked network (xECT2-AC, 8). Letters a, b, m and n indicate the number of respective repeats in the copolymers, whereas y and z denote the fraction of repeating units modified.
Experimental Section
Materials
ε-Caprolactone (CL), stannous octoate (Sn(Oct)2) 1,4-Cyclohexanedione monoethylene acetal, 3-chloroperoxybenzoic acid, triphenylcarbenium tetrafluoroborate (Ph3CBF4), 1,1-diphenylethylene (DPE), triethylamine (TEA), (Boc-aminooxy)acetic acid, hydroxyethyl acrylate, 4-dimethylaminopyridine (DMAP), dicyclohexylcarbodiimide (DCC), trifluoroacetic acid (TFA), 2, 2-dimethoxy-2-phenylacetophenone (DMPA), 1-vinyl-2-pyrrolidinone (NVP), α-hydroxyl ω-methoxy poly(ethylene glycol) (mPEG, Mn = 5,000 g/mol), Triton X-100 and PCL with a molecular weight in the range of 70~90 kDa were purchased from Sigma-Aldrich and were used as received, unless otherwise noted. mPEG was dried under vacuum at 100 °C in the presence of anhydrous phosphorous pentoxide (P2O5) overnight prior to use. CL was dried over calcium hydride (CaH2) for 48 h at room temperature and was distilled under reduced pressure just before use. Sn(Oct)2 was dried azeotropically with toluene, and then distilled under high vacuum. Anhydrous toluene and dichloromethane (DCM) were obtained from Fisher Scientific. Human bone marrow-derived mesenchymal stem cells (MSCs) were purchased from Lonza and were cultured in the MSC maintenance media. Dulbecco's phosphate-buffered saline (DPBS) was purchased from Gibco. Syto-13 and propidium iodide nucleic acid stains, as well as a the Alamar blue dye, were purchased from Invitrogen.
Instrumentation
1H NMR spectra were recorded on a Bruker AV400 NMR spectrometer under standard quantitative conditions in CDCl3. All the spectra were analyzed with Mestrenova software, and chemical shifts were calibrated using NMR solvent peak (7.27 ppm for CDCl3). Gel permeation chromatography (GPC) was carried out using THF as the eluent at a flow rate of 1.0 mL/min on Waters Styragel columns at ambient temperature at a polymer concentration of ~4 mg/mL. The GPC data were acquired using a refractive index detector (Waters 410) and analyzed by Waters Empower software using polystyrene standards (Polysciences Corporation). Water contact angle was measured with a Kruss Easy Drop contact angle goniometer. Differential Scanning Calorimetry (DSC) experiments were conducted on a TA Instruments Discovery series DSC. Polymer samples were cooled/heated in N2 from −80 °C to 100 °C at a rate of 10 °C/min and the thermogram of the second cycle was reported. The glass transition temperature (Tg), the cold crystallization temperature (Tcc) and the melting temperature (Tm) were determined from the heating curve while the crystallization temperature (Tc) was determined from the cooling curve.
Synthesis of poly(ethylene glycol)-b-poly(ε-caprolactone-ran-1,4,8-trioxaspiro-[4,6]-9-undecanone) (4, ECT2-CK, Scheme 1) [15]
PCL-based copolymers were prepared by mPEG-initiated ring opening polymerization of CL and TSU (3, Scheme 1) in the presence of Sn(Oct)2. TSU was synthesized following reported procedures [47] and was purified by repeated crystallization in diethyl ether and subsequent sublimation under reduced pressure before use. In our study, the weight ratio of TSU/CL in feed was maintained as 1/4. Specifically, mPEG (0.10 mmol), CL (53 mmol) and TSU (8.7 mmol) were dissolved in anhydrous toluene (12 mL) and the mixture was azeotropically distilled (3 times) under reduced pressure at 50 °C. Upon the addition of Sn(Oct)2 (10 mol% relative to the hydroxyl end group in mPEG) under N2 protection, the reaction vessel was sealed and subsequently heated at 120 °C for 6 h, with the reaction mixture being constantly stirred. The polymerization was terminated by cooling the reaction vessel to 0 °C and the product was solubilized in DCM and precipitated in a large excess of methanol twice and then in hexanes once. The white solid product was collected by filtration and was dried under vacuum at ambient temperature to a constant weight. Yield: 84%. 1H NMR (Figure S1A): δ = 1.39 (br, m, 2H, -CH2CH2-CH2-CH2CH2C(O)O-), 1.65 (br, m, 4H, -CH2-CH2-CH2-CH2-CH2C(O)O-), 1.99 (br, m, 4H, -CH2-CH2-C(-OCH2CH2O-)-CH2-CH2C(O)O-), 2.31 (t, 2H, 3J = 7.4Hz, -CH2CH2CH2-CH2-CH2-C(O)O-), 2.38 (t, 2H, 3J= 7.6Hz, -CH2CH2C(-OCH2CH2O-)CH2-CH2-C(O)O-), 3.39 (s, 3H, CH3O-PEG-), 3.65 (s, 4H, -O-CH2-CH2-O-), 3.95 (s, 4H, -CH2CH2C(-OCH2CH2O-)CH2CH2C(O)O-), 4.07 (t, 2H, 3J = 6.6Hz, -CH2-CH2CH2CH2CH2C(O)O-), 4.17 (t, 1H, 3J = 7.0 Hz, -CH2-CH2C(-OCH2CH2O-)CH2CH2C(O)O-).
Synthesis of ketone containing copolymers (5, ECT2-CO, Scheme 1)
A deep green-colored solution of Ph3CBF4 (10 mg, 0.030 mmol) and 1,1-diphenylethylene (DPE, 200 µL) in DCM (4 mL) was added slowly into a stirred DCM solution (8 mL) of ECT2-CK (0.50 g, 0.57 mmol of cyclic ketal). The resulting mixture was stirred at room temperature for 2 h before being the reaction was quenched by the addition of triethylamine (1 mL). After repeated precipitations in diethyl ether, the filtered product was dried under vacuum. Yield: 73%. 1H NMR (Figure S1B): δ = 1.39 (br, m, 2H, -CH2CH2-CH2-CH2CH2C(O)O-), 1.65 (br, m, 4H, -CH2-CH2-CH2-CH2-CH2C(O)O-), 2.00 (br, m,-CH2-CH2-C(-OCH2CH2O-)-CH2-CH2C(O)O-), 2.31 (t, 2H, 3J = 7.4Hz, -CH2CH2CH2-CH2-CH2-C(O)O-), 2.38 (t, 2H, 3J = 7.6Hz, -CH2CH2C(-OCH2CH2O-)CH2-CH2-C(O)O-), 2.61 (t, 2H, 3J = 6.4Hz, -CH2CH2C(O)CH2-CH2-C(O)O-), 2.76 (t, 2H, 3J = 6.6Hz, -CH2CH2C(O)-CH2-CH2C(O)O-), 2.81 (t, 2H, 3J = 6.2Hz, -CH2-CH2-C(O)CH2CH2C(O)O-), 3.39 (s, 3H, CH3O-PEG-), 3.65 (s, 4H, -O-CH2-CH2-O-), 3.95 (s, 4H, -CH2CH2C(-OCH2CH2O-)CH2CH2C(O)O-), 4.07 (t, 2H, 3J= 6.6Hz, -CH2-CH2CH2CH2CH2C(O)O-), 4.17 (t, 2H, 3J = 7.0Hz, -CH2-CH2C(-OCH2CH2O-)CH2CH2C(O)O-), 4.35 (t, 2H, 3J = 6.2Hz, -CH2-CH2C(O)CH2CH2C(O)O-).
Synthesis of 2-(2-(aminooxy)acetoxy)-ethyl acrylate (6, AEA, Scheme 1)
The heterodifunctional linker was synthesized following a previously reported procedure[48] with minor revisions. To a round bottle flask covered with alumina foil was added (Bocaminooxy) acetic acid (0.50 g, 2.6 mmol), hydroxyethyl acrylate (0.55 mL, 5.2 mmol), 4-dimethylaminopyridine (DMAP, 37 mg, 0.30 mmol) and anhydrous DCM (8 mL). The flask was sealed and cooled in an ice bath before dicyclohexylcarbodiimide (DCC, 0.60 g, 2.9 mmol) was slowly added. The reaction mixture was maintained at 0 °C for 5 min, then at ambient temperature for 12 h under constant agitation in the dark. The precipitated dicyclohexylurea (DCU) was filtered out, and the solvent in the filtrate was evaporated using a rotary evaporator. The residue was dissolved in DCM (50 mL) and the solution was sequentially washed with 0.5 N HCl (100 mL× 3), saturated NaHCO3 solution (100 mL× 3) and DI water (100 mL × 3). After being dried over anhydrous MgSO4, solvent was evaporated under vacuum to give the corresponding Boc-protected AEA. The Boc-protected AEA (0.32 g, 1.1 mmol) was subsequently dissolved in 14 mL of a 50/50 (v/v) mixture of trifluoroacetic acid/DCM to remove the Boc protecting group. Three hours later, all volatile was removed under vacuum and the final product AEA was produced in a yield of 79%. 1H NMR (Figure S2): δ = 4.30 (s, -CH2-ONH2), 4.42 (m, 4H, -O-CH2CH2-O-), 5.89 (d, H, 3J= 10.4Hz, -CH=CHH), 6.15 (dd, 1H, 3J= 10.4, 17.6Hz, -CH=CHH), 6.45 (d, H, 3J= 17.6Hz, -CH=CHH).
Synthesis of acrylate bearing copolymers (7, ECT2-AC, Scheme 1)
Freshly prepared AEA (0.055 g, 0.29 mmol) was added to a DCM solution (12 mL) of ECT2-CO (1.2 g, 0.15 mmol ketone groups) [49]. The reaction mixture was stirred for 12 h at room temperature in the dark. After repeated precipitations in diethyl ether, the product was dried under vacuum. Yield: 71%. 1H NMR (Figure S1C): δ = 1.39 (br, m, 2H, -CH2CH2-CH2-CH2CH2C(O)O-), 1.65 (br, m, 4H, -CH2-CH2-CH2-CH2-CH2C(O)O-), 2.00 (br, m,-CH2-CH2-C(-OCH2CH2O-)-CH2-CH2C(O)O-), 2.31 (t, 2H, 3J = 7.4Hz, -CH2CH2CH2-CH2-CH2-C(O)O-), 2.38 (t, 2H, 3J = 7.6Hz, -CH2CH2C(-OCH2CH2O-)CH2-CH2-C(O)O-), 2.55 (s, 2H, -CH2CH2C(=NOCH2C(O)O-CH2CH2OC(O)CH=CH2)-CH2CH2C(O)O-), 2.61 (t, 2H, 3J = 6.4 Hz, -CH2CH2C(O)CH2CH2C(O)O-), 2.64 (s, 2H, -CH2CH2C(=NOCH2C(O)OCH2CH2OC(O)CH=CH2)-CH2-CH2C(O)O-), 2.70 (t, 2H, 3J = 6.6Hz, -CH2-CH2-C(=NOCH2C(O)OCH2CH2OC(O)CH=CH2)CH2CH2C(O)O-), 2.76 (t, 2H, 3J = 6.6Hz, -CH2CH2C(O)-CH2-CH2C(O)O-), 2.81 (t, 2H, 3J = 6.4Hz, -CH2CH2C(O)CH2-CH2-C(O)O-), 3.39 (s, 3H, CH3O-PEG-), 3.65 (s, 4H, -O-CH2-CH2-O-), 3.95 (s, 4H, -CH2CH2C(-OCH2CH2O-)CH2CH2C(O)O-), 4.07 (t, 2H, 3J = 6.6Hz, -CH2-CH2CH2CH2CH2C(O)O-), 4.17 (t, 1H, 3J = 7.0Hz, -CH2-CH2C(O)CH2CH2C(O)O-), 4.30 (t, 2H, 3J= 7.4Hz, -CH2-CH2C(=NOCH2C(O)OCH2CH2OC(O)CH=CH2)CH2CH2C(O)O-), 4.35 (t, 2H, 3J = 6.2Hz, -CH2-CH2C(O)CH2CH2C(O)O-), 4.39 (s, 4H, -CH2CH2C(=NOCH2C(O)OCH2CH2OC(O)CH=CH2)CH2CH2C(O)O-), 4.60, 4.61 (1:1, syn: anti, s, 2H, -CH2CH2C(=NO-CH2-C(O)OCH2CH2OC(O)CH=CH2)CH2CH2C(O)O-), 5.88 (d, 1H, 3J= 10.4Hz -CH2CH2C(=NOCH2C(O)OCH2CH2OC(O)CH=CHH)CH2CH2C(O)O-), 6.15 (dd, 3J = 10.4, 17.6Hz, 1H, -CH2CH2C(=NOCH2C(O)OCH2CH2OC(O)CH=CH2)CH2CH2C(O)O-), 6.45 (d, 1H, 3J= 17.6 Hz, -CH2CH2C(=NOCH2C(O)OCH2CH2OC(O)CH=CHH)CH2CH2C(O)O-).
Synthesis of crosslinked networks (8, xECT2-AC, Scheme 1)
A photoinitiator stock solution was prepared by dissolving 2, 2-dimethoxy-2-phenylacetophenone (DMPA) in 1-vinyl-2-pyrrolidinone (NVP) to obtain a 30 wt% solution. A predetermined amount of initiator mixture was added to a DCM solution containing 0.3 g/mL ECT2-AC to afford a final DMPA concentration of 3 mg/mL. The precursor solution was exposed to UV light at 365 nm for 10 min to complete the gelation. Upon evaporation of DCM under vacuum, the initial dry weight (Wi) of xECT2-AC was noted. The dry samples were then soaked in DCM at ambient temperature for 24 h before the wet weight (Ws) was recorded. The swelling ratio (SW) was defined as Ws/Wi. After soaking in DCM for 24 h, followed by repetitive washing with fresh DCM, the swollen samples were vacuum dried and the final dry weight (Wf) was measured. The sol fraction (SF) was calculated based on SF = [(Wi-Wf)/Wi]×100%. Six specimens were tested for SW and SF determinations.
Atomic force microscopy (AFM)
A Bruker AFM (BioScope Catalyst™ BioAFM) operated in PeakForce™ tapping mode was used to investigate polymer morphology. The nanoscale mechanical properties of polymer films were analyzed using the same AFM employing Quantitative Nanomechanical Mapping (QNM). Samples were spun cast from a THF solution at a polymer concentration of 30 mg/mL on a clean glass slide at 2,500 RPM for 30 s. The residual solvent was allowed to evaporate under vacuum. UV-crosslinked polymer films were similarly prepared from an ECT2-AC solution containing the dissolved photoinitiator. The spun-cast film was immediately subject to a 10-min UV exposure prior to vacuum drying. Fused silica (FSILICA, PeakForce QNM sample kit, Bruker) was used to calibrate the AFM tip (Model: MPP-12120-10, Bruker). Low density polyethylene (LDPE, PeakForce QNM Sample kit, Bruker) with a Young’s modulus of ~200 MPa was used as the standard. The tip calibration routine gave a tip radius of 20 nm, a spring constant of 8 N/m, a Poission ratio of 0.4 and a deformation depth of 10 nm. During all subsequent measurements, the deformation depth was maintained as 10 nm by adjusting the “PeakForce Setpoint”. Images were acquired at a scanning rate of 0.2 Hz and a scale of 100 µm × 100 µm and 30 µm × 30 µm. Force-volume data were collected simultaneously with a total of 65,536 force-separation curves (Figure S4) generated in each captured image. The data was then analyzed with Nanoscope Analysis software (Version 1.40, Bruker).
Tensile tests
Uniaxial tensile tests were performed using a Rheometrics Dynamic Mechanical Analyzer (RSA G2, TA Instruments). Solvent-cast polymer samples were cut into a dumbbell shape with ASTM D412-06a standardized sizes (1.5 mm×2.0 mm×10 mm). The initial grip separation was 5.0 mm and the stretching speed was 0.333 mm/s. The Young’s modulus (E) was calculated as the slope of the initial linear portion of the stress-strain curve. Five cyclic loading and unloading tests were conducted on the crosslinked samples at a rate of 0.333 mm/s at 37 °C with a maximum strain of ~170% and a 1000 s waiting time between each cycle. Stress decay with time between consecutive cycles was also monitored (Figure S5). At least 3 specimens were tested for each composition.
Characterization of shape memory properties
The shape-memory properties were characterized by cyclic thermo-mechanical tensile tests [50] (Figure S6) on a RSA-G2 DMA using the dumbbell-shaped specimen. Samples were first stretched to a maximum strain (εm) of >300% at a rate of 0.333 mm/s at a relatively high temperature (Thigh) (step 1). Samples were subsequently cooled to a lower temperature (Tlow) while keeping the strain constant (step 2). Samples were then completely unloaded at Tlow and strain was recorded as εu (step 3). Finally, samples were heated to Thigh at zero stress to recover the residual strain (εp, step 4). The shape fixity (Rf) and shape recovery ratio (Rr) were calculated using the following equations for strain controlled cyclic testing, where N is the number of cycles:
For visual inspections, the xECT2-AC samples were crosslinked into a spirals or a flat stripe as their permanent shapes. After heating the samples above the thermal transition temperature (Ttrans) of the switching domains (in our case, it is the melting temperature of xECT2-AC), samples were deformed to the desired temporary shapes (flat stripe or spiral) at 4°C. The permanent shape was recovered by heating the samples above Ttrans at 37 °C in a water bath. The programmed processing of the samples was photographed by using a digital camera (Lumix DMC-LX3, Panasonic).
Cytotoxicity
Mesenchyam stem cells (MSCs) were sub-cultured at a seeding density of 4,000–5,000 cells/cm2 on a T150 tissue culture flask (Corning) in the MSC maintenance media. After reaching ~80% confluency, cells (passages 3–5) were trypsinized, centrifuged and re-suspended in fresh MSC media. MSCs were subsequently seeded on a 24-well plate at a density of 104 cells/well in 500 µL media and were allowed to grow overnight. Separately, purified xECT2-AC films (~600 µm thick) were immersed in 70% ethanol and water under agitation for 48 h and 24 h, respectively. Samples were subsequently exposed to a bactericidal UV lamp for 10 min. Ten milligrams of xECT2-AC films were used in each experimental group. PCL films similarly prepared were included as the positive controls while Triton X-100 (0.2 mg/mL) was added as the negative controls. Cell culture inserts (Millicell®) were used to hold the polymer films, in the media above the adherent cells in 24-well plate. At day 1 and 3, the cell culture inserts were removed and Alamar blue dye was added. After 1-h incubation at 37 °C, 100 µL of the media/dye mixture was transferred into a 96-well plate and the fluorescence intensity of the mixture was monitored using a plate reader (Victor3 Wallac, Perkin Elmer) at Ex/Em of 535/590 nm. The fluorescence intensity of the test sample was normalized to that obtained from cells cultured without any additives (NO samples). Results were expressed as mean ± S.D. Statistical analysis was conducted using a two-tailed, equal variance Student’s t-test, where a p-value of ≤0.05 was considered to be statistically different. Separately, live/dead staining was performed at day 3 on different experimental groups. Briefly, cells were rinsed with cold DPBS, stained with propidium iodide (1:2000 in DPBS) and Syto-13 (1:1000 in DPBS) for 5 min, and imaged with a multiphoton confocal microscope (Zeiss 510 NLO).
Results and Discussion
We have previously synthesized amphiphilic block copolymers [15] consisting of hydrophilic PEG and hydrophobic polyester bearing pendent cyclic ketals (CK) via mPEG-initiated ring opening copolymerization of CL and TSU using Sn(Oct)2 as the catalyst. In our initial investigations, the amount of mPEG (5,000 g/mol) and the total amount of monomer were maintained constant while the monomer feed ratio was varied systematically. The resulting copolymers are abbreviated as ECTx-CK, with×indicating the relative amount of TSU added to the reaction mixture. Compositional analyses by 13C NMR indicate that TSU was randomly distributed in the hydrophobic block [15, 51]. Differential Scanning Calorimetry (DSC) analysis revealed that when the TSU content in the copolymers increased, the glass transition temperature increased accordingly. The wide angle X-ray diffraction (WAXD) analyses confirm the disruption of the regularity of PCL molecular structure by the random insertion of TSU units in the hydrophobic block, hindering the ability of PCL to crystallize. Nanoparticles prepared from these copolymers have been utilized as drug delivery vehicles for controlled release of camptothecin [15] and dexamethasone [52].
The goal of the current investigation is to develop synthetic methodologies for the fabrication of elastomeric materials with robust mechanical properties, suitable for use as tissue engineering scaffolds and biomedical implants. The cyclic ketals along the polymer backbone not only serve as chain softening units but also are masked reactive handles that, when selectively deprotected, provide reactive sites for further chemical derivation. Although the crystalline PCL domains in ECTx copolymers have the potential to function as physical crosslinks, due to the random placement of TSU units along the backbone [15, 51] and the low melting transition of the microcrystals, ECTx without covalent crosslinking is likely to creep under long term or cyclic mechanical deformation. Thus, we aim to create synthetic elastomers by covalent crosslinking using a rubbery prepolymer with low crystallinity. In this study, ECTx with a pre-determined composition and molecular weight was subjected to multiple chemical transformations to include reactive acrylates for radical crosslinking purposes. We report an in-depth investigation on the derivation of the parent PEG-polyester copolymers, as well as a systematic investigation on the structure-property relationship of the crosslinked networks.
Synthesis of acrylated ECT2 (7, ECT2-AC)
ECT2-CK was synthesized at a TSU/CL feed ratio of ¼ (w/w). 1H NMR spectrum for ECT2-CK (Figure S1A) shows representative methylene signals from the CL repeating units (-CH2CH2CH2CH2CH2C(O)O- at 1.65 ppm and -CH2CH2CH2CH2CH2C(O)O- at 4.07 ppm). Characteristic peaks from the TSU repeating units, including the ethylene ketal signals (-OCH2CH2O- at 3.95 ppm), the methylene protons adjacent to the cyclic ketal (e+d in Figure S1A) at 1.99 ppm, and the methyl ester protons (f, Figure S1A) at 4.17 ppm. Representative methylene protons (-CH2CH2O-, 3.65 ppm) from mPEG repeating units are clearly visible. By comparing the relative intensity of the methylene protons from mPEG (b in Figure S1A, 454 H) at 3.65 ppm and the typical proton signals for each monomer (TSU: 4.17 ppm, f, 2H; CL: 4.07 ppm, l, 2H), Mn of the copolymer and the TSU content were determined to be 77 kDa and 15 mol%, respectively. Thus, ECT2-CK contains an average 89 TSU units per polymer chain.
Next, oxidative deacetylation of the cyclic ketals was attempted by reacting ECT2-CK with triphenylcarbenium tetrafluoroborate (Ph3CBF4) in DCM, as previously reported [47]. Oxidative deacetylation of ketals by hydride transfer using sterically hindered carbeniums via an oxonium intermediate is a standard method for unmasking of the ketone functionality [53]. Our initial attempt for ketone deprotection frequently resulted in the production of polymers with progressively reduced molecular weights as compared to the parent ECT2-CK. The rapid reaction of Ph3CBF4 with residual water, if any, produces a strong acid (HBF4) that facilitates the hydrolytic cleavage of the ester bonds in the polymer backbone. The unfavorable backbone degradation was suppressed by the addition of 1,1-diphenylethylene (DPE). A proton typically released from a Bronsted acid can convert DPE to Ph2MeC+ that is chemically similar to triphenylmethyl carbenium (Ph3C+) and therefore is capable of participating in the oxidative deacetylation reaction. Being only slightly less sterically hindered than Ph3C+, Ph2MeC+ cannot electrophilically attack the ester carbonyl to cleave the polymer chains. The hydrolytic polymer degradation was therefore dramatically suppressed to a negligible level in our anhydrous and proton-free reaction system employing excess DPE. These collective properties justify the utility of DPE as a proton scavenger in our deacetylation reaction system. The deacetylation reactions were optimized by reducing reaction time to 2 h to avoid any potential side reactions from reactive cationic species.
As a result of partial deacetylation, two new diagnostic signals at 2.76 ppm and 2.81 ppm (Figure S1B) were detected on the 1H NMR spectrum for ECT2-CO. These two peaks are characteristic methylene protons next to the ketones (e’ and d’ in Figure S1B, –CH2-C(O)CH2-) in the deprotected TSU units [54]. The alteration of the chemical environment also resulted in a peak shift for methylene protons neighboring the ester linkages. Specifically, methylene protons adjacent to the oxygen atom of the ester group (-CH2-OC(O)-, f and f’, Figure S1A, S1B) shifted from 4.17 ppm in TSU repeats to 4.35 ppm in the deprotected counterpart. In addition, the methylene protons next to the carbonyl carbons of the ester groups (-CH2-C(O)O-, c and c’, Figure S1A, S1B) shifted from 2.38 ppm in TSU repeats to 2.61 ppm in the deprotected TSU repeats. The presence of peaks associated with TSU, such as the ethylene ketal protons (-OCH2CH2O-) (g, Figure S1B) at 3.95 ppm, is a clear evidence for the maintenance of significant amount of TSU units in ECT2-CO. Under the experimental condition employed here, the deprotection percentage, calculated by comparing the integration of peaks corresponding to f and f’ and using the methylene protons (b, Figure S1B, 454 H) in mPEG as the internal reference, was estimated to be around 10 mol%. It is worth mentioning that the quantitative conversion of ketals to ketones is possible by controlling the reaction time (data not shown). The low percent deacetylation was needed to maintain the copolymer in a dynamic rubbery state prior to covalent crosslinking. Previous work from Jerome and coworkers showed that a complete conversion of ketones from the ketal groups in a random copolymer of CL and TSU (without PEG) resulted in an increase in melting enthalpy (ΔHm) as compared to the ketal carrying parent copolymer and PCL of a similar molecular weight [51]. Thus, a high degree of deacetylation would defeat the purpose of incorporating the chain-softening TSU units. Moreover, studies from the same group on ketone containing random copolyesters of CL and 2-oxepane-1,5-dione (OPD, an equivalent to deacetylated TSU repeating units in ECT2-CO), revealed that copolymers with an OPD content of ca. 30% were hydrolytically less stable than the PCL homopolymers [55].
The unmasked ketone carbonyl carbons are inherently electrophilic, susceptible to orthogonal reaction with aminooxy derivatives to form stable ketoxime ether linkage under mild reaction conditions [49]. For the introduction of reactive acrylates, a heterodifunctional crosslinker, 2-(2-(aminooxy)acetoxy)-ethyl acrylate (AEA), was successfully synthesized via a Boc-protected intermediate (Figure S2). Comparing the 1H NMR spectra of ECT2-CO (Figure S1B) and ECT2-AC (Figure S1C), the incorporation of AEA grafts was confirmed by the new signals associated AEA, including the characteristic vinyl protons at 5.88, 6.15 and 6.45 ppm, the methylene protons adjacent to the ketoxime group (–CH2-O-N=C, p, Figure S1C) and the methylene protons between the ester oxygens (-(O)CO-CH2-CH2-OC(O)- (q and r, Figure S1C). All other signals characteristics of the PEG block and the CL and TSU repeating units are clearly present. Peak f” at 4.30 ppm was assigned to methylene proton (-(O)CO-CH2-) in the AEA grafted repeating units. The conversion of ketone to ketoxime was calculated based on the integrations of f” and f’. An upfield shift of peak f’ at 4.35 ppm in ketone repeats to peak f” at 4.30 ppm in the ketoxime repeats implies a 31% consumption of ketones during the oxime reaction. Based on the NMR peak integrations and using mPEG methylene protons (b in Figure S1C, 3.65 ppm) as an internal reference, ECT2-AC was calculated to have a Mn of 79 kDa.
GPC analysis of the parent ECT2-CK substrate, the ketone-containing intermediate (ECT2-CO) and the acrylated precursor (ECT2-AC) showed no substantial alteration of molecular weight and molecular weight distribution (Figure 1 and Table 1). While the traces for ECT2-CO and ECT2-AC are perfectly overlapping, the minor variation of the elution time between ECT2-CK and ECT2-AC (or ECT2-CO) might be a result of altered hydrodynamic volume in the same GPC mobile phase (THF) due to the change of polymer composition. Overall, our GPC results confirm that little to no degradation to the polyester backbone occurred during the sequential chemical transformation process. [56]
Figure 1.
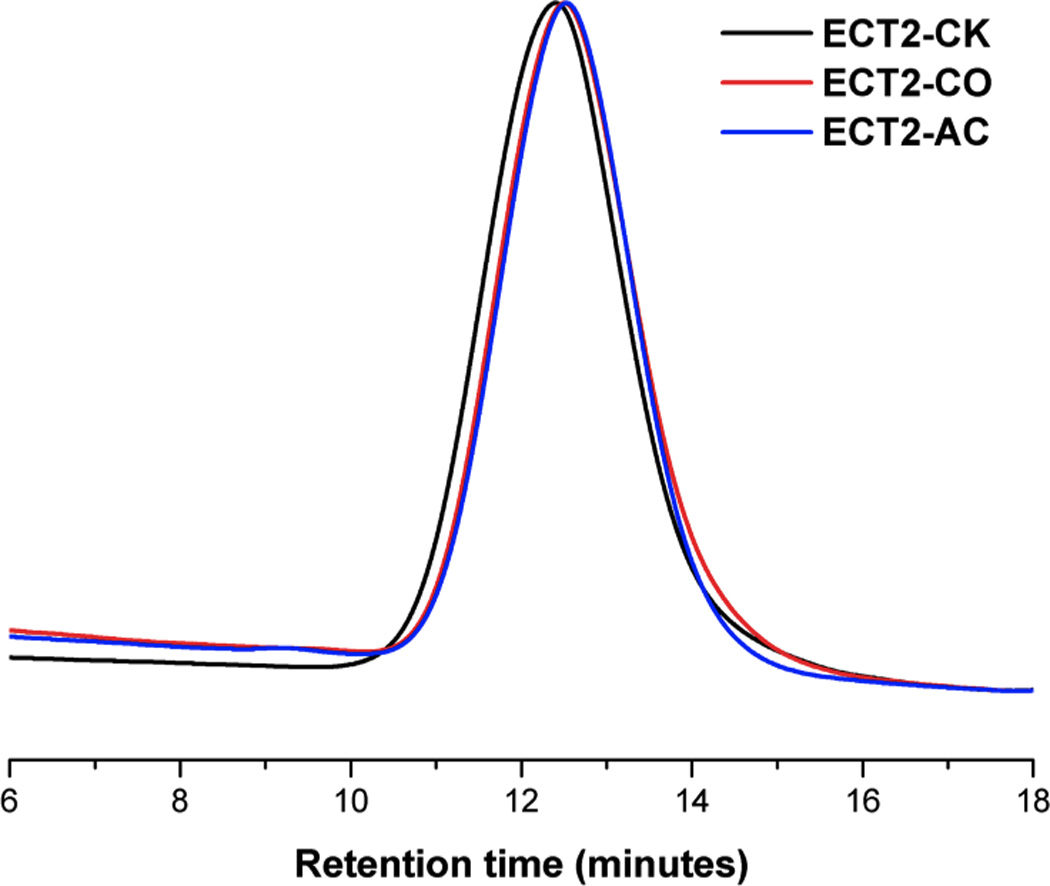
Overlaid GPC traces for ECT2-CK (black), ECT2-CO (red), and ECT2-AC (blue). Mobile phase: THF; Detector: refractive index.
Table 1.
Characterization of ECT2-CK, ECT2-CO, and ECT2-AC by 1H NMR and GPC.
| Polymer ID | Functional Group |
1H NMR1 | GPC2 | |||
|---|---|---|---|---|---|---|
| Mn (kg/mol) | Fn (mol%) | Mn (kg/mol) | Mw (kg/mol) | PDI | ||
| ECT2-CK | cyclic ketal | 77 | 15.1 | 52 | 84 | 1.6 |
| ECT2-CO | ketone | 80 | 1.6 | 46 | 74 | 1.6 |
| ECT2-AC | acrylate | 79 | 0.2 | 48 | 74 | 1.5 |
Determined via the integration of 1H NMR spectra acquired in CDCl3. Fn: mole percent functional groups in the hydrophobic block.
Determined by GPC using a refractive index detector and PS standards. Polymers were eluted using THF.
Thermal, morphological and mechanical characterizations
UV irradiation of a DCM solution of ECT2-AC (0.30 g/mL) in the presence of a photoinitiator resulted in the formation of solid-like networks (Figure S3) through the radical polymerization of the dangling acrylate groups in ECT2-AC. Of note, under the experimental conditions employed, additional radical species, potentially generated via the UV-induced fragmentation of the ester linkages [57], could participate in the crosslinking reaction with the conjugated acrylate groups, contributing to the overall network. The resultant gel had an average sol fraction (SF) of 33 ± 8%. This SF value is comparable to other radically crosslinked networks.[58, 59] The crosslinked network did not dissolve in DCM, instead, it swelled 20.4 ± 1.6 times of its original dry weight. The incorporation of reactive handles, chain softening units and a relatively short mPEG block to PCL did not significantly alter the hydrophobicity of the polymer, as evidenced by similar water contact angle values for PCL (76 ± 3), ECT2-CK (66 ± 8) and xECT2-AC (65 ± 3).
Because the physical and mechanical properties of crosslinked polymer networks are strongly dependent on their thermal and morphological characteristics, polymers were characterized by DSC prior to the mechanical analysis. The semicrystalline nature of ECT2-CK, ECT2-AC and xECT2-AC was evidenced by the presence of a glass transition and multiple melting peaks in the DSC thermograms (Figure 2). A broad transition centered around −55 °C, slightly higher than the glass transition temperature (Tg) of PCL [12] and ECT2-CK (Table 2), was detected for xECT2-AC. Our previous investigation revealed a progressive increase of Tg with an increase TSU content in the copolymer, and copolymers with 100% TSU (ECT10) had a Tg of −25 °C [15]. The presence of a single glass transition confirms the miscibility of the amorphous phase and reinforces the notion that the CL and TSU repeats are randomly distributed in the copolymers. An exothermic cold crystallization peak at −23°C was also observed for xECT2-AC (Figure 2A) where the polymers underwent a moderate crystallization during the heating cycle. All samples exhibited two melting peaks (Tm1, Tm2, Table 2) attributed to the PCL segments. Due to its higher molecular weight in comparison to mPEG, the PCL block was anticipated to crystallize first, effectively restricting the crystallization of the PEG blocks. Consequently, no melting endotherm was observed for PEG. The presence of two PCL melting peaks can be attributed to the difference in the chain fold number in the lamellae and/or to the formation of imperfect crystal structures of varying sizes [15]. Compared to ECT2-CK and ECT2-AC, the melting temperatures for xECT2-AC were lower and the melting enthalpy is smaller. This trend was further reflected by the crystallization temperature detected from the cooling curves (Figure 2B). This observation is consistent with the other semicrystalline polymers and can be attributed to impeded folding and reorganization of polymer chains imposed by the crosslink junctions. Consequently, imperfect crystallite with smaller size and less in content was created [60].
Figure 2.
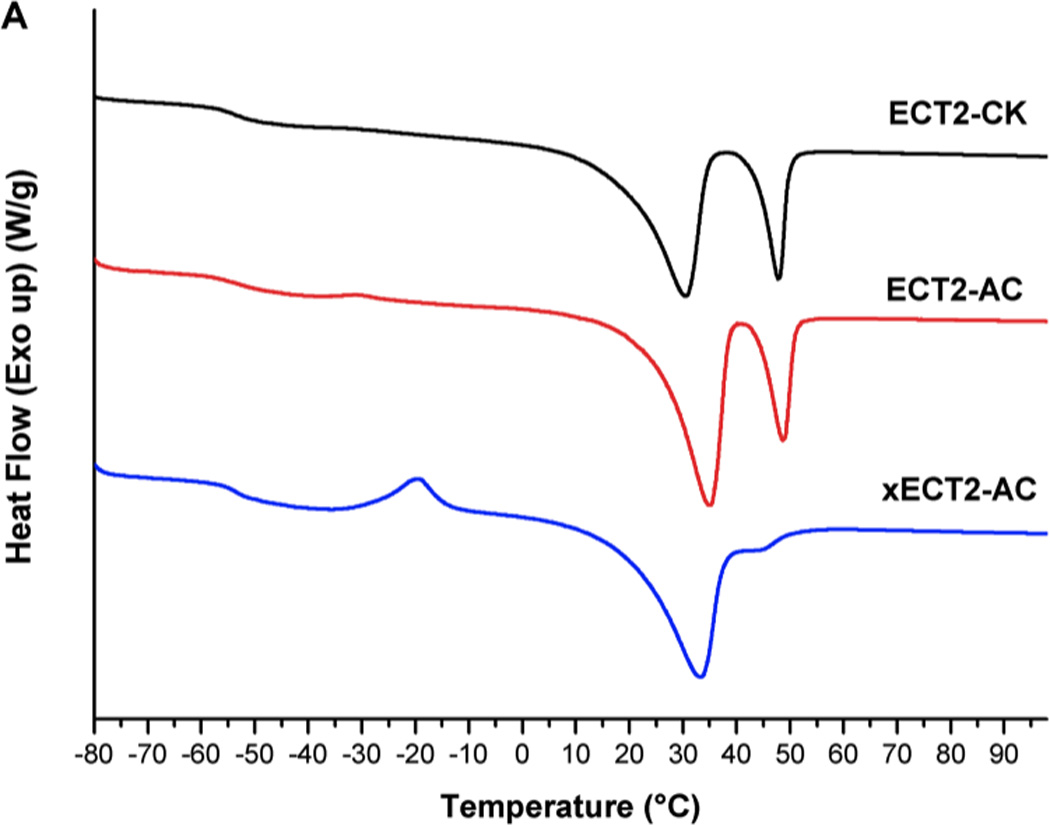

DSC thermographs (second scan) of ECT2-CK (black), ECT2-AC (red) and xECT2-AC (blue) from 80 °C to 100 °C at a heating rate of 10 °C/min. (A): heating cycle; (B): cooling cycle. Graphs have been shifted vertically for clarity.
Table 2.
Thermal properties of ECT2-CK, ECT2-AC and xECT2-AC.
| Polymer ID | Tg (°C)1 | Tcc (°C)2 | Tc (°C)3 | Tm1 (°C)4 | Tm2 (°C)5 | ΔHm (J/g)6 |
|---|---|---|---|---|---|---|
| ECT2-CK | −53.0 | - | −15.8 | 30.7 | 48.1 | −62.61 |
| ECT2-AC | −52.5 | - | −14.6 | 34.8 | 48.6 | −42.18 |
| xECT2-AC | −55.3 | −23.2 | −20.8 | 29.6 | 43.1 | −35.18 |
Glass transition temperature;
Cold crystallization temperature detected during the heating cycle;
Crystallization temperature detected during the cooling cycle;
First melting transition temperature;
Second melting transition temperature;
Melting enthalpy.
AFM, operated in PeakForce Tapping mode, was employed to obtain maps of sample modulus and the surface topography simultaneously using precisely controlled forces and carefully calibrated probes [61]. The height images (Figure 3A) of ECT2-CK, ECT2-AC and xECT2-AC show classical spherulite morphology for semicrystalline polymers, with the spherulites growing outwards from single nucleating points and meeting each other at the boundary (impingement) to give polygonal microstructure. In each spherulitic domain (Figure 3B), alternating amorphous interstitial and crystalline lamellae growing out from the crystal nucleus were also resolved distinctly. Such morphological features were further confirmed by the modulus map, where the crystalline lamellae appeared brighter (stiffer) than the surrounding amorphous regions. The boundary between individual spherulites appears more distinct in the height images (lower amplitude) than in the modulus maps. The spun-cast ECT2-CK films contained smaller spherulites surrounded by a larger planner structure. While the smaller domains appear brighter in the height image, their modulus is at the same magnitude as the surrounding regions. The spherulite morphology was preserved during crosslinking. Comparing the images acquired on ECT2-AC and xECT2-AC, one can clearly see that the average size of the spherulites is larger in ECT2-AC samples than in xECT2-AC films. The grain size differences between ECT2-AC and xECT2-AC samples can be attributed to the restricting power of the crosslinking reaction that prevents efficient polymer chain folding [62].
Figure 3.
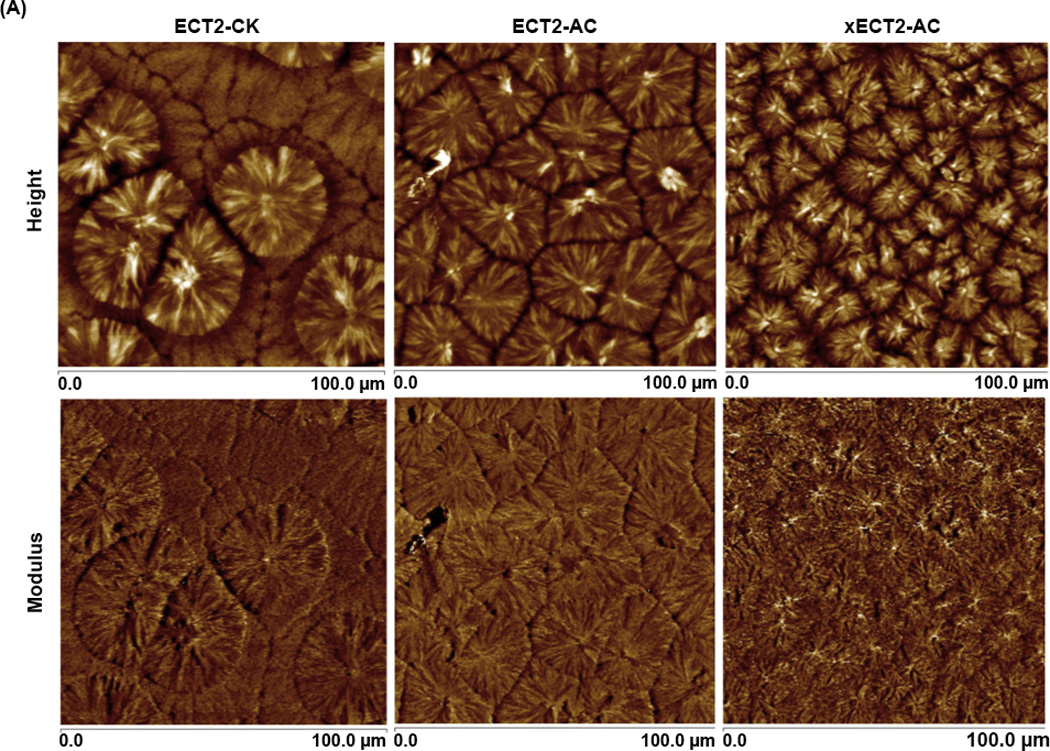
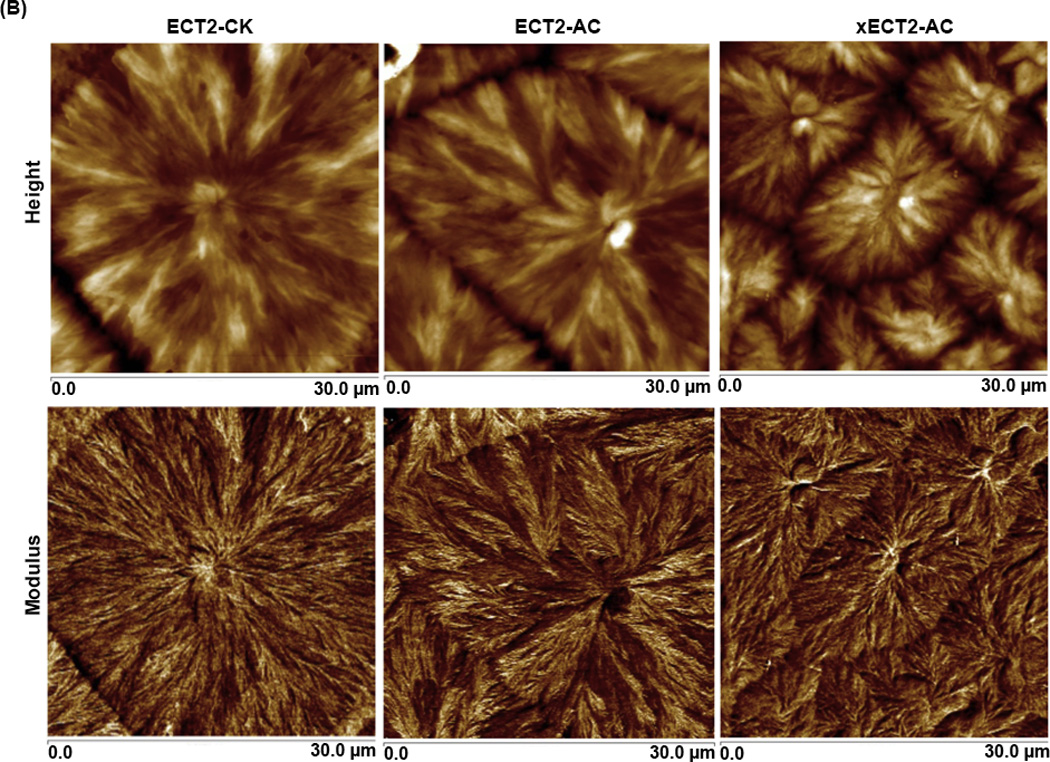
AFM height and modulus images of spun-cast polymer films at different magnifications (A: 100 µm×100 µm; B: 30 µm×30 µm).
The advantage of PeakForce QNM is the ability of the system to acquire and analyze the individual force curves from each tap that occurs during the imaging process, providing the real time mapping of materials properties. Obviously, local topography or roughness can have an impact on the measured moduli due to variation in contact area and all samples were spun cast to minimize the topographical variations [63]. The force-separation curves (Figure S4) for PCL, ECT2-CK and xECT2-AC were similar in shape, although ECT2-CK and xECT-AC are more energy dissipative, as evidenced by the area below the zero force reference. The force-separation curves were analyzed to obtain the Young’s modulus by fitting the 30%~90% of the retraction curve using the Hertzian model, taking into consideration the adhesive forces. Because of the effect of polymer chain orientation and semicrystallinity at the nanoscale, it is reasonable that a distribution of Young’s moduli would be measured (representative histograms are shown in Figure S4). From these data, the mean and associated standard deviations were calculated and these values are shown in Table 3. The significant decrease of AFM Young’s modulus from PCL (218.7 ± 41.6 MPa) to ECT2-CK (56.3 ± 1.7 MPa) can be attributed to the disruption of crystallinity by the bulky cyclic ketals. Compared to ECT2-CK, the commercial PCL is of a higher molecular weight, thus physical entanglement in PCL samples also contributes to the higher stiffness. xECT2-AC, on the other hand, had an AFM modulus value (47.3 ± 4.1) comparable to ECT2-CK.
Table 3.
Young’s modulus of PCL, ECT2-CK and xECT2-AC determined microscopically by PeakForce QNM analyses and macroscopically by uniaxial tensile tests.
| Young’s Modulus (MPa) | ||||
|---|---|---|---|---|
| Test Method | PCL | ECT2-CK | xECT2-AC1 | xECT2-AC2 |
| Tensile test | 123.0 ± 2.7 | 23.5 ± 2.8 | 10.0 ± 2.0 | 0.5 ± 0.1 |
| QNM | 218.7 ± 41.6 | 56.3 ± 1.7 | 47.3 ± 4.1 | N/A |
Tested at ambient temperature;
Tested at 37 °C.
The mechanical properties of various polymer samples were also evaluated macroscopically using DMA (Table 3). Consistent with the AFM observation, ECT2-CK was 5 times softer (Young’s modulus: 23.5 ± 2.8 MPa) than PCL (Young’s modulus: 123.0 ± 2.7 MPa). The crosslinked networks exhibit a Young’s modulus of 10.0 ± 2.0 MPa at room temperature, and become significantly softer at 37 °C (Young’s modulus: 0.7 ± 0.1 MPa) due to the partial melting of the microcrystals that contribute to the overall mechanics (Tm1=30 °C).
Comparing the Young’s moduli calculated from AFM QNM analyses with those obtained from macroscopic scale tensile tests, one can see that the former are consistently higher than the latter for all samples tested. Such discrepancies are not unreasonable considering that the QNM tests were conducted microscopically with deformation as little as 10 nm in compression mode (as opposed to 18~40 mm used in tension mode in macroscopic tests) on thin film samples with fewer defects (such as microscopic cracks) and free volume (related to the elastic modulus of polymers). Because the Young’s modulus is highly affected by the strain rate in many materials[64], the different loading rates in tensile and QNM tests may also contribute to the observed variations. Moreover, the finite size of the AFM tip used resulted in the crystalline and amorphous phases being probed separately; the reported modulus values are averaged mathematically. In all the samples, the nanoscale modulus, with respect to the polymer composition, followed the same trend as that obtained from the macroscale tensile tests, with PCL being the stiffest and xECT2-AC being the softest.
When subjected to uniaxial tension, the PCL films (Figure 4B) undergo a plastic deformation (a transition point from elastic to plastic deformation) with a distinct yield point at a strain of ~15% and an extended cold-drawing region (extension of folded chains in the crystalline domain) from 20–230%. ECT2-CK films are significantly more pliable although a similar plastic deformation (Figure 4A, yield point: 22%; cold drawing: 40–220%) was observed. When tested at ambient temperature, xECT2-AC is characterized by a gradual change of the slope from 9.6 MPa in the low strain region (<30%) to 1.6 MPa at the high strain region (>100%); the polymer is capable of sustaining large deformation until 500% strain when the instrument limit is reached. Because the crosslinked network is intended for use in human body temperature, tensile test was carried out at 37 °C. No distinct yield point or cold drawing region was detected. Instead, a compliant stress-strain response characteristic of elastomeric deformation was observed. A close inspection of the stress-strain curve shows a smooth change of slopes from a strain of 47% reaching a stress of 0.23 MPa at a strain of 400% (height limit of the temperature control chamber). At the body temperature when the crystalline domain was mostly melted, the rubbery polymer chains were held together by covalent crosslinks, effectively preventing polymer chains from sliding past one another, thus the yield point would disappear.
Figure 4.
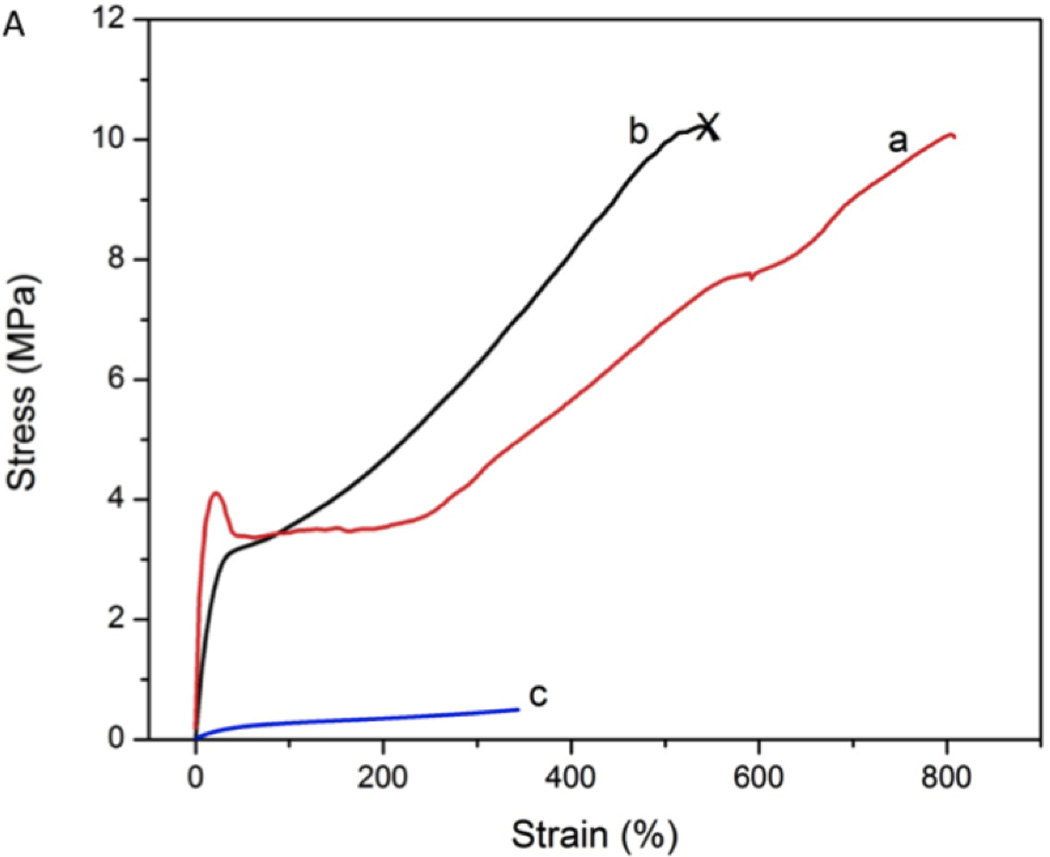

Tensile properties of ECT2-CK, ECT2-AC, xECT2-AC and commercial PCL as assessed by DMA. (A): Stress-strain curves for ECT2-CK (a), xECT2-AC at room temperature (b) and xECT2-AC at 37 °C (c). Except for c, all other testing materials reached the gap limit/chamber limit. (B): Stress-strain curve for PCL (d) plotted at a different scale. The test was terminated when the sample length reached the gap limit.
The Young’s modulus and the stress-strain behavior of xECT2-AC at 37°C are similar to previously reported biodegradable elastomers made from poly(glycerol-sebacate) (PGS) [65]. The Young’s modulus of xECT2-AC is also within the range of articular cartilage, skeletal and cardiac muscle [66]; while the strain to failure (at room temperature) is within the range of arteries and veins, higher than most of the tissues [67, 68]. xECT2-AC can be prepared using a well-defined macromer precursor, allowing facial fabrication of porous scaffolds via a photochemical process. These results collectively underscore the profound effects of the cyclic ketal groups and the covalent crosslinking on the physical and mechanical properties of the copolymers; a decrease in crystallinity is associated with an increase in chain flexibility, thus a lower modulus and more pliable behavior. Our synthetic procedures can be readily manipulated to obtain a variety of copolymers with different compositions and desired materials properties for targeted biomedical applications.
In cyclic tests at 37 °C, the xECT2-Ac samples were subjected to continuous loading and unloading processes up to 170% strain. The unloading curves followed a different path from the loading curves, creating a hysteresis loop (Figure 5) while consecutive cycles are perfectly overlapping and are independent of the cycle number. The network contains dynamically flexible chains that effectively dissipate energy during cyclic testing through the extension and elongation of the rubbery polymer chains between crosslinks. The unfolding of polymer chains in the crystalline domains is also possible, especially at large strains. At the end of each cycle when the sample was unloaded to zero strain, a negative stress developed in the sample and a waiting period had to be applied for the stress to return to zero. The residue stress on the xECT2-AC decays to half the original value within 12 s, and then slowly decays to zero over 1000 s in the absence of any changes (such as temperature) in the environment (Figure S5). After the samples were unloaded, the amorphous components immediately reverted to their original dimensions due to the covalent crosslinks, whereas the crystalline domains experienced semi-permanent deformations. The fact that the majority of the residual stress was recovered in a short time frame implies that only a small portion of polymer chains had to be refolded into the crystalline lamellae phase [50]. Alternatively, the retraction force generated by the crosslinked network may overcome the small permanent deformation of the crystalline regions, ultimately returning the samples to the original dimension over time. Overall, the chemical modifications have effectively converted a stiff, plastic material (PCL) to a soft network with elastomeric properties.
Figure 5.
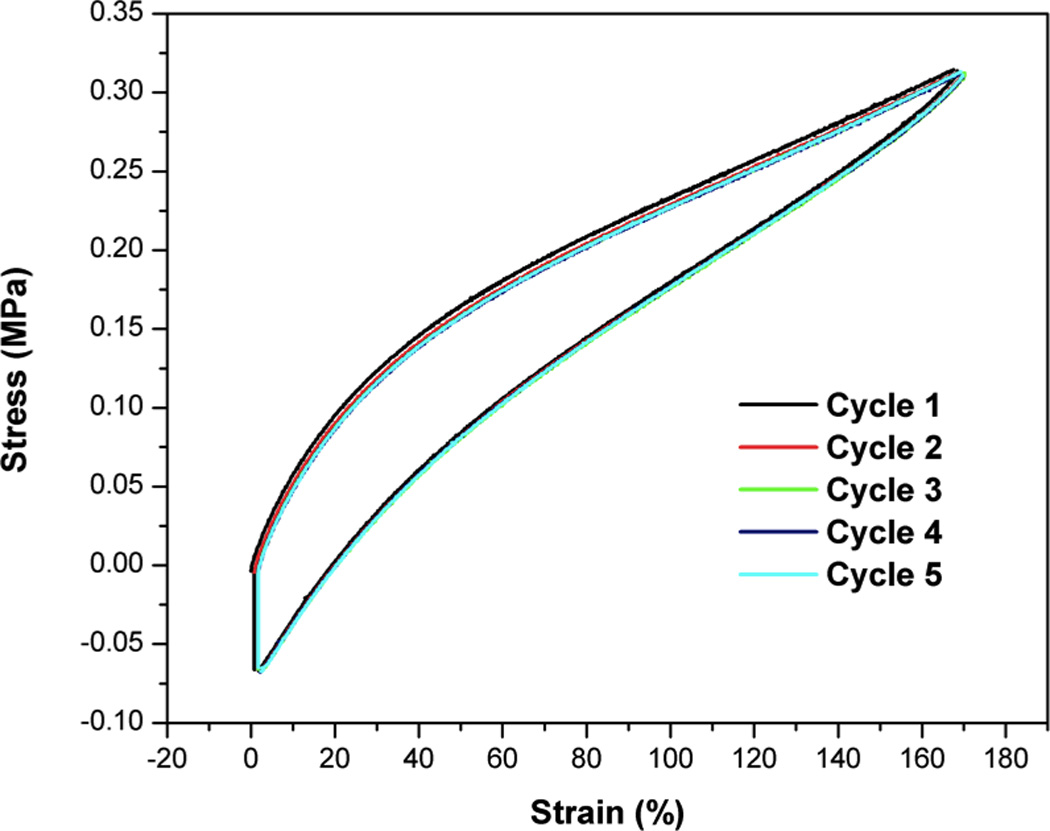
Cyclic tensile tests on xECT2-AC at 37°C. Five cycles are shown in this figure. After each unloading step, the sample was held at a constant strain for 1000 s.
Shape memory properties and cytocompatibility
The covalently crosslinked ECT2-AC networks exhibit elastomeric properties above Tm and contain microscopic crystalline domains that are temperature sensitive. Thus, xECT2-AC is expected to exhibit shape memory properties, with the memory effects originating from the covalent crosslinking points and the switchable segments being the microcrystals. Cyclic thermo-mechanical tensile tests were performed in the strain-controlled mode [40] to obtain shape recovery ratio and shape fixity ratio. In addition to the chemical composition and the density of covalent crosslinks in the network, the programming parameters, thermal conditions, kinetics, and type of mechanical deformation, all have a profound effect on the fixity and recoverability. In our initial screening studies, Thigh and εm were kept constant at 37 °C and 170%, respectively while Tlow was varied from −25 °C to 25 °C. Our results show that Rr was independent of Tlow and is generally >99%. Rf, on the other hand, was >97% if Tlow was ≤20 °C. At a Tlow of 25 °C, Rf deteriorated to as low as 24%. Clearly, the greater the difference between Tm and Tlow, the higher the thermodynamic driving force for crystallization is. Therefore, good shape-memory properties with strain recovery rates close to 100% could be obtained under stress-controlled programming at Tlow of <20 °C.
After the fine-tuning of the programming parameters, we chose Tlow of 10 and 20 °C for repetitive cyclic testing and the results are shown in Figure S6. Samples were stretched to a maximum strain (εm) of >300% during step 1. The deformation was fixed during the cooling cycle (step 2). In our system at such a large deformation, εu was remarkably close to εm. In step 4, a significant elastic recoil of the molten chains occurred and the network responded to the temperature change so rapidly that the DMA force transducer could not follow properly. As a result, a sudden jump of stress was observed. The stress eventually returned to zero when the system was fully equilibrated. Table 4 lists the Rf and Rr values for the crosslinked ECT2-AC networks. Both values were close to 100%, and were independent of the cycle times. The shape memory performance showed no deterioration over the 4–5 cycles performed, in that the curves are almost identical. Although a large number of shape memory polymers have been developed, very few of the existing materials are soft and elastomeric at the application temperature, albeit the clear evidence for demand. Many well-explored shape memory polymer networks can sustain very small deformation (<50%) and experience irreversible bond breakage during the shape programming process [69]. The unprecedented shape recovery and fixity of our materials at a relatively high strain can be attributed to the elastomeric nature of the polymer network.
Table 4.
Rr and Rf values for xECT2-AC network, determined by strain-controlled cyclic thermomechanical tests conducted at different Tlow. Thigh was maintained at 37 °C for all tests. εm was 360% for Tlow=10 °C and 353% for Tlow=20 °C.
| Cycle no. | Tlow=10 °C | Tlow=20 °C | ||
|---|---|---|---|---|
| Rf (%) | Rr (%) | Rf (%) | Rr (%) | |
| 1 | 99.5 | 99.9 | 97.6 | 99.9 |
| 2 | 99.5 | 99.5 | 97.6 | 99.8 |
| 3 | 99.9 | 100 | 97.9 | 99.9 |
| 4 | 100 | 99.6 | 98.1 | 99.7 |
| 5 | 100 | 100 | ||
The shape memory effect was further characterized by visual inspection of the shape memory process. The xECT2-AC polymer was firstly fixed in a flat stripe/coil shape as the temporary shape, and then it was immersed in a water bath at 37 °C. As seen in Figure 6, the xECT2-AC returned to its permanent shape (spiral/flat stripe) within a very short time (4s). A video recording of the respective recovery processes can be found in the supporting information. To our knowledge, our polyester network recovers faster and more completely at body temperature than the fastest self-expandable stents reported to date [41].
Figure 6.


Time series photographs showing the shape memory effect. Samples were dropped into a water batch that was maintained at 37 °C. (A): Recovery of the permanent spiral shape from a temporary rod shape in 3.3 s; (B): Recovery of the permanent rod shape from a temporary spiral shape in 1.1 s.
Finally, the cytocompatibility of xECT2-AC was evaluated by culturing MSCs in the presence of 20 mg/mL polymer samples in an indirect contact mode. Alamar blue assay results (Figure 7) showed that on either day 1 or day 3, except for negative control groups treated with Triton, all other group exhibit similar cell viability was the control group with no additives (NO samples). No statistical difference in terms of cell viability was detected for MSCs cultured in the presence of equal amount of PCL and xECT2-AC. xECT2-AC did not compromise the ability of MSCs to proliferate, and the cell number increased from day 1 to day 3 by 1.6 fold, compared to the NO and PCL controls. Note that the culture media was not refreshed during the culture. Also included in Figure 7 are representative live/dead staining results of all experimental groups at day 3. It is clear that cells exposed to xECT2-AC were highly viable and adopted a healthy, spindle-shaped morphology as the control cells. Collectively, our results confirmed non-toxic nature of the crosslinked elastomers.
Figure 7.
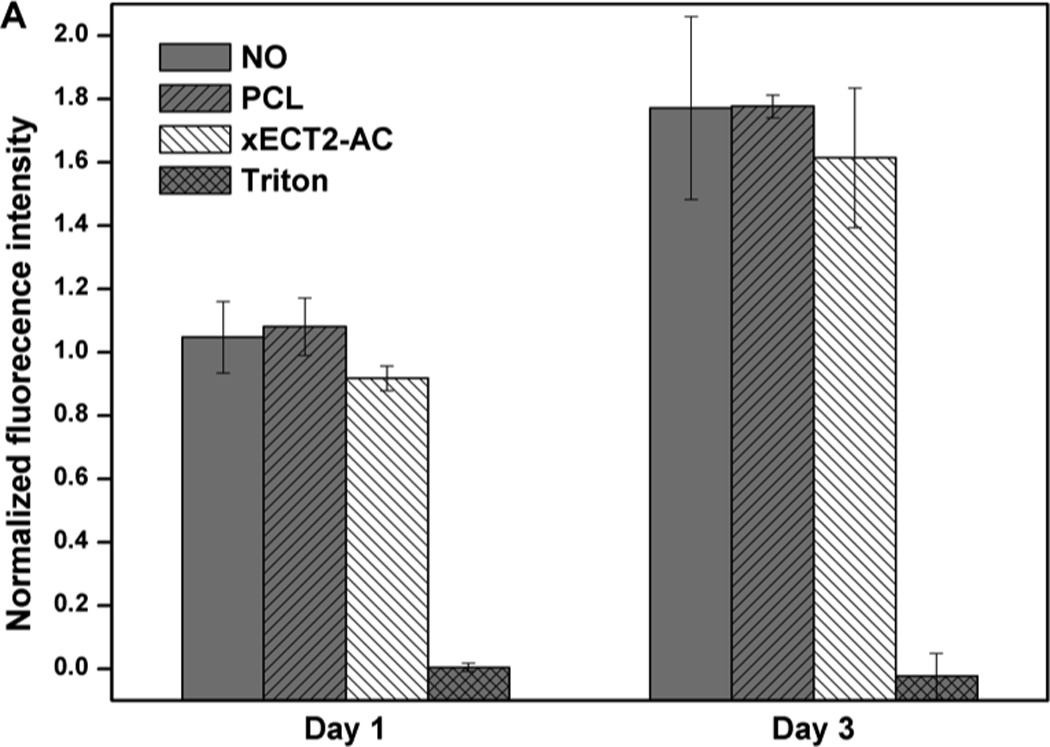
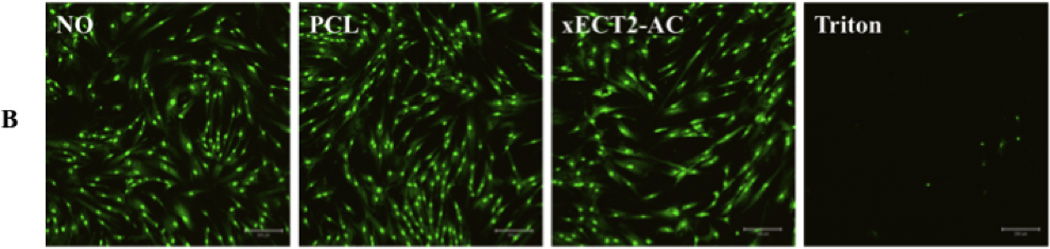
Cytotoxicity of xECT2-AC as assessed by Alamar blue assay (A) and live/dead assay (B). MSCs were cultured on tissue culture polystyrene without any additive (NO) or in the presence of 20 mg/mL PCL and xECT2-AC. Triton (0.2 mg/mL) was added as the negative control. (A): Alamar blue fluorescence intensity for each sample was normalized to the NO control on day 1. (B): Confocal microscopy images showing representative live/dead staining of MSCs cultured without any additive (NO), in the presence of 20 mg/mL PCL and xECT2-AC or 0.2 mg/mL Triton.
Conclusion
Functional PCL/PEG copolymers were synthesized by ring opening polymerization of CL and TSU using mPEG as the initiator and Sn(Oct)2 as the catalyst. Partial side chain deacetalization revealed the reactive ketone moieties without backbone scission. Reaction of hydroxylamine-functionalized acrylate with electrophilic ketones resulted in radically crosslinkable macromers. UV irradiation of ECT2-AC in DCM in the presence of photoinitiator resulted in pliable and elastomeric networks. DSC and AFM characterizations revealed the semicrystalline nature of the network. The inclusion of cyclic ketals and covalent crosslinking significantly reduced the polymer stiffness. While ECT2-CK underwent plastic deformation with a distinct yield point, xECT2-AC is elastomeric at body temperature. The presence of covalent crosslinking and microcrystalline domains endowed the network with shape memory properties. When properly programmed, shape fixity and recoverability as close as 1 were obtained. The viability and the proliferation of MSCs were not compromised in the presence of xECT2-AC. The novel materials are promising in versatile applications such as tissue engineering and minimal-invasive, endoscopic surgery.
Supplementary Material
Acknowledgments
We thank Roddel Remy for his help with DSC analysis, Longxi Xiao for his insight into the NMR results, and Tianzhi Luo for digital recording the shape memory behavior. Research reported in this paper was supported in part by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under grant number P20GM103541. C.R.S. was supported by the National Science Foundation EPSCoR Grant No. EPS-0814251 and the State of Delaware. The authors also thank the National Science Foundation (DMR-1206310) for financial support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Griffith LG. Polymeric biomaterials. Acta Materialia. 2000;48:263–277. [Google Scholar]
- 2.Williams DF. On the nature of biomaterials. Biomaterials. 2009;30:5897–5909. doi: 10.1016/j.biomaterials.2009.07.027. [DOI] [PubMed] [Google Scholar]
- 3.Jagur-Grodzinski J. Polymers for tissue engineering, medical devices, and regenerative medicine. Concise general review of recent studies. Polymers for Advanced Technologies. 2006;17:395–418. [Google Scholar]
- 4.Butler DL, Goldstein SA, Guilak F. Functional tissue engineering: The role of biomechanics. Journal of Biomechanical Engineering. 2000;122:570–575. doi: 10.1115/1.1318906. [DOI] [PubMed] [Google Scholar]
- 5.Tong Z, Jia X. Biomaterial-based strategies for the engineering of mechanically active soft tissues. MRS Communications. 2012;2:31–39. doi: 10.1557/mrc.2012.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim BS, Mooney DJ. Scaffolds for engineering smooth muscle under cyclic mechanical strain conditions. Journal of Biomechanical Engineering-Transactions of the Asme. 2000;122:210–215. doi: 10.1115/1.429651. [DOI] [PubMed] [Google Scholar]
- 7.Niklason LE, Gao J, Abbott WM, Hirschi KK, Houser S, Marini R, et al. Functional arteries grown in vitro. Science. 1999;284:489–493. doi: 10.1126/science.284.5413.489. [DOI] [PubMed] [Google Scholar]
- 8.Waldman SD, Spiteri CG, Grynpas MD, Pilliar RM, Hong J, Kandel RA. Effect of biomechanical conditioning on cartilaginous tissue formation in vitro. Journal of Bone and Joint Surgery-American Volume. 2003;85A:101–105. doi: 10.2106/00004623-200300002-00013. [DOI] [PubMed] [Google Scholar]
- 9.Webb AR, Yang J, Ameer GA. Biodegradable polyester elastomers in tissue engineering. Expert Opinion on Biological Therapy. 2004;4:801–812. doi: 10.1517/14712598.4.6.801. [DOI] [PubMed] [Google Scholar]
- 10.Amsden BG, Misra G, Gu F, Younes HM. Synthesis and characterization of a photo-crosslinked biodegradable elastomer. Biomacromolecules. 2004;5:2479–2486. doi: 10.1021/bm049578h. [DOI] [PubMed] [Google Scholar]
- 11.Dash TK, Konkimalla VB. Poly-ε-caprolactone based formulations for drug delivery and tissue engineering: A review. Journal of Controlled Release. 2012;158:15–33. doi: 10.1016/j.jconrel.2011.09.064. [DOI] [PubMed] [Google Scholar]
- 12.Woodruff MA, Hutmacher DW. The return of a forgotten polymer-Polycaprolactone in the 21st century. Progress in Polymer Science. 2010;35:1217–1256. [Google Scholar]
- 13.Gunatillake P, Mayadunne R, Adhikari R. Recent developments in biodegradable synthetic polymers. Biotechnology Annual Review. 2006;12:301–347. doi: 10.1016/S1387-2656(06)12009-8. [DOI] [PubMed] [Google Scholar]
- 14.Albertsson AC, Varma IK. Recent developments in ring opening polymerization of lactones for biomedical applications. Biomacromolecules. 2003;4:1466–1486. doi: 10.1021/bm034247a. [DOI] [PubMed] [Google Scholar]
- 15.Wang XY, Gurski LA, Zhong S, Xu XA, Pochan DJ, Farach-Carson MC, et al. Amphiphilic block co-polyesters bearing pendant cyclic ketal groups as nanocarriers for controlled release of camptothecin. Journal of Biomaterials Science-Polymer Edition. 2011;22:1275–1298. doi: 10.1163/092050610X504260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pitt CG, Gratzl MM, Kimmel GL, Surles J, Schindler A. Aliphatic polyesters II. The degradation of poly (DL-lactide), poly (epsilon-caprolactone), and their copolymers in vivo. Biomaterials. 1981;2:215–220. doi: 10.1016/0142-9612(81)90060-0. [DOI] [PubMed] [Google Scholar]
- 17.Pitt CG, Chasalow FI, Hibionada YM, Klimas DM, Schindler A. Aliphatic polyesters. I. The degradation of poly(ε-caprolactone) in vivo. Journal of Applied Polymer Science. 1981;26:3779–3787. [Google Scholar]
- 18.Eastmond GC. Poly(epsilon-caprolactone) blends. Biomedical Applications: Polymer Blends. 1999;149:59–223. [Google Scholar]
- 19.Cipitria A, Skelton A, Dargaville TR, Dalton PD, Hutmacher DW. Design, fabrication and characterization of PCL electrospun scaffolds-a review. Journal of Materials Chemistry. 2011;21:9419–9453. [Google Scholar]
- 20.Detrembleur C, Mazza M, Lou X, Halleux O, Lecomte P, Mecerreyes D, et al. New functional aliphatic polyesters by chemical modification of copolymers of epsilon-caprolactone with gamma-(2-bromo-2-methylpropionate)-epsilon-caprolactone, gamma-bromo-epsilon-caprolactone, and a mixture of beta- and gamma-ene-epsilon-caprolactone. Macromolecules. 2000;33:7751–7760. [Google Scholar]
- 21.Lou XD, Detrembleur C, Jerome R. Novel aliphatic polyesters based on functional cyclic (di)esters. Macromolecular Rapid Communications. 2003;24:161–172. [Google Scholar]
- 22.Lenoir S, Riva R, Lou X, Detrembleur C, Jerome R, Lecomte P. Ring-opening polymerization of alpha-chloro-is an element of-caprolactone and chemical modification of poly(alpha-chloro-is an element of-caprolactone) by atom transfer radical processes. Macromolecules. 2004;37:4055–4061. [Google Scholar]
- 23.Riva R, Schmeits S, Jerome C, Jerome R, Lecomte P. Combination of ring-opening polymerization and "click chemistry": Toward functionalization and grafting of poly(epsilon-caprolactone) Macromolecules. 2007;40:796–803. [Google Scholar]
- 24.Tian D, Dubois P, Jerome R, Teyssie P. Macromolecular engineering of polylactones and polylactides .18. Synthesis of star-branched aliphatic polyesters bearing various functional end-groups. Macromolecules. 1994;27:4134–4144. [Google Scholar]
- 25.Kim HW, Knowles JC, Kim HE. Hydroxyapatite/poly(epsilon-caprolactone) composite coatings on hydroxyapatite porous bone scaffold for drug delivery. Biomaterials. 2004;25:1279–1287. doi: 10.1016/j.biomaterials.2003.07.003. [DOI] [PubMed] [Google Scholar]
- 26.Rhee SH. Bone-like apatite-forming ability and mechanical properties of poly(epsilon-caprolactone)/silica hybrid as a function of poly(epsilon-caprolactone) content. Biomaterials. 2004;25:1167–1175. doi: 10.1016/j.biomaterials.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 27.Chen BQ, Evans JRG. Poly(epsilon-caprolactone)-clay nanocomposites: Structure and mechanical properties. Macromolecules. 2006;39:747–754. [Google Scholar]
- 28.Habibi Y, Goffin AL, Schiltz N, Duquesne E, Dubois P, Dufresne A. Bionanocomposites based on poly(epsilon-caprolactone)-grafted cellulose nanocrystals by ring-opening polymerization. Journal of Materials Chemistry. 2008;18:5002–5010. [Google Scholar]
- 29.Cohn D, Salomon AF. Designing biodegradable multiblock PCL/PLA thermoplastic elastomers. Biomaterials. 2005;26:2297–2305. doi: 10.1016/j.biomaterials.2004.07.052. [DOI] [PubMed] [Google Scholar]
- 30.Kylma J, Seppala JV. Synthesis and characterization of a biodegradable thermoplastic poly(ester-urethane) elastomer. Macromolecules. 1997;30:2876–2882. [Google Scholar]
- 31.Lee SH, Kim BS, Kim SH, Choi SW, Jeong SI, Kwon IK, et al. Elastic biodegradable poly(glycolide-co-caprolactone) scaffold for tissue engineering. Journal of Biomedical Materials Research Part A. 2003;66A:29–37. doi: 10.1002/jbm.a.10497. [DOI] [PubMed] [Google Scholar]
- 32.Pego AP, Poot AA, Grijpma DW, Feijen J. Biodegradable elastomeric scaffolds for soft tissue engineering. Journal of Controlled Release. 2003;87:69–79. doi: 10.1016/s0168-3659(02)00351-6. [DOI] [PubMed] [Google Scholar]
- 33.Iha RK, Van Horn BA, Wooley KL. Complex, Degradable Polyester Materials via Ketoxime Ether-Based Functionalization: Amphiphilic, Multifunctional Graft Copolymers and Their Resulting Solution-State Aggregates. Journal of Polymer Science Part a-Polymer Chemistry. 2010;48:3553–3563. [Google Scholar]
- 34.Barrett DG, Yousaf MN. Preparation of a class of versatile, chemoselective, and amorphous polyketoesters. Biomacromolecules. 2008;9:2029–2035. doi: 10.1021/bm800271f. [DOI] [PubMed] [Google Scholar]
- 35.Koenig MF, Huang SJ. Biodegradable blends and composites of polycaprolactone and starch derivatives. Polymer. 1995;36:1877–1882. [Google Scholar]
- 36.Broz ME, VanderHart DL, Washburn NR. Structure and mechanical properties of poly(D,L-lactic acid)/poly(epsilon-caprolactone) blends. Biomaterials. 2003;24:4181–4190. doi: 10.1016/s0142-9612(03)00314-4. [DOI] [PubMed] [Google Scholar]
- 37.Kweon H, Yoo MK, Park IK, Kim TH, Lee HC, Lee HS, et al. A novel degradable polycaprolactone networks for tissue engineering. Biomaterials. 2003;24:801–808. doi: 10.1016/s0142-9612(02)00370-8. [DOI] [PubMed] [Google Scholar]
- 38.Wang SF, Lu LC, Gruetzmacher JA, Currier BL, Yaszemski MJ. Synthesis and characterizations of biodegradable and crosslinkable poly(epsilon-caprolactone fumarate), poly(ethylene glycol fumarate), and their amphiphilic copolymer. Biomaterials. 2006;27:832–841. doi: 10.1016/j.biomaterials.2005.07.013. [DOI] [PubMed] [Google Scholar]
- 39.Wang SF, Yaszemski MJ, Gruetzmacher JA, Lu LC. Photo-crosslinked poly(epsilon-caprolactone fumarate) networks: Roles of crystallinity and crosslinking density in determining mechanical properties. Polymer. 2008;49:5692–5699. doi: 10.1016/j.polymer.2008.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lendlein A, Kelch S. Shape-memory polymers. Angewandte Chemie International Edition. 2002;41:2034–2057. [PubMed] [Google Scholar]
- 41.Xue L, Dai S, Li Z. Biodegradable shape-memory block co-polymers for fast self-expandable stents. Biomaterials. 2010;31:8132–8140. doi: 10.1016/j.biomaterials.2010.07.043. [DOI] [PubMed] [Google Scholar]
- 42.Tessmar JK, Göpferich AM. Customized PEG-Derived Copolymers for Tissue-Engineering Applications. Macromolecular Bioscience. 2007;7:23–39. doi: 10.1002/mabi.200600096. [DOI] [PubMed] [Google Scholar]
- 43.Takeshita H, Fukumoto K, Ohnishi T, Ohkubo T, Miya M, Takenaka K, et al. Formation of lamellar structure by competition in crystallization of both components for crystalline-crystalline block copolymers. Polymer. 2006;47:8210–8218. [Google Scholar]
- 44.McPherson T, Kidane A, Szleifer I, Park K. Prevention of Protein Adsorption by Tethered Poly(ethylene oxide) Layers: Experiments and Single-Chain Mean-Field Analysis. Langmuir. 1998;14:176–186. [Google Scholar]
- 45.Sheth SR, Leckband D. Measurements of attractive forces between proteins and end-grafted poly(ethylene glycol) chains. Proceedings of the National Academy of Sciences. 1997;94:8399–8404. doi: 10.1073/pnas.94.16.8399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bhat R, Timasheff SN. Steric exclusion is the principal source of the preferential hydration of proteins in the presence of polyethylene glycols. Protein Science. 1992;1:1133–1143. doi: 10.1002/pro.5560010907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tian D, Dubois P, Grandfils C, Jerome R. Ring-opening polymerization of 1,4,8-trioxaspiro 4.6 -9-undecanone: A new route to aliphatic polyesters bearing functional pendent groups. Macromolecules. 1997;30:406–409. [Google Scholar]
- 48.Neises B, Steglich W. 4-Dialkylaminopyridines as acylation catalysts .5. Simple method for esterification of carboxylic-acids. Angewandte Chemie-International Edition in English. 1978;17:522–524. [Google Scholar]
- 49.Van Horn BA, Iha RK, Wooley KL. Sequential and single-step, one-pot strategies for the transformation of hydrolytically degradable polyesters into multifunctional systems. Macromolecules. 2008;41:1618–1626. [Google Scholar]
- 50.Wagermaier W, Kratz K, Heuchel M, Lendlein A. Shape-memory polymers. Berlin: Springer-Verlag Berlin; 2010. Characterization methods for shape-memory polymers; pp. 97–145. [Google Scholar]
- 51.Tian D, Dubois P, Jerome R. Macromolecular engineering of polylactones and polylactides .22. Copolymerization of epsilon-caprolactone and 1,4,8-trioxaspiro 4.6 -9-undecanone initiated by aluminum isopropoxide. Macromolecules. 1997;30:2575–2581. [Google Scholar]
- 52.Krishnan V, Xu X, Barwe SP, Yang X, Czymmek K, Waldman SA, et al. Dexamethasone-loaded block copolymer nanoparticles induce leukemia cell death and enhance therapeutic efficacy: A novel application in pediatric nanomedicine. Molecular Pharmaceutics. 2012 doi: 10.1021/mp300350e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Smith MB. Organic chemistry: An acid-base approach. Boca Raton: CRC press; 2010. [Google Scholar]
- 54.Tian D, Dubois P, Jerome R. Macromolecular engineering of polylactones and polylactides .23. Synthesis and characterization of biodegradable and biocompatible homopolymers and block copolymers based on 1,4,8-trioxa 4.6 spiro-9-undecanone. Macromolecules. 1997;30:1947–1954. [Google Scholar]
- 55.Dwan'Isa JPL, Lecomte P, Dubois P, Jerome R. Hydrolytic and thermal degradation of random copolyesters of epsilon-caprolactone and 2-oxepane-1,5-dione. Macromolecular Chemistry and Physics. 2003;204:1191–1201. [Google Scholar]
- 56.Lynd NA, Meuler AJ, Hillmyer MA. Polydispersity and block copolymer self-assembly. Progress in Polymer Science. 2008;33:875–893. [Google Scholar]
- 57.Bei JZ, He WS, Hu XZ, Wang SG. Photodegradation behavior and mechanism of block copoly(caprolactone-ethylene glycol) Polymer Degradation and Stability. 2000;67:375–380. [Google Scholar]
- 58.Moreau JL, Kesselman D, Fisher JP. Synthesis and properties of cyclic acetal biomaterials. Journal of Biomedical Materials Research Part A. 2007;81A:594–602. doi: 10.1002/jbm.a.31104. [DOI] [PubMed] [Google Scholar]
- 59.Martens P, Anseth KS. Characterization of hydrogels formed from acrylate modified poly(vinyl alcohol) macromers. Polymer. 2000;41:7715–7722. [Google Scholar]
- 60.Khonakdar HA, Morshedian J, Wagenknecht U, Jafari SH. An investigation of chemical crosslinking effect on properties of high-density polyethylene. Polymer. 2003;44:4301–4309. [Google Scholar]
- 61.Schön P, Bagdi K, Molnár K, Markus P, Pukánszky B, Julius Vancso G. Quantitative mapping of elastic moduli at the nanoscale in phase separated polyurethanes by AFM. European Polymer Journal. 2011;47:692–698. [Google Scholar]
- 62.Sperling LH. Introduction to physical polymer science. 4 ed. Hoboken, NJ: John Wiley & Sons, Inc.; 2005. [Google Scholar]
- 63.Walheim S, Böltau M, Mlynek J, Krausch G, Steiner U. Structure Formation via Polymer Demixing in Spin-Cast Films. Macromolecules. 1997;30:4995–5003. [Google Scholar]
- 64.Mulliken AD, Boyce MC. Mechanics of the rate-dependent elastic–plastic deformation of glassy polymers from low to high strain rates. International Journal of Solids and Structures. 2006;43:1331–1356. [Google Scholar]
- 65.Wang YD, Ameer GA, Sheppard BJ, Langer R. A tough biodegradable elastomer. Nature Biotechnology. 2002;20:602–606. doi: 10.1038/nbt0602-602. [DOI] [PubMed] [Google Scholar]
- 66.Levental I, Georges PC, Janmey PA. Soft biological materials and their impact on cell function. Soft Matter. 2007;3:299–306. doi: 10.1039/b610522j. [DOI] [PubMed] [Google Scholar]
- 67.Lee MC, Haut RC. Strain rate effects on tensile failure properties of the common carotid-artery and jugular veins of ferrets. Journal of Biomechanics. 1992;25:925–927. doi: 10.1016/0021-9290(92)90232-p. [DOI] [PubMed] [Google Scholar]
- 68.Haut RC. The effect of a lathyritic diet on the sensitivity of tendon to strain rate. Journal of Biomechanical Engineering-Transactions of the Asme. 1985;107:166–174. doi: 10.1115/1.3138537. [DOI] [PubMed] [Google Scholar]
- 69.Choi NY, Lendlein A. Degradable shape-memory polymer networks from oligo (L-lactide)-ran-glycolide dimethacrylates. Soft Matter. 2007;3:901–909. doi: 10.1039/b702515g. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.