

Abstract
The variable efficacy of tuberculosis (TB) vaccines and the emergence of drug resistant strains of Mycobacterium tuberculosis (Mtb) emphasize the urgency for not only generating new and more effective vaccines against TB, but also understanding the underlying mechanisms that mediate vaccine-induced protection. We demonstrate that mucosal adjuvants, such as type II heat labile enterotoxin (LT-IIb), delivered through the mucosal route induce pulmonary Mtb-specific T helper 17 (Th17) responses and provide vaccine-induced protection against Mtb infection. Importantly, protection is Interferon-γ(IFNγ)-independent but Interleukin (IL-17)-dependent. Our data show that IL-17 mediates CXCL13 induction in the lung for strategic localization of pro-inflammatory cytokine-producing CXCR5+ T cells within lymphoid structures, thereby promoting early and efficient macrophage activation and the control of Mtb. Our studies highlight the potential value of targeting the IL-17-CXCL13 pathway rather than the IFNγ pathway as a new strategy to improve mucosal vaccines against TB.
Keywords: IL-17, Mucosal-induced immunity, Tuberculosis, Homeostatic chemokines, LT-IIb
Introduction
Mycobacterium tuberculosis (Mtb), the causative agent of tuberculosis (TB), infects one third of the world’s population and kills more than 1.7 million people worldwide every year. Although most infected individuals develop latent TB, the disease will reactivate in 5 to 10% of latently infected individuals. Therefore, efforts to design improved vaccines against TB are a research priority. Primary immunity to Mtb infection in humans and mice is thought to be dependent on type 1 T helper (Th1) cells that produce Interferon gamma (IFNγ) and Tumor Necrosis Factor alpha (TNFα); both cytokines which activate macrophages within the granulomas to control intracellular Mtb growth1. However, current vaccination approaches that target Th1 responses have not been successful in enhancing protection against Mtb2–4, suggesting that additional pathways may need to be targeted to improve vaccine-induced immunity to TB.
The TB granuloma is formed in response to Mtb infection in the lung and is an essential component of immune protection. Interestingly, active TB granulomas contain organized lymphoid structures known as inducible Bronchus Associated Lymphoid Tissue (iBALT) and induction of iBALT is mediated in part by the expression of CXCL135–8, which controls the formation of B cell follicles, T cell placement and the optimal activation of macrophages for Mtb control6,8. Furthermore, pulmonary CXCL13 expression and iBALT formation are controlled by IL-17 7. Therefore, despite a dispensable role for IL-23 and IL-17 in primary immunity to TB9, IL-23-dependent T helper type 17 (Th17) cells play a key role in Th1 recruitment and vaccine-induced protection in a parenteral model of vaccine-induced immunity to Mtb infection10. Some mucosal vaccines are thought to provide superior protection against Mtb when compared to parenteral vaccination11–13; however the absolute requirement of the Th1 and Th17 pathways and the mechanism that mediates mucosal vaccine-induced protection against Mtb challenge is not known. In the current study, we demonstrate that the mucosal adjuvant, type II heat labile enterotoxin (LT-IIb)14, when delivered mucosally with an Mtb-specific antigen, induces pulmonary Th17 responses and Th1 responses and provides vaccine-induced protection against Mtb. Importantly, we show that vaccine-induced immunity against Mtb infection is IFNγ-independent and in Ifng−/− mice, is dependent on IL-17. We also show that Th17 responses in vaccinated mice are associated with IL-17-dependent induction of CXCL13 within the lung parenchyma, promoting lymphoid structure formation and organization, strategic positioning of T cells within the lung and maximal macrophage activation. These findings demonstrate that the immune mechanisms involved in successful primary and recall responses to TB are distinct and provide new evidence that targeting the IL-17-CXCL13 pathway by mucosal vaccination has the potential to improve vaccine-strategies against TB.
Results
IL-17, but not IFNγ, is critical for mucosal vaccine-induced immunity against Mtb
Given that some mucosal vaccines are thought to mediate superior protection against Mtb when compared to parenteral vaccination routes11–13; it is critical to determine if Th1 or Th17 responses or both are required to mediate mucosal vaccine-induced protection against TB. Wild type C57BL/6J (B6) mice intranasally vaccinated with an Mtb immunodominant I-Ab restricted peptide, Early Secreted Antigenic Target (ESAT61–20) in combination with LT-IIb, a potent and well-characterized mucosal adjuvant14, exhibited protection when challenged with Mtb when compared to sham vaccinated Mtb-challenged mice (Figure 1a). Importantly, protection was maintained following Mtb challenge in mucosally vaccinated mice that were rested long term (>100 days) (Figure 1b). We found that protection in mucosally vaccinated Mtb-challenged lungs was associated with inflammatory lesions associated with well organized lymphoid structures harboring distinct B cell follicles interspersed with CD3+ T cells (Figure 1c). Furthermore, protection detected in mucosally vaccinated Mtb-challenged mice was dependent on generation of Mtb-specific CD4+ T cells, since mice mucosally vaccinated with ESAT61–20 peptide with LT-IIb, but not sham vaccinated mice were protected when challenged with Mtb (Figure 1a). Consistent with this, we found that mucosal boost-regimes with ESAT61–20 in LT-IIb primed robust lung-resident ESAT61–20-specific Th17 cell responses, while inducing a small but detectable lung-resident ESAT61–20-specific Th1 response (Figure 1d). In addition, enhanced lung-resident memory Th17 responses were maintained until day 100 post immunization in lungs of mucosally vaccinated mice (Figure 1e). Upon challenge with Mtb, we found that a significant population of activated CD4+ T cells that accumulated early in the lungs of mucosally vaccinated mice produced IL-17 (Figure S1a), TNFα (Figure S1b), and coproduced IFNγ and TNF-α (Figure S1c); interestingly, the activated CD4+ IFNγ+ cell producing population did not increase significantly in the lung (Figure S1d). In addition, we found that activated CD4+ T cells that accumulated early in the lungs of vaccinated Mtb-challenged mice also expressed the chemokine receptors CXCR3 and CXCR5 (Figure S1e–f). On day 15 post Mtb challenge, we found higher ESAT61–20-specific Th17 cell responses when compared to Th1 cell responses (Figure 1f), and this coincided with higher induction of IL-17 mRNA when compared to IFNγ mRNA in cells isolated from mucosally vaccinated Mtb-challenged lungs (Figure 1g). Together, these data suggest that mucosal vaccination with ESAT61–20 and LT-IIb induces robust Th17 recall responses, enhances lymphocytic infiltration into lung parenchyma and mediates vaccine-induced protection against Mtb challenge.
Figure 1. Mucosal vaccination induces vaccine-induced protection and robust Th17 responses following Mtb challenge.
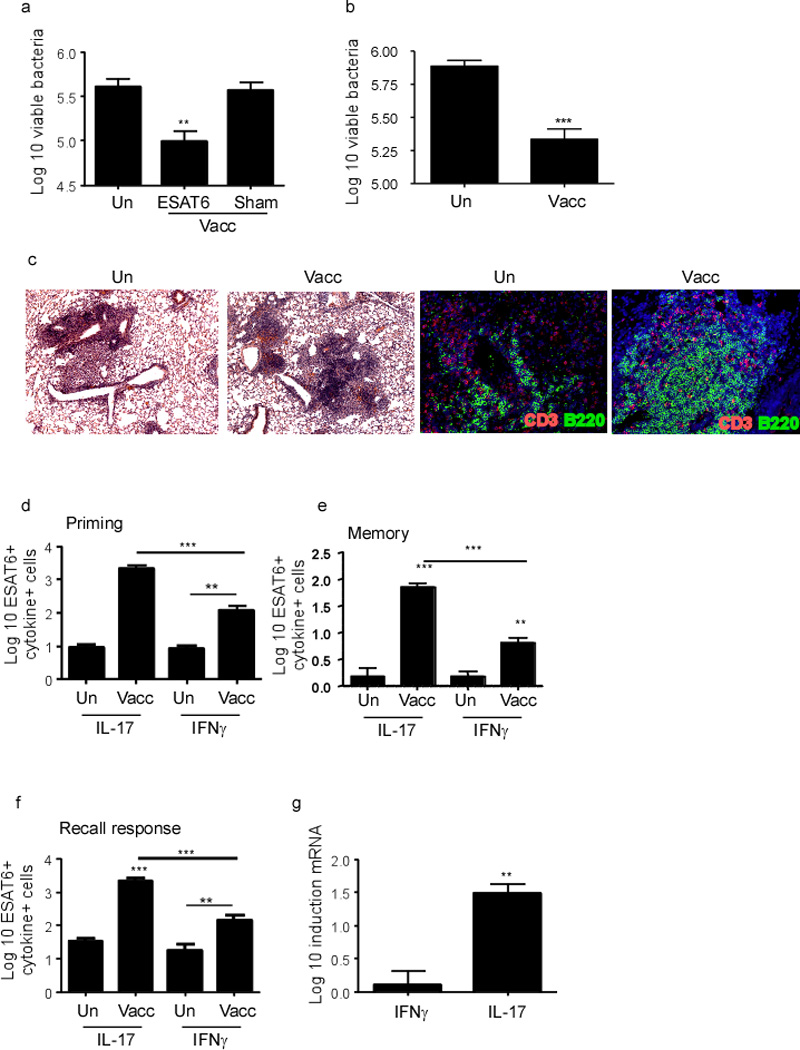
B6 mice were vaccinated mucosally vaccinated and boosted with ESAT61–20 in combination with LT-IIb (Vacc-ESAT6) or sham vaccinated (Vacc-Sham) via the intranasal route. Control unvaccinated mice were also included (Un). Mucosally vaccinated B6 mice were rested for 30 (a) days or 100 days (b), infected with aerosolized Mtb (100 cfu) and lung bacterial burden was determined on day 30 post infection. On day 30 post infection, lungs were fixed in 10% formalin, embedded in paraffin and inflammatory lesions and lymphoid structure formation were assessed in formalin-fixed lungs by staining with H&E (c-left panel); or CD3 (red) and B220 (green) (c-right panel). Original magnification for H&E sections, 100X and immunofluorescence staining, 200X. B6 mice were mucosally vaccinated as described before and the number of cytokine-producing ESAT61–20-specific CD4+ T cells in the lungs were determined by ELISpot assay on day 14 (d) or day 100 post-vaccination (e) or in day 15 post Mtb-challenge (f) by antigen-driven ELISpot assay. The Log10 fold induction of IFNγ and IL-17 mRNA was determined in cells isolated from mucosally vaccinated Mtb-challenged lungs when compared to levels expressed in cell isolated from unvaccinated Mtb-challenged lungs by RT-PCR (g). The data points represent the mean (±SD) of values from 4–6 mice (a–g). **≤0.005, ***p≤0.0005. One experiment representative of two is shown.
To determine whether IL-17 or IFNγ production by vaccine-induced CD4+ T cells was important for mucosal vaccine-induced protection, we next mucosally vaccinated Ifng−/− and Il17−/− mice and challenged them with Mtb. We found that, even though Ifng−/− mice had higher Mtb burden in the lungs, mucosal vaccination provided protection upon challenge at day 30 post challenge (Figure 2a). In addition, vaccine induced protection was maintained in Ifng−/− vaccinated mice until day 45 (Figure 2b), at which point the unvaccinated Ifng−/− mice died. Vaccine-induced protection was maintained until day 60 in B6 vaccinated mice (Figure 2c), contrasting with the lack of protection at both early and time points in Il17−/− mucosally vaccinated mice (Figure 2a,c). Furthermore, vaccinated Ifng−/− mice exhibited robust Th17 recall responses (Figure 2d). In contrast, even though Il17−/− mice generated a normal Th1 recall response (Figure 2e), they did not generate vaccine-induced immunity against Mtb infection (Figure 2a,c). A role for IL-17 in vaccine-induced immunity to Mtb was also confirmed by neutralizing IL-17 in mucosally vaccinated B6 mice (Figure 2f). In addition, mucosal vaccination of mice that lack STAT3 in CD4+ T cells (Stat3.Cd4−/− mice) resulted in induction of antigen-specific Th1 responses (IFNγ and TNFα) but not lung-resident Th17 responses (Figure 2g). Coincident with lack of induction of Th17 responses in Stat3.Cd4−/− mice, mucosal vaccination did not confer vaccine-induced immunity upon Mtb challenge (Figure 2h). Finally, we show that neutralization of IL-17 in Ifng−/− vaccinated mice also resulted in loss of vaccine-induced protection (Figure 2i). Therefore, our data indicate that, despite a requirement for IFNγ in protection against primary Mtb infection15–16, targeting IFNγ in mucosal vaccine-strategies may not be beneficial for improving protection against Mtb infection. Instead, our data show that IL-17 is a critical mediator of vaccine-induced immune protection following Mtb infection, and that targeting the IL-17 pathway is likely to improve existing vaccine strategies against Mtb.
Figure 2. IL-17, but not IFNγ is crucial for vaccine-induced mucosal immunity against Mtb infection.
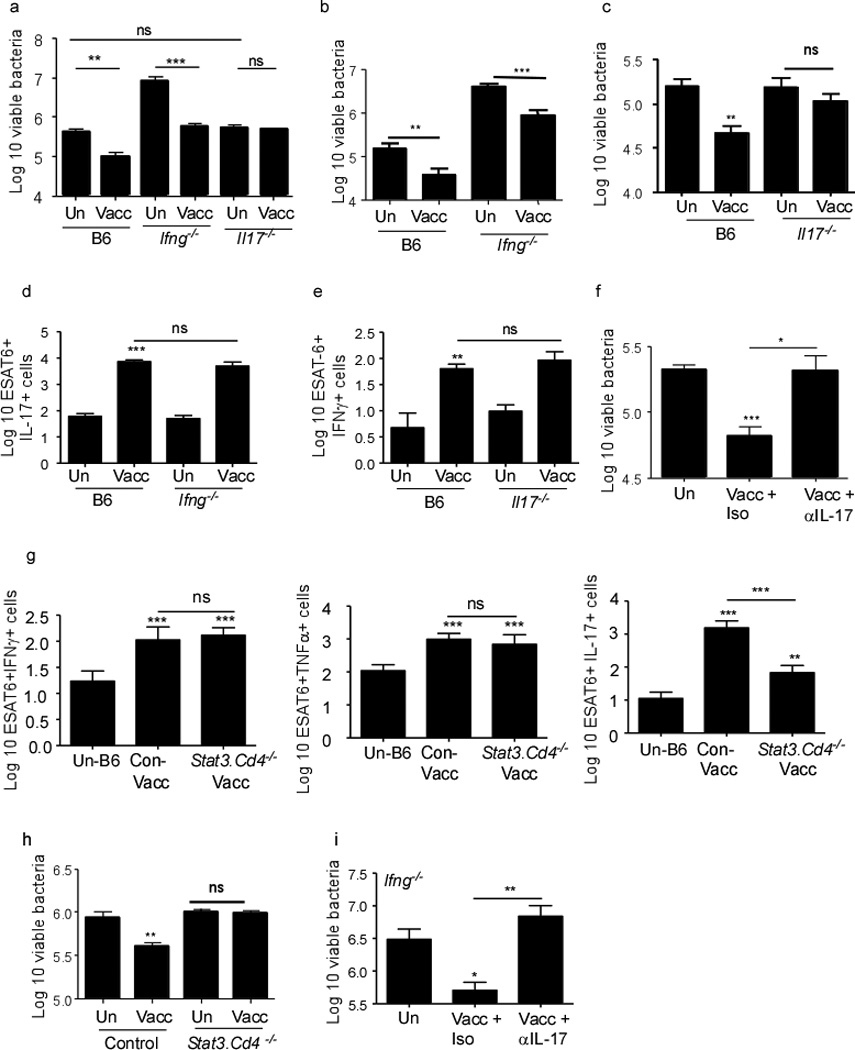
B6, Ifng−/− and Il17−/− were left unvaccinated (Un) or mucosally vaccinated (Vacc) with ESAT61–20 in combination with LT-IIb and rested for 30 days. Subsequently mice were challenged with ~100 CFU Mtb by the aerosol route and lung bacterial burden was determined on day 30 post infection (a), day 45 (b) or day 60 (c). The number of cytokine-producing ESAT61–20-specific CD4+ T cells in the lungs were determined by ELISpot in B6 and Ifng−/− (d), B6 and Il17−/− (e) mucosally vaccinated Mtb-challenged mice on day 15 post infection. B6 mice were mucosally vaccinated, rested for 30 days and challenged with Mtb, following which they were treated with isotype control antibody or IL-17 neutralizing antibody between day 5 and 21 (100 µg/mouse every 48 hours), and lung bacterial burden was determined on day 30 post-infection (f). Stat3.Cd4−/− mice or control littermates (Control) were vaccinated with ESAT61–20 in HLTs or left unvaccinated and number of cytokine-producing ESAT61–20-specific CD4+ T cells in the lungs were determined by ELISpot on day 15 post mucosal vaccination (g). Mucosally vaccinated Stat3.Cd4−/− mice or control littermate (Control) mice were rested for 30 days and challenged with ~100 CFU Mtb by the aerosol route and lung bacterial burden was determined on day 30 post infection (h). Ifng−/− mice were vaccinated, rested for 30 days, challenged with Mtb following which they were treated with isotype control antibody or IL-17 neutralizing antibody between day 5 and 21 (100 µg/mouse every 48 hours), and the lung bacterial burden was determined on day 30 post infection (i). The data points represent the mean (±SD) of values from 4–6 mice (a–i). *, p ≤0.05. **, p ≤0.005. ***, p ≤0.0005, ns-not significant. One of two independent experiments shown.
IL-17 drives lung CXCL13 expression and mediates vaccine-induced protection against TB
Since protection in mucosally vaccinated mice was associated with ectopic lymphoid structure formation (Figure 1), we next determined whether IL-17-dependent vaccine-induced protection following Mtb challenge was associated with the development of organized lymphoid structures populated with lymphocytes. We found that mucosally vaccinated Il17−/− mice formed small inflammatory lesions with disorganized lymphocytic infiltrates (Figure 3a, c-upper panel) and containing small B cell follicles (Figure 3b,c-lower panel). In contrast, B6 and Ifng−/− vaccinated mice developed distinct inflammatory lesions with rich lymphocytic infiltrates (Figure 3a, c-upper panel), featuring well-organized extensive B cell follicles (Figure 3a, c-lower panel), both of which were lost upon neutralization of IL-17 (Figure 3d–f). Furthermore, in support of the role of IL-17 produced by CD4+ Th17 cells in induction of organized iBALT structures, both lymphocytic infiltrates (Figure S2a, c-upper panel) and B cell follicles (Figure S2b, c-lower panel) were poorly organized in mucosally vaccinated Mtb-challenged Stat3.Cd4−/− mice. These data strongly suggest that IL-17 production by Th17 cells is required to efficiently form and organize lymphocytic infiltrates within inflammatory lesions for control of Mtb in mucosally vaccinated mice.
Figure 3. IL-17, but not IFNγ is crucial for lymphocytic infiltration, B cell lymphoid follicle and granuloma formation in lungs of mucosally vaccinated Mtb-challenged mice.
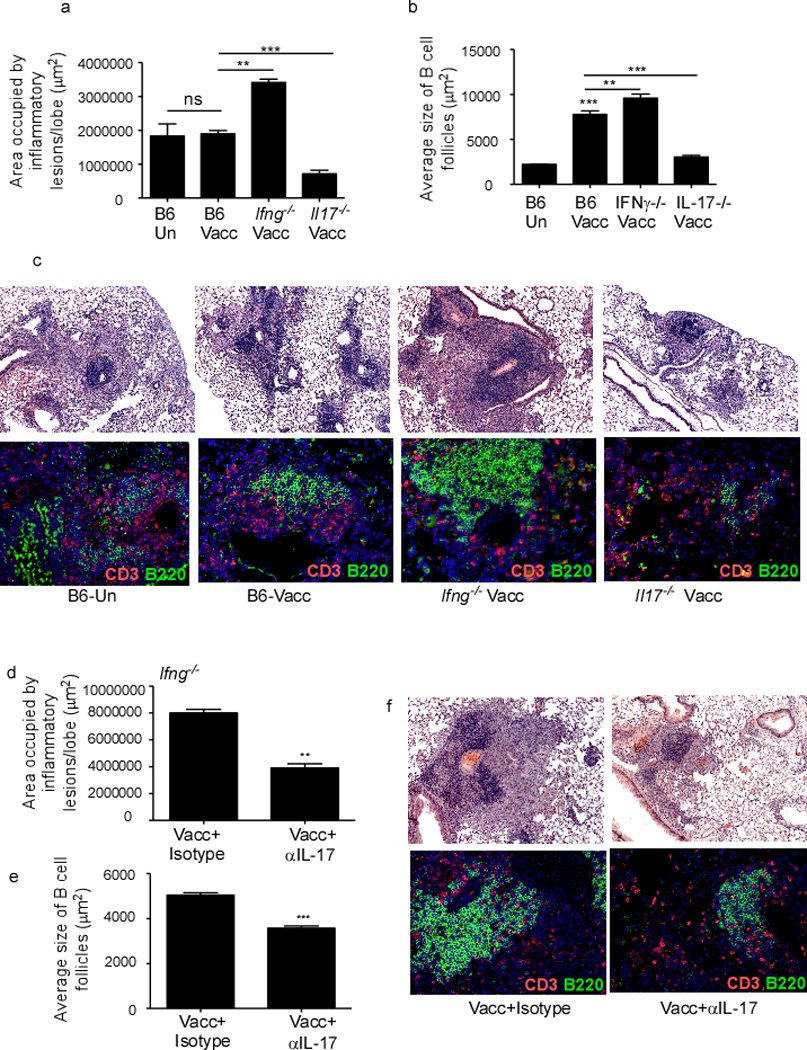
B6, Ifng−/− and Il17−/− were mucosally vaccinated with ESAT61–20 in combination with LT-IIb (Vacc) and rested for 30 days, then challenged with ~100 CFU Mtb by the aerosol route. On day 30 post challenge, formalin fixed lung samples were stained with H&E or CD3 (red) B220 (green) and the area occupied by granuloma as inflammatory lesions (a) and B cell lymphoid follicles (b) quantified using the morphometric tool of the Zeiss Axioplan microscope. Representative pictures of inflammatory lesions (c-top panel) and B cell lymphoid follicles (c-bottom panel) are shown. Original magnification for H&E sections, 100X, B cell follicles, 200X. Ifng−/− mice were vaccinated, rested, Mtb-infected and treated with isotype control antibody or treated with IL-17 neutralizing antibody between day 5 and 21 (100 µg/mouse every 48 hours), and sacrificed on day 30 post infection. Formalin fixed samples were stained with H&E or CD3 (red) B220 (green) and the area occupied by inflammatory lesions /lung lobe (d) and average size of B cell lymphoid follicles harboring CD3+ lymphocytes (e) was quantified by using the morphometric tool of the Zeiss Axioplan microscope. Representative figures showing inflammatory lesions (f-top panel) and B cell lymphoid follicles (f-bottom panel) are shown. The data points represent the mean (±SD) of values from 4–6 mice (a–f). **≤0.005, ***p≤0.0005. One experiment representative of two is shown.
We recently showed that IL-17 was required for formation of iBALT structures in neonate lungs via the induction of CCL19 and CXCL13 following LPS-induced inflammation7. In addition, CXCL13 is induced in the lung following Mtb infection, and absence of CXCL13 results in impaired T cell localization within the inflammatory lesions, decreased activation of lung macrophages and increased susceptibility to Mtb infection6. Therefore, we hypothesized that the critical role for IL-17 in mucosally vaccinated mice was linked to efficient CXCL13-induction and the subsequent attraction of CXCR5-bearing T cells within TB inflammatory lesions. In support of our hypothesis, we found that CXCL13 mRNA (Figure 4a-upper panel) and protein (Figure 4a-lower panel) was expressed and localized within iBALT found in lungs of vaccinated B6 and Ifng−/− mice, but was poorly expressed within the loosely organized cellular aggregates formed in lungs of vaccinated Il17−/− mice. Quantification of CXCL13 mRNA within inflammatory lesions further confirmed these findings (Figure 4b). Furthermore, the expression of CXCL13 mRNA (Figure 4c-upper panel, d) and protein (Figure 4c-lower panel) in the inflammatory lesions of vaccinated Ifng−/− mice was lost upon neutralization of IL-17. In addition, reduced levels of CXCL13 mRNA (Figure 4e-upper panel), reduced area of granulomas containing CXCL13 mRNA (Figure 4f), and reduced expression of CXCL-13 protein (Figure 4e-lower panel) were detected within poorly organized inflammatory lesions of vaccinated Stat3.Cd4−/− mice. Vaccine-induced IL-17 is required for early CXCL9 induction and CXCR3-expressing Th1 recruitment into lungs of parenterally vaccinated Mtb-challenged mice10. However, we observed similar pulmonary CXCL9 mRNA expression in mucosally vaccinated B6 and Il17−/− Mtb-challenged lungs (Figure S3). As expected, pulmonary CXCL9 mRNA expression was lost in mucosally vaccinated Ifng−/− Mtb-challenged lungs (Figure S3). Altogether, these data suggest that unlike parenteral immunization models, mucosal vaccine-induced IL-17 is critical for induction of CXCL13 but not CXCL9 during TB recall responses.
Figure 4. IL-17 promotes CXCL13 expression within TB inflammatory lesions and mediates vaccine-induced protection against Mtb infection.
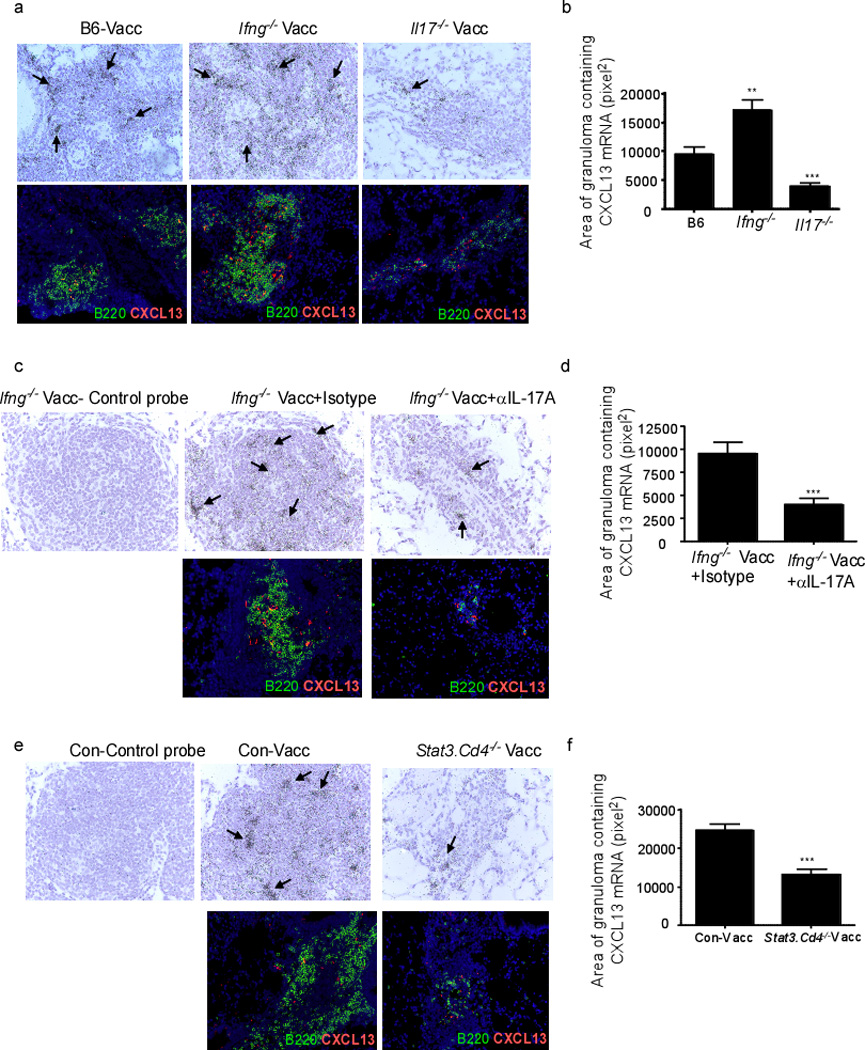
B6, Ifng−/− and Il17−/− were mucosally vaccinated with ESAT61–20 in combination with LT-IIb, rested, challenged with Mtb and sacrificed on day 30 post infection (a–d). Ifng−/− mucosally vaccinated mice rested for 30 days were challenged with Mtb and treated with isotype antibody or IL-17 neutralizing antibody as described in Figure 2 (c,d). Littermate control mice (Con-Vacc), Stat3.Cd4−/− mice were mucosally vaccinated, rested, challenged with Mtb and sacrificed on day 30 post infection (e,f). Formalin-fixed, paraffin-embedded lung sections from the above groups were assayed for CXCL13 mRNA localization by ISH using a murine CXCL13 mRNA probe or control probe (a,c,e upper panels). Areas containing CXCL13 mRNA expression within inflammatory lesions was quantified as described under methods (b,d,f). Formalin-fixed, paraffin-embedded lung sections from the above groups were assayed using immunofluorescence for spatial detection of CXCL13 protein (a,c,e-lower panels). Original magnification for CXCL13 ISH sections 400X, immunofluorescent sections, 200X. Pictures are representative of staining observed in lungs of mice within the group (n= 4–6 mice) (a,c,e). The data points represent the mean (±SD) of values from 4–6 mice (b,d,f). **≤0.005, ***p≤0.0005. One experiment representative of two is shown.
Absence of IL-17 did not impact accumulation of ESAT61–20-specific Th1 cells in the lungs (Figure 2). In addition, we found similar accumulation of activated CD4+ T cells (Figure S4a) and similar numbers of activated CD4+ T cells expressing the chemokine receptors, CXCR5 and CXCR3 (Figure S4b,c) in vaccinated Mtb-challenged lungs of B6 and Il17−/− mice. Comparable numbers of activated CD4+ T cells producing proinflammatory cytokines such as IFNγ (Figure S4d), TNFα (Figure S4e), IL-2 (Figure S4f), as well as the ability of activated CD4+ T cells to coproduce these cytokines (data not shown) was also similar between B6 and Il17−/− vaccinated Mtb-challenged lungs. As expected, IL-17-producing activated CD4+ T cells (Figure S4g) and IL-17/IFNγ coproducers (Figure S4h) were absent in lungs of vaccinated Mtb-challenged Il17−/− mice. Importantly, despite similar accumulation of activated CD4+ T cells producing proinflammatory cytokines to the lung, T cells failed to localize within lymphoid structures and resulted in increased T cell perivascular cuffing (Figure 5a) and coincided with decreased T cell localization within the granulomas (Figure 5b) in Il17−/− vaccinated Mtb-infected lungs. Based on these data we hypothesized that vaccine-induced, activated cytokine-producing CD4+ T cells accumulate in the Mtb-challenged lungs, but since they do not localize within lung inflammatory lesions, this may result in reduced activation of lung macrophages and therefore loss of vaccine-induced control. Consistent with this hypothesis, despite similar numbers of macrophages at early time points in B6 unvaccinated (2.1×105 ± 2.8×104) and vaccinated (2.6×105±2.4×104, p=0.2138 between unvaccinated and vaccinated B6 mice) Mtb-challenged lungs, macrophages were less activated in unvaccinated Mtb-challenged lungs when compared to macrophages in vaccinated Mtb-challenged lungs (Figure 5c). Importantly, despite comparable numbers of lung macrophages in Il17−/− vaccinated Mtb-challenged lung (2.8×105±3.6×104, p=0.6130 between B6 and Il17−/− vaccinated lungs), early macrophage activation was compromised in Il17−/− vaccinated Mtb-challenged lungs (Figure 5c). Consequently, fewer inducible nitric oxide synthase (iNOS) expressing macrophages were found within the lungs in vaccinated Mtb-challenged Il17−/− mice (Figure 5d). We observed similar results in Stat3.Cd4−/− vaccinated Mtb-infected mice (data not shown). These data show that IL-17 induces CXCL13 expression and promotes correct T cell localization for optimal macrophage activation, two key events required for early Mtb control in mucosally vaccinated mice.
Figure 5. Absence of IL-17 results in impaired lung macrophage activation during vaccine-induced immunity.
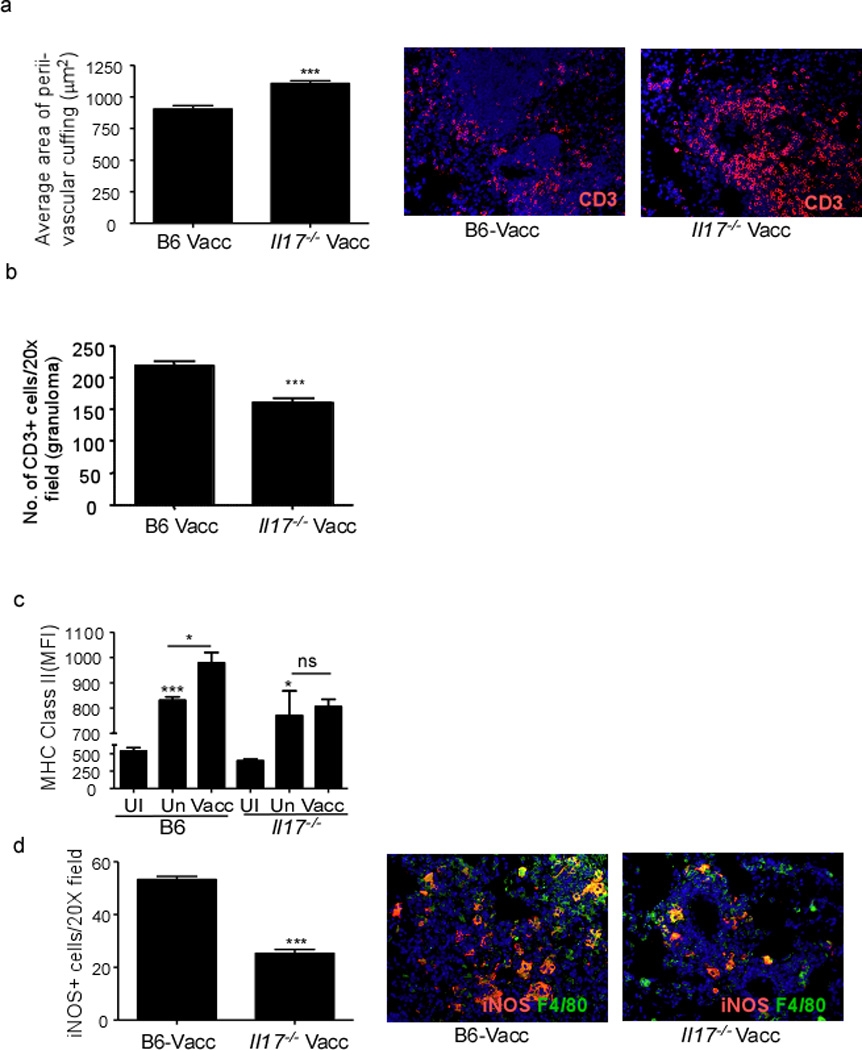
B6, and Il17−/− were mucosally vaccinated with ESAT61–20 in combination with LT-IIb, rested, challenged with Mtb as described in Figure 1. Formalin-fixed, paraffin embedded lung sections from day 30 Mtb-challenged lungs were analyzed by immunofluorescence for T cell perivascular cuffing (CD3+) and quantified using the morphometric tool of the Zeiss Axioplan microscope and a representative image of typical T cell perivascular cuffing shown (a). The number of CD3+ T cells within the granuloma were quantitated as described under method (b). The mean fluorescent intensity (MFI) of MHC Class II I-Ab expression on lung macrophages was determined by flow cytometry on day 15 from mucosally vaccinated Mtb-infected mice (Vacc), unvaccinated Mtb-infected mice (Un) or uninfected mice (UI) (c). The number of F4/80 macrophages producing iNOS were enumerated on day 30 post infection in formalin fixed lungs by immunofluorescence and a representative picture shown (d). The data points represent the mean (±SD) of values from 4–6 mice (a–d). *, p ≤0.05, ***p≤0.0005. One experiment representative of two is shown. ns-not significant.
To confirm that CXCL13-dependent recruitment of T cells within inflammatory lesions was critical for vaccine-induced immunity, we mucosally vaccinated Cxcl13−/− mice and found that absence of CXCL13 resulted in loss of vaccine-induced immunity against TB (Figure 6a). Importantly, lack of CXCL13 did not result in impairment in generation of Th17 cells in the lung following mucosal vaccination (Figure 6b), but influenced lung inflammatory lesion size and lymphocytic infiltration (Figure 6c,e-upper panel), resulting in smaller B cell follicles (Figure 6d,e-lower panel) in mucosally vaccinated Mtb-challenged Cxcl13−/− mice when compared to B6 vaccinated Mtb-challenged mice. Importantly, we found that T cells did not localize within lymphoid structures and accumulated as perivascular cuffs (Figure 6e,f) and coincidently exhibited decreased localization within the tubercle granuloma (Figure 6g). This decreased localization of T cells within the granuloma also coincided with decreased activation of macrophages in inflammatory lesions of Cxcl13−/− vaccinated Mtb-infected mice (Figure 6h). These data together suggest that vaccine-induced IL-17-mediated induction of CXCL13 is essential for correct T cell localization within TB inflammatory lesions, early macrophage activation and vaccine-induced Mtb control.
Figure 6. CXCL13 is required for mucosal vaccine-induced immunity against TB.
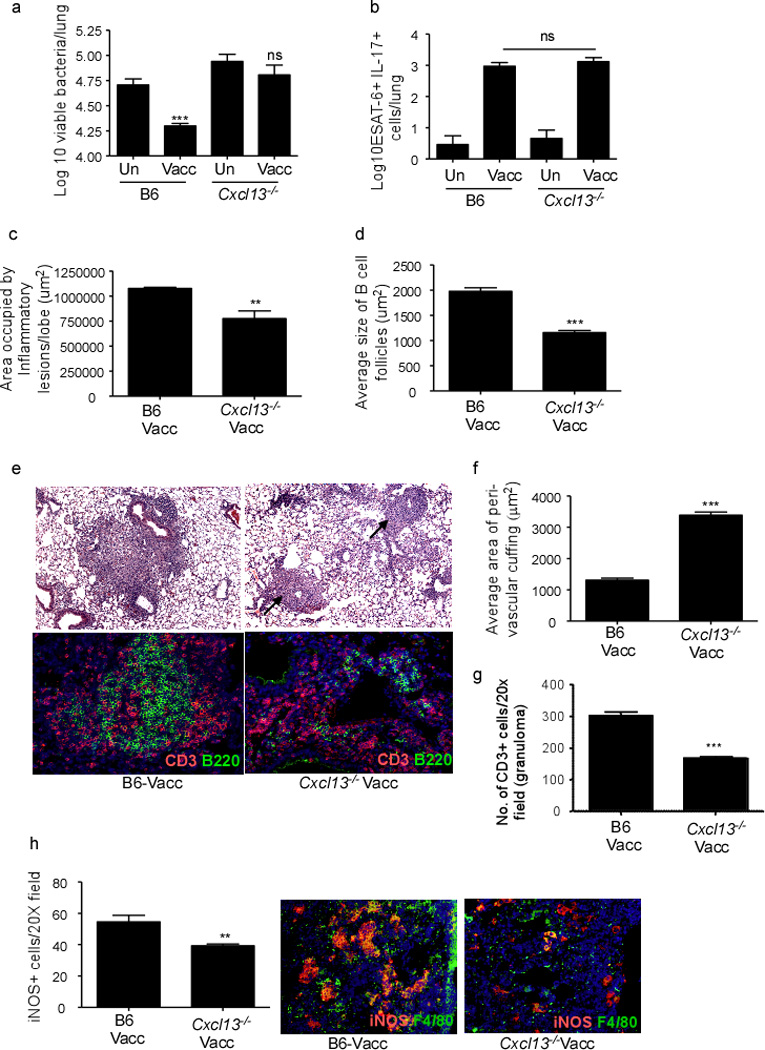
B6 and Cxcl13−/− mice were vaccinated with ESAT61–20 in Lt-IIb, rested for 30 days and challenged with Mtb as described in Figure 1 and lung bacterial burden was determined (a). (n=8–10 mice combined over two separate experiments). B6 and Cxcl13−/− mice were mucosally vaccinated, and number of ESAT61–20-specific IL-17 producing CD4+ T cells in the lungs were determined by ELISpot on day 15 post vaccination (b). Area occupied by inflammatory lesions/lung lobe (c) and average size of B cell lymphoid follicles (d) was quantified using the morphometric tool of the Zeiss Axioplan microscope in formalin-fixed day 30 Mtb-challenged lungs. Lung sections from B6 and Cxcl13−/− mucosally vaccinated Mtb-infected mice were stained with H&E (e-top panel) or CD3 (red), and B220 (green) (e-bottom panel) on day 30 post infection. Arrows point to perivascular cuffs in Cxcl13−/− mice. Formalin-fixed, paraffin embedded lung sections were analyzed by immunofluorescence for T cell perivascular cuffing (CD3+ staining) (f), number of CD3+ T cells that localized within the granuloma (g) and number of iNOS+ cells per inflammatory lesions (h). T cell perivascular cuffing was quantified in formalin fixed lung sections (CD3+) using the morphometric tool of the Zeiss Axioplan microscope (f). CD3+ T cells and F4/80 expressing macrophages producing INOS were enumerated by immunofluorescence and representative pictures are shown (g,h). The data points represent the mean (±SD) of values from 4–6 mice (b–h). **≤0.005, ***p≤0.0005. One experiment representative of two is shown. ns-not significant.
IL-17 but not IFNγ treatment can improve vaccine-induced protection against Mtb challenge
Our data demonstrate that the protection afforded in mucosally vaccinated mice is mediated through the enhanced production of IL-17, local CXCL13 production and improved lymphoid structure formation. This is a novel and important observation, since efforts to improve Th1 responses alone have not improved vaccine-induced protection following Mtb challenge2. Moreover, most studies only measure antigen-specific IFNγ responses following Mtb challenge12,17, and are thus likely to miss the critically important IL-17-producing Th17 cells. Based on these data, we treated mucosally vaccinated mice with recombinant IL-17 or IFNγ, following Mtb challenge and determined if this would further enhance immune protection. Accordingly, we found that early exogenous IL-17 treatment in mucosally vaccinated B6 mice following Mtb challenge, further decreased lung bacterial burden, when compared to bacterial burden in control mucosally vaccinated mice, and mucosally vaccinated mice that received recombinant IFNγ (Figure 7a). The improved protection in mucosally vaccinated mice that received exogenous IL-17 also coincided with improved granuloma formation (Figure 7b,d-upper panels), enhanced B cell follicle formation (Figure 7c,d-middle panels) and enhanced production of CXCL13 (Figure 7d-lower panels). This also coincided with decreased T cell perivascular cuffing in B6 mucosally vaccinated mice that received early IL-17 therapy (Figure 7e). Similarly, adenoviral over-expression of IL-23 in mucosally vaccinated B6 mice also improved protective outcomes upon Mtb challenge, when compared to mucosally vaccinated Mtb-infected mice that received control vector (Figure 7f). We then addressed if mucosal boosting in mice parenterally vaccinated with M.bovis BCG would improve vaccine-induced immunity upon Mtb challenge. Despite similar levels of vaccine-induced responses seen in mice parenterally vaccinated with M.bovis BCG and mucosally vaccinated with Mtb antigen and adjuvant, we found that mucosal boosting in M.bovis BCG vaccinated mice further improved upon vaccine induced immunity in Mtb challenged mice (Figure 7g). These data, therefore, demonstrate that experimentally manipulating IL-17 for improvement of vaccine efficacy against Mtb can be an effective strategy that can lead to improved vaccines against TB.
Figure 7. IL-17 improves the protective efficacy of mucosal vaccination following Mtb challenge.
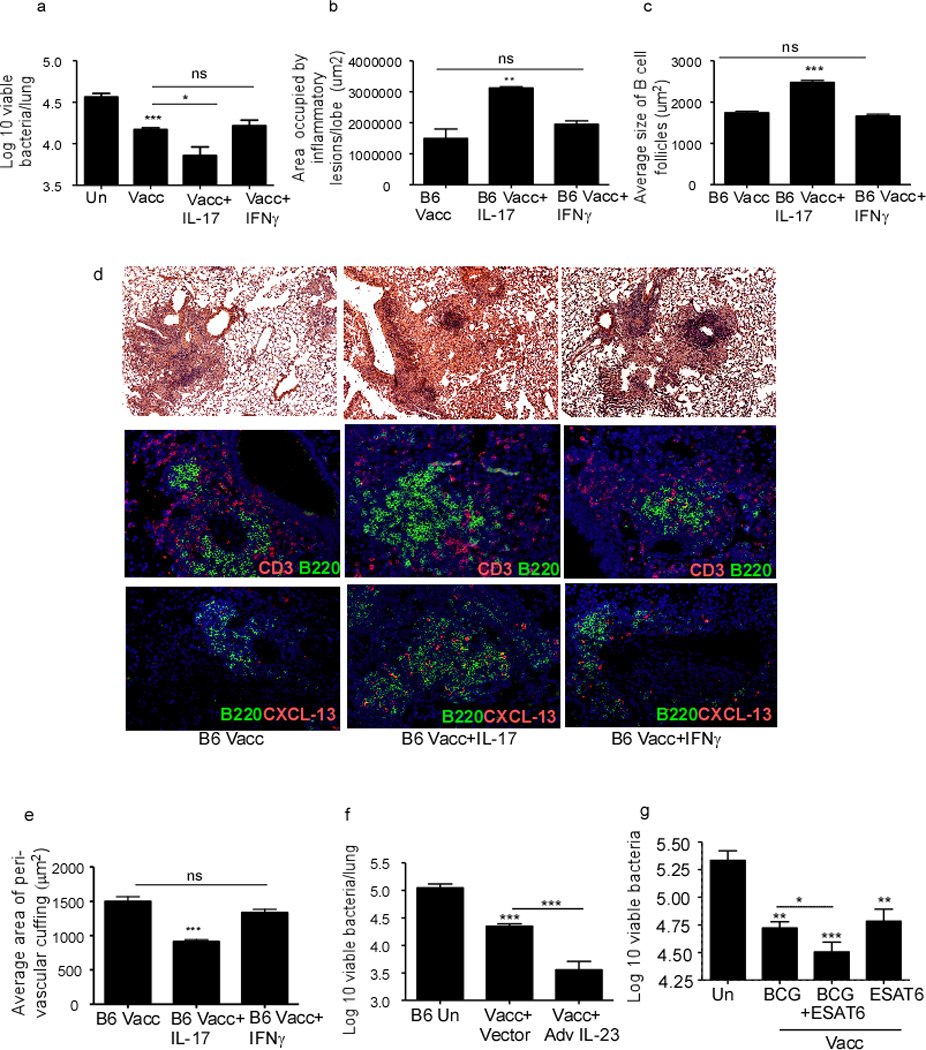
B6 mice were mucosally vaccinated with ESAT61–20 in LT-IIb, rested and challenged with Mtb as described in Figure 1. Vaccinated Mtb-challenged mice either received PBS, rIL-17 or rIFNγ (1.5 µg per mouse) intratracheally from day 5 to day 17 post-infection during recall response and the lung bacterial burden was determined on day 30 post vaccination (a). On day 30 post-infection, formalin-fixed, paraffin embedded lung sections were stained with H&E or CD3 (red), and B220 (green). Area occupied by inflammatory lesions/lung lobe (b), B cell lymphoid follicles (c) was quantified using the morphometric tool of the Zeiss Axioplan microscope. The representative figure showing a typical inflammatory lesions (d-top panel), B cell follicle (d-middle panel) and CXCL13 expression within B cell follicles (d-bottom panel) is included. The average area of perivascular cuffing from the above mentioned groups were quantified (e). One group of mucosally vaccinated B6 mice received control adenovirus expressing luciferase vector (Vacc+Vector) while a second group received adenovirus overexpressing IL-23 (Vacc+Ad IL-23) (5×108pfu) on the day of vaccination. Mucosally vaccinated and unvaccinated mice were rested and challenged with Mtb and lung bacterial burden was determined on day 30 post infection (f). B6 mice were either subcutaneously vaccinated with 1×106 M.bovis BCG, mucosally vaccinated, or subcutaneously vaccinated with M.bovis BCG followed by a period of rest for 30 days and boosted mucosally as described under methods. All groups of mice were then rested for 30 days and challenged with Mtb as described in Figure 1 and lung bacterial burden was determined on day 30 post infection (g). Original magnification for H&E sections, 100X; immunofluorescent sections, 200X. The data points represent the mean (±SD) of values from 4–6 mice (a–g). *p≤0.05, **≤0.005, ***p≤0.0005. One experiment representative of two is shown. ns-not significant.
Discussion
Mtb latently infects one third of the world’s population, of which 10% (~200 million people) will develop active TB during their lifetime. Therefore it is critical that we design effective vaccine strategies to limit rising numbers of infected individuals. Immunity to TB has been associated with induction of effective Th1 responses1; however, an IFNγ-independent mechanism of protection against Mtb infection exists, since Mtb-specific Th cells lacking IFNγ or TNFα still confers protection in Mtb-infected mice4,18. Instead, IFNγ may limit IL-17 production, chemokine induction, neutrophil accumulation and coincident lung inflammation during TB18. Our new data show that the IFNγ-independent mechanism of protection functional in a mucosal vaccination model against Mtb infection is an IL-17-dependent mechanism, a surprising and novel finding, considering that IL-17 is dispensable for primary immunity to TB8. These data suggest that protective immune molecules in primary and memory responses to mucosal infections are distinct and need to be clearly defined before exploring ways to experimentally target them in vaccine strategies. Furthermore, our data projects the new finding that the role for IL-17 in mediating vaccine-induced protective responses in parenteral versus mucosal vaccine models are also likely different. For example, previously, we had described that following parenteral vaccination, IL-23 is required to prime a population of lung-resident Th17 cells that upon Mtb infection induce CXCR-3 ligating chemokines such as CXCL9, CXCL10 and CXCL11 responsible for the attraction of Th1 cells to the lung and subsequent activation of macrophages for Mtb control10. When compared to this parenteral model of vaccination, our new data show that potent mucosal adjuvants and mucosal routes of vaccination can also elicit enhanced generation of lung-resident Th17 cells; but in this model the protection is IFNγ-independent, but instead is dependent on IL-17 induced CXCL13 expression and strategic localization of T cells within inflammatory lesions to activate macrophages to control Mtb. This is an important and novel finding, since absence of IL-17 completely abrogated vaccine-induced protection in the mucosal model. We recently showed that IL-17 induces expression of chemokines CCL19 and CXCL13 in the neonate lung and mediates generation of iBALT structures following LPS-induced inflammation7. Furthermore, lung fibroblasts can directly respond to IL-17 and induce CXCL13 expression in vitro8. Consistent with these previous observations, our new findings presented here show that vaccine-induced Th17 cells produce IL-17 that drives CXCL13 induction within the lung parenchyma, promoting the correct T cell localization within the inflammatory lesions, in order to maximally activate macrophages and optimize immune protection. It is interesting that absence of IL-17 does not impact primary immunity to TB8, while absence of CXCL13 increases susceptibility to primary Mtb infection6. These observations suggest the existence of IL-17-independent mechanisms of CXCL13 induction during primary Mtb infection. In mucosally vaccinated Ifng−/− mice, despite loss of CXCL9 expression, IL-17-dependent production of CXCL13 is intact, and helps to localize T cells within TB inflammatory lesions for adequate vaccine-induced control. We have detected CXCR5+ CD4+ T cells that express IFNγ, TNF-α and IL-2, which potentially could accumulate inside inflammatory lesions (data not shown). In this location, CXCR5-expressing T cells can produce a variety of Th1 effector cytokines to induce iNOS-dependent and iNOS-independent mechanisms, critical for macrophage activation and vaccine-induced protection. We observed that depletion of IL-17 in Ifng−/− vaccinated mice notably reduced CXCL13 expression, impaired T cell localization within inflammatory lesions and severely compromised vaccine-induced protection. These collective data confirms that production of IL-17, but not IFNγ, is the critical first step in the downstream events that mediate vaccine-induced immunity against TB.
Recently, several adjuvants including oil-in-water nanoemulsions19, cholera toxin20–21, polyelectrolyte microcapsules22 have been shown to induce potent Th17 responses when delivered mucosally. Similarly, our data demonstrate that delivery of Mtb-specific antigens in the presence of LT-IIb as adjuvant enhances lung-resident Th17 responses. Recently, it has become clear that following exposure to adjuvants, both αβ T cells as well as innate cells such as γδ T cells can produce IL-1723. However, we found that following mucosal vaccination of ESAT61–20 with LT-IIb, αβ T cells were the primary cellular source of IL-17 in the lung (data not shown). These data suggest that mucosal vaccination with LT-IIb may induce production of polarizing cytokines such as IL-6, TGFβ, IL-23 in mucosal DCs to induce Th17 responses20–22. Accordingly, DCs isolated from lungs of mice mucosally administered ESAT61–20 in LT-IIb induce mRNA for IL-12p40 (data not shown). These data suggest that the mucosal route of vaccination in combination with an appropriate mucosal adjuvant can be an effective strategy to induce lung-resident Mtb-specific vaccine-induced Th17 responses and improve upon vaccine-induced immunity against TB. However, excess IL-17 induced in response to repeated mycobacterial vaccinations24 or in the absence of IFNγ signaling18 can result in severe lung pathology. Therefore, the pathological versus protective role of IL-17 should be carefully explored before targeting IL-17 in vaccine strategies for TB.
Recent studies show that pulmonary infection with Mtb triggers the formation of lymphoid aggregates associated with TB granuloma25. Our data in the current paper expand these findings and show that mucosal vaccine-induced Th17 cells induce enhanced formation and organization of lymphoid structures within TB inflammatory lesions, suggesting that protection in mucosally vaccinated mice coincides with the increased cellular complexity of iBALT structures. Consistent with these data, absence of vaccine-induced immunity in Il17−/− mice correlates with the reduced expression of CXCL13, poor generation of B cell follicles, poorly formed lymphocytic infiltrates and increased T cell perivascular cuffing. These data together suggest that IL-17-dependent iBALT formation and organization is required to facilitate the cross-talk between T cells and macrophages inside granulomas and to enhance Mtb control. Together, our recent studies in primary Mtb infection6,8 and in mucosal vaccine-induced immunity described here, support the idea that iBALT is critical for protection against TB. In both models, CXCL13 expression and accumulation of CXCR5+ T cells within the granuloma is fundamental for quick and optimal macrophage activation. Interestingly, in primary Mtb infection, IL-17 is not required for protection8, suggesting that other factors can induce CXCL13 and mediate protection. Based on our data, it is tempting to speculate that mucosal boosting may enhance Th17 lung-resident cells in pre-immunized individuals, improve iBALT formation and provide protection against TB, thereby decreasing global TB burdens. Taken together, these results have far reaching implications for the design of future vaccines for TB.
Materials and Methods
Mice
C57BL/6 (B6), Cxcl13−/− and Ifng−/− mice were purchased from The Jackson Laboratory (Bar Harbor, ME), Il17−/− mice were obtained on the B6 background26, Stat3.Cd4−/− mice were obtained from Indiana University School of Medicine27. Experimental mice were age-and sex-matched and used between the ages of 6 to 8 wks. All mice were maintained and used in accordance with approved University of Pittsburgh IACUC guidelines.
Cloning and purification of HLTs
Engineering of plasmid encoding wildtype LT-IIb has been described14. Plasmid-encoded, his-tagged holotoxins were purified from the periplasmic extracts of cultures of Escherichia coli DH5αF’kan (Life Technologies, Inc., Gaithersburg, MD) as described before14. Precipitated proteins were dialyzed to remove salts and low molecular weight molecules. His-tagged holotoxins in the dialysate were initially resolved by nickel affinity chromatography using His•Bind resin (Novagen, Madison, WI). Peak fractions from the nickel affinity chromatography demonstrated by SDS-PAGE of the eluted fractions were pooled and subsequently applied to a Sephacryl S-100 gel filtration column (Pharmacia, Piscataway, NJ) connected to an AKTAfplc (Pharmacia). Holotoxins obtained from gel filtration were resolved by SDS-PAGE to confirm homogeneity and by immunoblotting using anti-LT-IIb antibodies to confirm identity. Finally, purified proteins were analyzed for potentially contaminating endotoxin (lipopolysaccharide) using a quantitative Limulus amoebocyte lysate assay (Charles River Endosafe, Charleston, SC). Holotoxins were essentially free of lipopolysaccharide (<0.03 ng/µg of protein).
Vaccinations and experimental infections
ESAT61–20 peptide (133 mg) was mixed with LT-IIb holotoxin (1µg) and unanesthetized mice were mucosally vaccinated intranasally 3 times at 2-week intervals. In some experiments, adenovirus over-expressing IL-23 or control adenovirus expressing luciferase vector28 was delivered once (5×108pfu) intratracheally on the day of vaccination. M.bovis BCG Pasteur was grown in Proskauer Beck (PB) medium containing 0.05% Tween 80 to mid-log phase and then frozen in 1-ml aliquots at −80°C. Bacterial stocks were plated on 7H11 agar plates to calculate colony forming units (CFU). Mice were vaccinated subcutaneously with 1×106 CFU of M.bovis BCG in PBS and after 30 days some mice were mucosally vaccinated with two doses of Ag85B240–254 peptide (133 mg) mixed with LT-IIb holotoxin (1µg). All vaccinated mice were rested for 30 days following which they were challenged with Mtb H37Rv. The H37Rv strain of Mtb was grown in Proskauer Beck medium containing 0.05% Tween-80 to mid-log phase and frozen in 1 ml aliquots at −70°C. For Mtb aerosol infections, animals were infected with 100 CFU of bacteria using a Glas-Col airborne infection system as described before10. In some experiments mice were treated intraperitoneally (i.p.) with either 100 µg of αIL-17A (clone 50104) or Isotype control (clone 54447; R&D Systems) from day 9 through day 21 post-infection. In other experiments mice were treated with recombinant IL-17A (rIL-17A) or IFNγ (rIFNγ) (R&D Systems) intratracheally (i.t.) at a dose of 1.5 µg in 50 µl saline per mouse10 from day 5 to day 17 post-infection every 48 hours. Bacterial burden was estimated by plating the lung homogenates on 7H11 agar plates.
Lung cell preparation
Lung cell suspensions were prepared as described and single cells were used for ELISpot or flow cytometric analyses9–10.
Flow Cytometry
Single cell suspensions were stained with fluorochrome-labeled antibodies specific for CD3 (145-2C11), CD4 (RM4-5), CXCR5 (2G8), IFNγ (XMG1.2), IL-17 (TC11-18H10), TNFα (MP6-XT22), IL-2 (JES6-5H4), CD44 (IM7), CD11c (HL3), CD11b (M1/70), CXCR3 (173), MHC Class II (M5/114.15.2) or isotype control antibodies. For intracellular analyses, cells were stimulated with Phorbol myristate acetate (PMA-50ng/ml) and ionomycin (750 ng/ml; Sigma Aldrich) in the presence of Golgistop (BD Pharmingen). Cells were then surface stained, permeabilized with Cytofix-Cytoperm solution (BD Pharmingen) and stained for relevant cytokines. Cells were collected using a Becton Dickinson FACS Aria flow cytometer using FACS Diva software. Cells were gated based on their forward by side scatter characteristics and the frequency of specific cell types was calculated using FlowJo (Tree Star Inc, CA). The mean fluorescent intensity was also calculated to determine expression levels of molecules using FlowJo (Tree Star Inc, CA).
Detection of cytokine producing cells by ELISpot assay
Detection of antigen-specific IFNγ- and IL-17-producing cells was carried out by using an ELISpot assay10. In brief, cell culture plates were coated overnight with monoclonal purified anti-mouse IFNγ (Clone R4-6A2; eBiosciences) or monoclonal purified anti-mouse IL-17 (clone 50101; R&D Systems) in PBS. Cells were seeded at an initial concentration of 2–5×106 cells/well and doubling dilutions made. Irradiated B6 splenocytes were used as APCs. ESAT-61–20 peptide was used as antigen for vaccinated mice and mouse rIL-2 (Sigma-Aldrich; 10U/ml) was added to all wells. Plates were washed and biotinylated anti-mouse IFNγ (clone XMG 1.2; eBiosciences) or biotynylated anti-mouse IL-17 antibody (clone eBio17B7) was used to detect the captured cytokine. Spots were enumerated by using CTL-Immuno Spot analyzer. The frequency of responding cells were determined and applied to the number of cells per sample to generate the total number of responding cells per organ.
Immunohistochemistry
Lung lobes were instilled with 10% neutral buffered formalin and embedded in paraffin. Lung sections were stained with hematoxylin and eosin stain and inflammatory features were evaluated by light microscopy (Research Histology Core, University of Pittsburgh). For immunofluorescent staining, formalin-fixed, lung sections were cut, immersed in xylene to remove paraffin and then hydrated in alcohol, 96% alcohol and PBS. Antigens were unmasked with a DakoCytomation Target Retrieval Solution and non-specific binding was blocked with 5% (v/v) normal donkey serum and Fc block (BD Pharmingen, San Diego, CA). Endogenous biotin (Sigma Aldrich) was neutralized by adding first avidin, followed by incubation with biotin. Sections were probed with, anti B220 to detect B cells (Clone RA3-6B2, BD Pharmingen, San Diego, CA), anti-CXCL13 (Clone Ile22Ala109, R and D Biosystems) and anti CD3 to detect T cells (Clone M-20, Santa Cruz Biotechnology. Santa Cruz, CA) in the inflammatory lesions. iNOS producing macrophages were identified using goat anti-NOS2 (M-19-G, Santa Cruz Biotechnology) and rat F4/80 (MCA497GA, Serotec). Inflammatory lesions and B cell follicles were outlined with the automated tool of the Zeiss Axioplan 2 microscope (Carl Zeiss) and average size in squared microns calculated. iNOS+ cells in three random 20× fields were enumerated per lung (n= 5 lungs) and the average was calculated. 3–5 granulomas per lobe section in each group were randomly chosen to quantify local CXCL13 mRNA expression or granuloma T cell infiltration. ISH signal was quantified inside granulomas with Image J, using the same threshold for all the analysis (100) and differences in the average area occupied by CXCL13 mRNA were graphed in GraphPad Prism 5. Infiltrating T cells in granulomatous structures were counted with the automated tool of the Axioplan Zeiss microscope in 3–5 randomly picked 200X fields. Samples were analyzed in a blinded fashion.
In situ hybridization
Mouse CXCL13 cDNA was RT-PCR amplified with primers BFJ. mCXCL13_F1 (59-GAACTCCACCTCCAGGCAGA-39) and BFJ. mCXCL13_R1 (59-CTTTTGA GATGATAGTGGCT-39). PCR products were ligated to the pGEM-T vector (Promega) and DNA sequenced. The GEMT-CXCL13 plasmid was linearized by restriction digest. Gene-specific riboprobes were synthesized by in vitro transcription using a Maxiscript SP6/T7 kit (Ambion), and unincorporated nucleotides were removed using RNA Mini Quick Spin Columns (Roche). Paraffin embedded tissue specimens were immersed with xylene for deparaffinization and rinsing in ethanol. In situ hybridization with 35S-labeled riboprobes was performed at 50°C overnight with 0.1 M DTT included in the hybridization mix. CXCL9 mRNA was detected as previously described29. Tissue sections were coated with NTB-2 emulsion (Kodak) and exposed at 10°C for 10 days. The sections were counterstained with hematoxylin (Vector Laboratories) and mounted with Permount (Fisher). Images were visualized using an Olympus BX41 microscope (Olympus) and captured using a SPOT RT3 digital camera (Diagnostics Instruments).
Statistical analysis
Differences between the means of groups were analyzed using the two tailed Student’s t-test in GraphPad Prism 5 (La Jolla, CA).
Supplementary Material
B6 mice were either uninfected (UI), unvaccinated (Un) or mucosally vaccinated with ESAT61–20 in LT-IIb, rested, Mtb-infected (Vacc) and sacrificed on day 15 post infection. The number of activated CD4+ T cells (CD3+, CD4+, CD44+) expressing IL-17 (a), TNFα (b), IFNγ and TNFα (c) IFNγ alone (d), or chemokine receptors CXCR5 (e) and CXCR3 (f) was determined by ex vivo stimulation of cells with PMA/Ionomycin followed by intracellular staining and flow cytometry. The data points represent the mean (±SD) of values from 4–6 mice (a–f). *p≤0.05, ns-not significant.
Littermate controls (Con-Vacc) and Stat3.Cd4−/− mice were mucosally vaccinated with ESAT61–20 in combination with LT-IIb, rested and Mtb-infected. Mice were sacrificed on day 30 post infection and formalin fixed samples were stained with H&E or CD3 (red), and B220 (green) and the area occupied by inflammatory lesions/lobe (a) and average size of B cell lymphoid follicles (b) was quantified using the morphometric tool of the Zeiss Axioplan microscope. Representative figures showing a typical inflammatory lesions (c-top panel) and B cell lymphoid follicle (c-bottom panel) is shown. Original magnification for H&E sections, 100X; immunofluorescent sections, 200X. The data points represent the mean (±SD) of values from 4–6 mice (a–c). *p≤0.05, ***p≤0.0005
B6, Ifng−/− and Il17−/− were mucosally vaccinated with ESAT61–20 in combination with LT-IIb, rested, challenged with Mtb and sacrificed on day 30 post infection. Formalin-fixed, paraffin-embedded lung sections were assayed for CXCL9 mRNA localization by ISH using a murine CXCL9 mRNA probe or control probe. Original magnification 400X. Data shown from one representative lung, but similar staining as observed in all lungs within the group (n= 4–6 mice).
B6 and Il17−/− mice were either uninfected (UI) or mucosally vaccinated with ESAT61–20 in LT-IIb, rested, Mtb-infected (Vacc) and sacrificed on day 30 post infection. The number of activated CD4+ T cells (CD3+, CD4+, CD44+) (a), expressing CXCR5 (b), CXCR3 (c), or producing IFNγ (d), TNFα (e), IL-2 (f), IL-17 (g) or co-producing IL-17 and IFNγ (h) was determined by ex vivo stimulation of cells with PMA/Ionomycin followed by intracellular staining and flow cytometry. The data points represent the mean (±SD) of values from 4–6 mice (a–h). *p≤0.05, ** p≤0.005, ***p≤0.0005, ns-not significant.
Acknowledgements
This work was supported by Children’s Hospital of Pittsburgh, NIH grants AI083541 and HL105427 to S.A.K., HL69409 to T.D.R., DE13833 to T.D.C. and AI060422 to T.A.R., Department of Medicine, University of Rochester and AI91036 to J.R.-M, a Research Advisory Committee Grants from Children’s Hospital of Pittsburgh of the UPMC Health System to S.R.S and Y.L. The authors thank Dr. Iwakura, University of Japan, for Il17−/− for breeders and Dr. Kaplan, Indiana University School of Medicine for Stat3.Cd4−/− breeders and Hillary Cleveland for mice breeding. We thank Dr. John Alcorn for critical reading of the manuscript.
Abbreviations
- Th
T helper
- DC
Bone-marrow Dendritic Cells
- IL
Interleukin
- IFNγ
Interferon gamma
Footnotes
All authors have no other conflicting financial interests.
References
- 1.Cooper AM, Khader SA. The role of cytokines in the initiation, expansion, and control of cellular immunity to tuberculosis. Immunol Rev. 2008;226:191–204. doi: 10.1111/j.1600-065X.2008.00702.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Leal I, Smedegard B, Andersen P, Appelberg R. Failure to induce enhanced protection against tuberculosis by increasing T-cell-dependent interferon-gamma generation. Immunology. 2001;104:157–161. doi: 10.1046/j.0019-2805.2001.01305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cowley SC, Elkins KL. CD4+ T cells mediate IFN-gamma-independent control of Mycobacterium tuberculosis infection both in vitro and in vivo. J Immunol. 2003;171:4689–4699. doi: 10.4049/jimmunol.171.9.4689. [DOI] [PubMed] [Google Scholar]
- 4.Gallegos AM, et al. A gamma interferon independent mechanism of CD4 T cell mediated control of M. tuberculosis infection in vivo. PLoS Pathog. 2011;7:e1002052. doi: 10.1371/journal.ppat.1002052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rangel-Moreno J, Moyron-Quiroz JE, Hartson L, Kusser K, Randall TD. Pulmonary expression of CXC chemokine ligand 13, CC chemokine ligand 19, and CC chemokine ligand 21 is essential for local immunity to influenza. Proc Natl Acad Sci U S A. 2007;104:10577–10582. doi: 10.1073/pnas.0700591104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Khader SA, et al. In a murine tuberculosis model, the absence of homeostatic chemokines delays granuloma formation and protective immunity. J Immunol. 2009;183:8004–8014. doi: 10.4049/jimmunol.0901937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rangel-Moreno J, et al. The development of inducible bronchus-associated lymphoid tissue depends on IL-17. Nat Immunol. 2011;12:639–646. doi: 10.1038/ni.2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Khader SA, et al. IL-23 Is Required for Long-Term Control of Mycobacterium tuberculosis and B Cell Follicle Formation in the Infected Lung. J Immunol. 2011;187:5402–5407. doi: 10.4049/jimmunol.1101377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Khader SA, et al. IL-23 compensates for the absence of IL-12p70 and is essential for the IL-17 response during tuberculosis but is dispensable for protection and antigen-specific IFN-gamma responses if IL-12p70 is available. J Immunol. 2005;175:788–795. doi: 10.4049/jimmunol.175.2.788. [DOI] [PubMed] [Google Scholar]
- 10.Khader SA, et al. IL-23 and IL-17 in the establishment of protective pulmonary CD4+ T cell responses after vaccination and during Mycobacterium tuberculosis challenge. Nat Immunol. 2007;8:369–377. doi: 10.1038/ni1449. [DOI] [PubMed] [Google Scholar]
- 11.Goonetilleke N, et al. Enhanced Immunogenicity and Protective Efficacy Against Mycobacterium tuberculosis of Bacille Calmette-Guerin Vaccine Using Mucosal Administration and Boosting with a Recombinant Modified Vaccinia Virus Ankara. Journal of Immunology. 2003;171:1602–1609. doi: 10.4049/jimmunol.171.3.1602. [DOI] [PubMed] [Google Scholar]
- 12.Chen L, Wang J, Zganiacz A, Xing Z. Single intranasal mucosal Mycobacterium bovis BCG vaccination confers improved protection compared to subcutaneous vaccination against pulmonary tuberculosis. Infect Immun. 2004;72:238–246. doi: 10.1128/IAI.72.1.238-246.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang J, et al. Single mucosal, but not parenteral, immunization with recombinant adenoviral-based vaccine provides potent protection from pulmonary tuberculosis. J Immunol. 2004;173:6357–6365. doi: 10.4049/jimmunol.173.10.6357. [DOI] [PubMed] [Google Scholar]
- 14.Nawar HF, Arce S, Russell MW, Connell TD. Mucosal adjuvant properties of mutant LT-IIa and LT-IIb enterotoxins that exhibit altered ganglioside-binding activities. Infect Immun. 2005;73:1330–1342. doi: 10.1128/IAI.73.3.1330-1342.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cooper AM, et al. Disseminated tuberculosis in interferon gamma gene-disrupted mice. Journal of Experimental Medicine. 1993;178:2243–2247. doi: 10.1084/jem.178.6.2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Flynn JL, et al. An essential role for interferon gamma in resistance to Mycobacterium tuberculosis infection. Journal of Experimental Medicine. 1993;178:2249–2254. doi: 10.1084/jem.178.6.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Andersen CS, et al. The combined CTA1-DD/ISCOMs vector is an effective intranasal adjuvant for boosting prior Mycobacterium bovis BCG immunity to Mycobacterium tuberculosis. Infect Immun. 2007;75:408–416. doi: 10.1128/IAI.01290-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nandi B, Behar SM. Regulation of neutrophils by interferon-{gamma} limits lung inflammation during tuberculosis infection. J Exp Med. 2011;208:2251–2262. doi: 10.1084/jem.20110919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bielinska AU, et al. Induction of Th17 cellular immunity with a novel nanoemulsion adjuvant. Crit Rev Immunol. 2010;30:189–199. doi: 10.1615/critrevimmunol.v30.i2.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee JB, Jang JE, Song MK, Chang J. Intranasal delivery of cholera toxin induces th17-dominated T-cell response to bystander antigens. PLoS One. 2009;4:e5190. doi: 10.1371/journal.pone.0005190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Datta SK, et al. Mucosal adjuvant activity of cholera toxin requires Th17 cells and protects against inhalation anthrax. Proc Natl Acad Sci U S A. 2010;107:10638–10643. doi: 10.1073/pnas.1002348107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.De Koker S, et al. Biodegradable polyelectrolyte microcapsules: antigen delivery tools with Th17 skewing activity after pulmonary delivery. J Immunol. 2010;184:203–211. doi: 10.4049/jimmunol.0803591. [DOI] [PubMed] [Google Scholar]
- 23.Reynolds JM, Angkasekwinai P, Dong C. IL-17 family member cytokines: regulation and function in innate immunity. Cytokine Growth Factor Rev. 2010;21:413–423. doi: 10.1016/j.cytogfr.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cruz A, et al. Pathological role of interleukin 17 in mice subjected to repeated BCG vaccination after infection with Mycobacterium tuberculosis. J Exp Med. 2010;207:1609–1616. doi: 10.1084/jem.20100265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Randall TD. Bronchus-associated lymphoid tissue (BALT) structure and function. Adv Immunol. 2010;107:187–241. doi: 10.1016/B978-0-12-381300-8.00007-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakae S, et al. Antigen-specific T cell sensitization is impaired in IL-17-deficient mice, causing suppression of allergic cellular and humoral responses. Immunity. 2002;17:375–387. doi: 10.1016/s1074-7613(02)00391-6. [DOI] [PubMed] [Google Scholar]
- 27.Harris TJ, et al. Cutting edge: An in vivo requirement for STAT3 signaling in TH17 development and TH17-dependent autoimmunity. J Immunol. 2007;179:4313–4317. doi: 10.4049/jimmunol.179.7.4313. [DOI] [PubMed] [Google Scholar]
- 28.Happel KI, et al. Pulmonary interleukin-23 gene delivery increases local T-cell immunity and controls growth of Mycobacterium tuberculosis in the lungs. Infect Immun. 2005;73:5782–5788. doi: 10.1128/IAI.73.9.5782-5788.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aujla SJ, et al. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat Med. 2008;14:275–281. doi: 10.1038/nm1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
B6 mice were either uninfected (UI), unvaccinated (Un) or mucosally vaccinated with ESAT61–20 in LT-IIb, rested, Mtb-infected (Vacc) and sacrificed on day 15 post infection. The number of activated CD4+ T cells (CD3+, CD4+, CD44+) expressing IL-17 (a), TNFα (b), IFNγ and TNFα (c) IFNγ alone (d), or chemokine receptors CXCR5 (e) and CXCR3 (f) was determined by ex vivo stimulation of cells with PMA/Ionomycin followed by intracellular staining and flow cytometry. The data points represent the mean (±SD) of values from 4–6 mice (a–f). *p≤0.05, ns-not significant.
Littermate controls (Con-Vacc) and Stat3.Cd4−/− mice were mucosally vaccinated with ESAT61–20 in combination with LT-IIb, rested and Mtb-infected. Mice were sacrificed on day 30 post infection and formalin fixed samples were stained with H&E or CD3 (red), and B220 (green) and the area occupied by inflammatory lesions/lobe (a) and average size of B cell lymphoid follicles (b) was quantified using the morphometric tool of the Zeiss Axioplan microscope. Representative figures showing a typical inflammatory lesions (c-top panel) and B cell lymphoid follicle (c-bottom panel) is shown. Original magnification for H&E sections, 100X; immunofluorescent sections, 200X. The data points represent the mean (±SD) of values from 4–6 mice (a–c). *p≤0.05, ***p≤0.0005
B6, Ifng−/− and Il17−/− were mucosally vaccinated with ESAT61–20 in combination with LT-IIb, rested, challenged with Mtb and sacrificed on day 30 post infection. Formalin-fixed, paraffin-embedded lung sections were assayed for CXCL9 mRNA localization by ISH using a murine CXCL9 mRNA probe or control probe. Original magnification 400X. Data shown from one representative lung, but similar staining as observed in all lungs within the group (n= 4–6 mice).
B6 and Il17−/− mice were either uninfected (UI) or mucosally vaccinated with ESAT61–20 in LT-IIb, rested, Mtb-infected (Vacc) and sacrificed on day 30 post infection. The number of activated CD4+ T cells (CD3+, CD4+, CD44+) (a), expressing CXCR5 (b), CXCR3 (c), or producing IFNγ (d), TNFα (e), IL-2 (f), IL-17 (g) or co-producing IL-17 and IFNγ (h) was determined by ex vivo stimulation of cells with PMA/Ionomycin followed by intracellular staining and flow cytometry. The data points represent the mean (±SD) of values from 4–6 mice (a–h). *p≤0.05, ** p≤0.005, ***p≤0.0005, ns-not significant.