

Abstract
Background
Red blood cell (RBC) alloantibodies to non-self antigens may develop following transfusion or pregnancy, leading to morbidity and mortality in the form of hemolytic transfusion reactions or hemolytic disease of the newborn. A better understanding of the mechanisms of RBC alloantibody induction, or strategies to mitigate the consequences of such antibodies, may ultimately improve transfusion safety. However, such studies are inherently difficult in humans.
Study Design and Methods
We recently generated transgenic mice with RBC specific expression of the human KEL glycoprotein, with the KEL2 or KEL1 antigens. Herein, we investigate recipient alloimmune responses to transfused RBCs in this system.
Results
Transfusion of RBCs from KEL2 donors into wild type recipients (lacking the human KEL protein but expressing the murine KEL orthologue) resulted in dose dependent anti-KEL glycoprotein IgM and IgG antibody responses, enhanced by recipient inflammation with poly (I:C). Boostable responses were evident upon repeat transfusion, with morbid appearing alloimmunized recipients experiencing rapid clearance of transfused KEL2 but not control RBCs. Although KEL1 RBCs were also immunogenic following transfusion into wild type recipients, transfusion of KEL1 RBCs into KEL2 recipients or vice versa failed to lead to detectable anti-KEL1 or anti-KEL2 responses.
Conclusions
This murine model, with reproducible and clinically significant KEL glycoprotein alloantibody responses, provides a platform for future mechanistic studies of RBC alloantibody induction and consequences. Long term translational goals of these studies include improving transfusion safety for at risk patients.
INTRODUCTION
RBC alloimmunization is a clinically significant problem that affects men, women, and children alike. These RBC alloantibodies may increase morbidity and morality, putting patients at risk for acute and hemolytic transfusion reactions, hemolytic disease of the fetus and newborn, or delays in locating compatible blood. In fact, hemolytic transfusion reactions due to non-ABO antibodies were the 2nd leading cause of transfusion associated death reported to the US FDA last year1. 1/600 pregnancies are affected by RBC alloantibodies, with some women entering pregnancy with RBC alloantibodies due to prior transfusions and others becoming alloimmunized during pregnancy or after delivery2. Thus, RBC alloantibodies can be dangerous in transfusion and pregnancy settings alike.
Outside of transfusion avoidance or limited phenotypic/genotypic matching, very few therapeutic strategies exist to prevent or to minimize the dangers of RBC alloimmunization. To date, much has been learned about the functional characteristics and immunogenicity of individual human RBC antigens3,4. However, the numbers of variables involved in each transfusion make studying factors influencing the formation of alloantibodies difficult. Such variables may include donor and recipient health status, donor and recipient genetics and antigenic differences, and donor unit preparation/storage. Furthermore, ethical and practical considerations limit in depth studies of the mechanisms of RBC alloantibody induction in humans. Lastly, human studies involving RBC clearance patterns have historically been limited by the method (radioactivity) of RBC labeling, which allows a gross estimate of RBC clearance but limits in depth analyses of individual transfused RBCs.
The therapeutic void for prevention and treatment of RBC alloimmunization is due, in part, to a lack of existing experimental models. In fact, the mechanism of action of the sole targeted immunomodulatory therapy in existence (polyclonal anti-D or RhoGam) remains unknown5,6. Although a number of monoclonal anti-D preparations have been tested in humans7,8, no animal model with RBC specific expression of the complex D antigen exists to delve in depth into potential mechanisms. With the realization that native murine RBC antigens9,10 have not been thoroughly defined and are minimally immunogenic, our group has developed or worked with a number of transgenic murine models of RBC alloimmunization over the past decade11,12. Some have model antigen expression on RBCs (membrane bound hen egg lysozyme or mHEL)13, others have human RBC antigen expression (human glycophorin or hGPA)14,15 or Duffyb 16, and yet others have a hybrid of model and human antigens (hen egg lysozyme, ovalbumin, Duffyb or HOD) 17,18.
Each animal model has its own set of strengths and weaknesses; however, none recapitulates all aspects of a clinically significant human RBC antigen. The mHEL and Duffyb models have ubiquitous antigen expression, something not found in many human RBC antigens; stringent removal of contaminating WBCs and platelets is necessary for evaluation of RBC antigen responses. Anti-HEL antibodies generated in response to mHEL or HOD transfusions have been shown to result in selective removal of the HEL antigen instead of RBC clearance19; ongoing studies are further evaluating this phenomenon. The hGPA model results in anti-hGPA antibodies20 when donors and recipients are MHC matched and when recipients are pre-treated with poly (I:C), though anti-hGPA (e.g. anti-M family) antibodies in humans are rarely clinically significant. Interestingly, hGPA transfusion in the absence of inflammation leads to non-responsiveness and potentially antigen specific tolerance15. Passively infused anti-hGPA antibodies have been shown to lead to clearance of antigen positive RBCs21–24, with mechanistic studies ongoing. Thus, much has been learned and much remains to be learned from these models; however, none to date results in a reproducible, robust, and boostable alloantibody response in the absence of an adjuvant, leading to RBC clearance with clinically significant sequelae.
We sought to develop a new model of RBC alloimmunization with the above characteristics, ideally utilizing an existing human antigen. The “Kell factor” was initially described half a century ago, after the direct antiglobulin test was created and performed in instances following hydropic fetal complications25. Subsequently, anti-Kell family antibodies were shown to result in fatal hemolytic transfusion reactions26. Since that time, it has been shown that the Kell factor is actually a family of antigens, with Kell being a glycoprotein with endopeptidase activity4. Multiple epitopes on the Kell protein have been defined as clinically significant antigens, including Jsa/b and Kpa/b27. The most well known, however, are KEL1 (also known simply as Kell) and KEL2 (also known as Cellano). Approximately 10% of transfusions are mismatched for KEL1/KEL2, with anti-KEL1 as well as anti-KEL2 antibodies leading to hemolysis of incompatible transfused RBCs or of incompatible fetal RBCs. In fact, KEL alloantibodies are a leading cause of transfusion and pregnancy associated morbidity/mortality today28–33.
Herein, we describe recipient immune responses to our newly generated murine KEL model. This model involves the human KEL antigen expressed in an RBC specific fashion on murine RBCs34. Despite some homology between murine and human KEL35, nearly all wild type C57BL/6 recipients of KEL2 RBCs form anti-KEL glycoprotein antibodies after a single transfusion. This response is boostable upon repeat exposure, enhanced in the presence of recipient inflammation with poly (I:C), and clinically significant in that anti-KEL glycoprotein antibodies lead to clearance of antigen positive RBCs. However, antibody generation appears limited to recipients that lack the human KEL antigen altogether, with animals with RBCs expressing an antithetical antigen (e.g. KEL2 RBCs into KEL1 recipients and vice versa) failing to develop detectable anti-KEL2 or anti-KEL1 alloantibodies under the conditions tested. Taken together, these findings lay the groundwork for in depth analyses of factors influencing RBC alloantibody induction as well as targeted immunomodulatory therapies to prevent the formation or to mitigate the dangers of RBC alloantibodies.
METHODS
Mice
C57BL/6 mice were purchased from the National Cancer Institute (Frederick, MD); KEL2 and KEL1 transgenic mice expressing the human KEL glycoprotein (previously published as “KEL2B” and “KEL1A”) were generated in our laboratory 34 and bred at Emory. All animals were housed in the Emory University Department of Animal Resources facilities, and all procedures and protocols were approved by the Emory University Institutional Animal Care and Use Committee.
Murine blood collection, fluorescent labeling, and transfusion
RBCs were collected into acid-citrate-dextrose (ACD) and washed 3 times with phosphate buffered saline (PBS) to remove residual citrate. Anti-KEL2 (Clone LKL1, Alba, Edinburgh, UK) or anti-KEL1 (Mima 23, generously provided by Greg Halverson of the New York Blood Center) were utilized to confirm the presence of the KEL2 or KEL1 antigens on transfused RBCs. KEL2 RBCs were transfused in titrated amounts (0.5, 5, and 50 μL of RBCs, diluted in PBS to 300 μL total volume) via lateral tail vein; in some experiments, recipient animals were transfused every 2 weeks and in others, recipients were pretreated with 100 μg of poly (I:C) (Amersham, Piscataway, NY) given intraperitoneally (i.p.) 2 hours prior to transfusion. In a subset of experiments, RBCs were labeled with chloromethylbenzamido 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (CM-DiI) or 3,3′-dihexadecyloxacarbocyanine perchlorate (DiO) according to the manufacturer’s instructions as previously described (Molecular Probes, Eugene OR)24,36,37. After labeling, experimental and control RBCs were mixed at a 1:1 ratio and recipient animals were transfused via lateral tail vein with 50–75 μL of each blood type, diluted in PBS to 300 μL total volume.
Human RBC assays
Human RBCs from residual segments were tested for KEL1 and KEL2 expression by flow cytometry, using monoclonal reagents. Those with KEL2 but not KEL1 expression were selected, and some RBCs were treated with 0.2 M dithiothreitol (DTT). These DTT treated and untreated human KEL2 RBCs (alongside DTT treated and untreated transgenic murine KEL2 RBCs) were then utilized as targets for flow cytometric crossmatch with sera from alloimmunized mice.
Flow cytometry
Serum was analyzed for the presence of anti-KEL IgM and IgG utilizing indirect immunofluorescence (flow cytometric crossmatch) with KEL2, KEL1, or control C57BL/6 RBCs; an adjusted mean fluorescence intensity (MFI) was calculated by subtracting the signal of serum crossmatched with antigen negative targets from that of serum crossmatched with antigen positive targets. Transfused RBCs were analyzed for presence of bound IgM and IgG using APC-conjugated goat anti-mouse IgM and Alexafluor® 488-conjugated goat anti-mouse IgG, respectively (Jackson Immunoresearch, West Grove PA), after gating on the lipophilically labeled RBCs. Post-transfusion KEL2 RBC recovery and survival were determined by flow cytometry and lipophilic dye fluorescence, utilizing a ratio of transfused KEL2 RBCs to control C57BL/6 RBCs. All antibodies were used at 1:100 dilution and all samples were analyzed on a 4-color BD FACSCalibur.
Cytokine Analysis
Serum cytokines, including interleukin (IL)-6, keratinocyte-derived chemokine (KC, the murine equivalent to IL-8 in humans), monocyte chemoattractant protein (MCP)-1, macrophage inflammatory protein (MIP)-1β, TNF-α and IFN-γ, were evaluated 90–120 minutes post transfusion using the Cytometric Bead Array Mouse Flex Kit (BD Biosciences, San Diego, CA)
Statistical Analysis
Statistical significance was determined by performing a Students t-test for comparison of 2 groups or one-way analysis of variance (ANOVA) with Tukey’s posttest when 3 groups were compared using Graphpad Prism software (San Diego, CA). Error bars represent one standard deviation and differences were considered statistically significant with p-values ≤0.05.
RESULTS
Serologic responses of C57BL/6 recipients to transfused KEL2 RBCs
Blood was collected from KEL2 donors, and the presence of KEL2 expression on the pooled RBCs was determined prior to transfusion using monoclonal anti-KEL2 (Figure 1A). The equivalent of 1 human “unit” of KEL2 RBCs (50–75 μL) was then transfused into wild type C57BL/6 recipients. 14–21 days post-transfusion, recipient serum was crossmatched with KEL2 or wild type control C57BL/6 RBC targets, using anti-mouse IgG as a secondary reagent (Figure 1B shows a representative crossmatch after 3 transfusions). An adjusted mean fluorescence intensity (MFI) was calculated by subtracting the background signal of serum crossmatched with wild type RBCs from that of serum crossmatched with KEL2 RBCs. The only known difference between wild type C57BL/6 and KEL2 RBC targets is the KEL glycoprotein antigen34, thus eliminating MHC mismatch as a variable and leading one to hypothesize that the observed adjusted MFI signal is due to anti-KEL glycoprotein alloantibodies.
Figure 1. Transgenic murine RBCs express the human KEL2 antigen, and C57BL/6 recipients of KEL2 RBC transfusion make alloantibodies with KEL specificity.
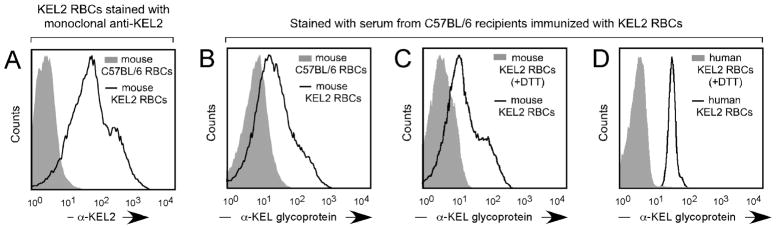
(A) Murine RBCs from KEL2 transgenic donors were collected and stained with anti-KEL2 prior to transfusion. (B) Serum from C57BL/6 recipients transfused with KEL2 RBCs was crossmatched with murine KEL2 RBCs (solid line) or wild type C57BL/6 RBCs (shaded histogram). (C) Serum from C57BL/6 recipients transfused with KEL2 RBCs was crossmatched with murine KEL2 RBCs (solid line) or with DTT treated murine KEL2 RBCs (shaded histogram). (D) Serum from C57BL/6 recipients transfused with KEL2 RBCs was crossmatched with human RBCs expressing KEL2 (solid line) or with DTT treated human RBCs (shaded histogram); representative results are shown.
To further evaluate the specificity of the anti-KEL glycoprotein antibody response, serum from immunized mice was crossmatched with murine KEL2 RBCs or murine KEL2 RBCs treated with 0.2M dithiothreitol (DTT) (Figure 1C). DTT is known to reduce disulfide bonds to free sulfhydryl groups, with disulfide bonds being a requirement for KEL antigen integrity38. These serum crossmatches were repeated using human RBCs expressing the KEL2 antigen or human RBCs expressing the KEL2 antigen that had been treated with 0.2M DTT as targets (Figure 1D). In both instances, the observed reactivity with intact KEL2 expressing murine or human target RBCs was essentially eliminated upon crossmatch with DTT treated target RBCs. These results lend further support to the KEL specificity of the alloantibody observed in the sera of C57BL/6 recipients following transfusion with transgenic murine KEL2 RBCs.
In order to evaluate dose responses to transfused KEL RBCs, 0.5, 5, or 50 μL of KEL2 RBCs were transfused into C57BL/6 recipients, with serum evaluated 2 weeks later by flow crossmatch with transgenic murine KEL2 or C57BL/6 RBCs. In 2/2 experiments (n=27 animals total), a dose titratable anti-KEL glycoprotein IgG response was observed (Figure 2A shows adjusted MFI). For the remainder of the experiments described herein, 50 μL of RBCs were thus utilized for transfusion. To more completely characterize the immune response of C57BL/6 recipients to transfused KEL2 RBCs, wild type or KEL2 syngeneic recipients were transfused with 50 μL of KEL2 RBCs, and anti-KEL IgM and IgG in the serum was measured by flow crossmatch from days 3 through 28 post-transfusion. In a compilation of 3 independent experiments (n=24 animals total), anti-KEL IgM was observed in the serum of C57BL/6 but not syngeneic recipients by day 3 post-transfusion, peaking by day 5. Anti-KEL IgG was observed in the serum of C57BL/6 but not syngeneic recipients by day 7 post-transfusion, peaking near days 14–21. (Figure 2B–C shows a representative experiment).
Figure 2. C57BL/6 recipients lacking human KEL have a dose dependent anti-KEL glycoprotein antibody response.

(A) Control (KEL2) or wild type C57BL/6 recipients were transfused with 0.5, 5, or 50 μL of KEL2 RBCs, with serum anti-KEL glycoprotein IgG evaluated 2 weeks post-transfusion. Serial evaluations of serum anti-KEL glycoprotein IgM (B) or IgG (C) were completed in KEL2 or wild type C57BL/6 recipients after a single transfusion of 50 μL of KEL2 RBCs. Results are representative of 2–3 independent experiments with at least 3–5 mice/group; *p<0.05.
Assessment of repeat antigen exposure and and cytokine responses
Given the reported evanescence patterns of RBC alloantibodies in humans39,40, antigen positive RBCs may be unintentionally transfused into humans with alloantibodies that fall below the level of detection by traditional blood bank antibody screening methodologies. Anamnestic responses may subsequently result, with a rapid increase in RBC alloantibody titer and premature clearance of transfused RBCs. In fact, some such transfusions may lead to such significant hemolysis of both transfused and “by-stander” RBCs that renal failure, DIC, and death result41. Past efforts by our laboratory to create murine models with antibody responses that are “boostable” in the absence of coexistent recipient inflammation have thus far not been successful, likely due to characteristics of the antigens previously studied. To examine potential boostable responses in our newly developed KEL system, KEL2 RBCs were transfused every 2 weeks for a total of 3 transfusions into C57BL/6 recipients, with total anti-KEL Igs measured 2 weeks after each transfusion. In 3/3 experiments (n=15 recipients total), significant increased responses were observed following each transfusion (Figure 3A shows a representative experiment). This increased alloantibody response was not simply due to a continued increase in anti-KEL titer over time, as control animals receiving a single transfusion had peak alloantibody responses at approximately 14–21 days post-transfusion.
Figure 3. C57BL/6 recipients have “boostable” responses to repeat KEL2 exposure, with a proinflammatory serum cytokine storm.

(A) C57BL/6 recipients were transfused every 2 weeks with KEL2 RBCs, with serum anti-KEL glycoprotein Igs evaluated on day 14 after each transfusion. (B–F) Serum cytokine responses in alloimmunized animals, 90–120 minutes after a 4th KEL2 RBC transfusion. Results are representative of 2–3 independent experiments with at least 3–5 mice/group; *p<0.05.
In humans, incompatible RBC transfusions are the 2nd leading cause of transfusion associated death1. To date, however, few animal models have been created in which repeat transfusions not only lead to high titer alloantibodies but also to adverse clinical outcomes. Two laboratories have reported an animal model in which anti-hGPA antibodies resulted in clearance of incompatible RBCs and a pro-inflammatory serum cytokine storm21–24,36; however, these antibodies were passively infused and not actively generated. In our current studies, C57BL/6 recipients were actively alloimmunized through repeat KEL2 transfusions. These alloimmunized recipients were then re-transfused a 4th time with “incompatible” KEL2 RBCs and observed. Within 5 minutes of the incompatible transfusion, recipients appeared ill and hunched over. Cytokine bead arrays were performed on recipient serum 90–120 minutes post-transfusion, revealing statistically significant elevations of IL-6, KC, MCP-1, MIP-1B, and TNF-α in alloimmunized animals receiving “incompatible” RBCs; no changes were observed in IFN-γ (Figure 3B–F shows representative data).
Anti-KEL responses in the presence of recipient inflammation with poly (I:C)
It is known that a “danger” signal of sorts leads to an augmented immune response in a number of settings, both clinical and experimental42. We have previously reported that recipient inflammation with the double stranded RNA poly (I:C) enhances RBC alloimmunization in multiple different murine RBC alloimmunization models, including the mHEL, hGPA, and HOD models15,43,44. To evaluate the effect of recipient inflammation on alloimmunization in our KEL model, C57BL/6 recipient animals were treated with 100 μg of poly (I:C) i.p.; control animals were treated with PBS i.p. Subsequently, all recipients were transfused with KEL2 RBCs and serum alloantibody responses were evaluated at multiple time points post-transfusion. In 3/3 experiments (n=27 animals total), poly (I:C) treated animals had significantly higher serum anti-KEL IgG levels post-transfusion than control animals (Figure 4A shows compilation data on day 14 post-transfusion, p<0.05). To determine boostable responses in the presence of poly (I:C), 3 separate cohorts of C57BL/6 recipients were transfused 1, 2, or 3 times in the presence of poly (I:C). Serum samples were evaluated every 2 weeks in each cohort, out 42 days from the time of the initial transfusion. Similar to what was observed in the absence of poly (I:C), boostable responses were observed in the presence of poly (I:C) with peak titers noted approximately 2 weeks post-transfusion (Figure 4B shows representative data). These responses were not an artifact of increased anti-KEL responses over time, as control animals receiving a single transfusion did not have significant increases in anti-KEL beyond 14–21 days post-transfusion (Figure 4B shows 3 distinct cohorts of animals receiving 1, 2, or 3 transfusions, with anti-KEL glycoprotein antibody responses measured over time in each cohort).
Figure 4. Recipient inflammation with poly (I:C) enhances anti-KEL responses, with “boostable” responses.
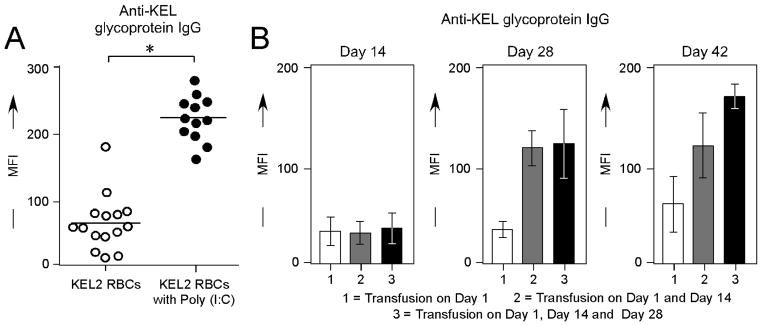
(A) Serum anti-KEL glycoprotein IgG responses in C57BL/6 animals transfused with KEL2 RBCs in the presence or absence of recipient poly (I:C) pretreatment; straight serum, 14 days post-transfusion. (B) Serum anti-KEL glycoprotein IgG responses after 1, 2, or 3 KEL2 RBC transfusions in the presence of poly (I:C); sera diluted 1:10, tested every 14 days. Results are representative of 2–3 experiments with 3–5 mice/group; *p<0.05
Anti-KEL RBC binding and clearance
The studies described to this point have focused on serologic evaluation of transfused recipients. Serum is the focus of clinical transfusion medicine evaluations, given difficulties in evaluating antibody bound to transfused RBCs. In fact, “mixed field” reactions, in which a subset of recipient RBCs (e.g. those transfused) have bound antibody whereas another subset of recipient RBCs (e.g. the patient’s own RBCs) lack bound antibody, can be quite technically challenging to evaluate. Practically speaking, however, an evaluation of the RBC antibody bound in vivo may be a better predictor of the fate of the transfused RBCs than an in vitro test of serum alloantibodies.
To allow visualization of cells in the recipient post-transfusion, KEL2 RBCs were labeled before transfusion with a lipophilic dye (DiI) that intercalates into the lipid RBC membrane; control C57BL/6 RBCs were labeled with a dye that fluoresces on a different channel (DiO). These dyes have been carefully dose titrated, such that the RBC circulatory half life remains unaffected34. KEL2 and C57BL/6 RBCs were mixed and transfused. Direct antiglobulin tests (DAT) for IgM and IgG were performed at multiple time points post-transfusion on transfused DiI positive KEL2 RBCs, comparing signal on transfused DiI positive KEL2 RBCs to that of control C57BL/6 RBCs (Figure 5A–B). In 3/3 experiments (n=24 animals total), anti-KEL glycoprotein IgM and IgG were detected on circulating KEL2 RBCs by approximately days 3 and 7, respectively. The timing of the initial detection of anti-KEL bound to circulating RBCs thus closely parallels that of anti-KEL detected in serum.
Figure 5. Circulating anti-KEL glycoprotein Igs bind to transfused KEL2 RBCs and are associated with KEL2 RBC clearance.

KEL2 and C57BL/6 RBCs were labeled with DiI and DiO, respectively, prior to transfusion. RBC bound IgM (A) and IgG (B) were evaluated serially post-transfusion on DiI positive KEL RBCs; shaded histograms are control antigen negative cells. Post-transfusion survival and recovery of KEL2 RBCs was determined by comparing a ratio of DiI KEL to DiO C57BL/6 RBCs (C). These studies were completed KEL2 and wild type C57BL/6 recipients (D); similar studies were also completed following a 2nd KEL2 transfusion in recipients initially transfused with or without poly (I:C) (E). Error bars represent standard deviation and results represent at least 3 independent experiments with 3–5 mice/group.
Transfused recipients were also serially evaluated post-transfusion for clearance of KEL2 RBCs. A ratio of DiI positive KEL2 RBCs to DiO positive C57BL/6 RBCs was determined at multiple time points post-transfusion (Figure 5C) in syngeneic KEL2 recipients or wild type naïve C57BL/6 recipients. Although little to no detectable KEL specific RBC clearance was observed in syngeneic KEL2 recipients to which KEL2 is a self-antigen, a slow relative decline in the ratio of transfused KEL2 to C57BL/6 RBCs was appreciated in wild type C57BL/6 recipients over a 28 day period (Figure 5D depicts a representative experiment with 3–5 animals/group).
Given the morbid appearance and serum cytokine storm observed in alloimmunized animals transfused with “incompatible” KEL2 RBCs, we hypothesized that KEL specific RBC clearance would rapidly occur. To test this hypothesis, alloimmunized or naïve C57BL/6 recipients were transfused with DiI labeled KEL2 and DiO labeled C57BL/6 RBCs, with post-transfusion KEL2 RBC recovery and survival tracked. As hypothesized, the “incompatible” KEL2 RBCs were cleared more rapidly post-transfusion in alloimmunized than naïve C57BL/6 recipients (Figure 5E depicts a representative experiment with 3–5 animals/group). Furthermore, recipients who were initially treated with poly (I:C) and transfused, with demonstrable higher titer anti-KEL glycoprotein alloantibodies, had subtly higher rates of KEL specific RBC clearance (Figure 5E). This clearance was not an artifact of decreased stability of the KEL2 RBCs in general, however, as these RBCs had a normal circulatory half life in syngeneic KEL2 recipients.
Serum responses to single amino acid polymorphisms on murine RBCs
Although some human RBC antigens (such as D) may be present on donor RBCs and absent on recipient RBCs, other human antigens differ by just a single amino acid polymorphism to their “antithetical” antigen. To test the immunologic effect of crossing this small antigenic barrier in mice, a separate strain of animals was created with RBC specific expression of the human KEL glycoprotein expressing the KEL1 antigen34 (also known as “Kell”). Unlike the robust anti-KEL glycoprotein response observed after 3 transfusions in wild type C57BL/6 recipients, no anti-KEL2 IgG could be detected in KEL1 recipients (Figure 6A). Furthermore, transfusions in the presence of poly (I:C) also failed to lead to detectable anti-KEL2 responses in KEL1 recipients.
Figure 6. No detectable alloimmune responses to KEL2 RBCs occur in KEL1 recipients (or vice versa), whose KEL RBC antigens differ by a single amino acid polymorphism.
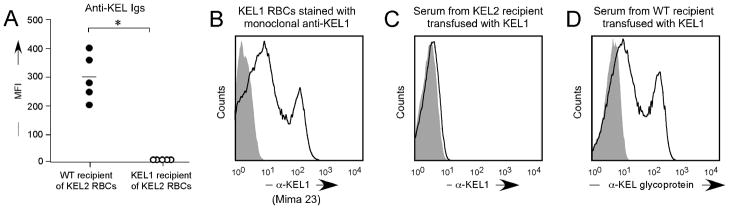
(A) Serum responses of KEL1 or C57BL/6 recipients after 3 transfusions of KEL2 RBCs RBCs. (B) KEL1 RBCs were stained with monoclonal anti-KEL1 (Mima 23) prior to transfusion. Representative serum response of KEL2 (C) or C57BL/6 (D) recipients after 3 transfusions with KEL1 RBCs. Results are representative of 2–3 independent experiments with 2–5 mice/group; *p<0.05.
Additional studies were completed using KEL1 RBCs transfused into KEL2 recipients, which is the scenario most likely to be encountered in human clinical setting. We have previously shown stable KEL1 antigen expression, epitope integrity, and post-transfusion recovery/survival in these animals34; Figure 6B shows KEL1 transgenic RBCs stained with a monoclonal anti-KEL1 reagent (Mima 23, generously provided by Greg Halverson of the New York Blood Center). KEL1 RBCs were transfused every 2 weeks x 3 into KEL2 recipients, in the presence of poly (I:C). As described in the KEL2 donor/KEL1 recipient setting, the KEL1 donor/KEL2 recipient setting also failed to generate detectable serum anti-KEL1 responses (Figure 6C). This lack of a response was not due to KEL1 RBCs being incapable of being immunogenic, as wild type C57BL/6 recipients made detectable anti-KEL glycoprotein antibodies after KEL1 RBC transfusion (Figure 6D).
DISCUSSION
Herein, we have described our initial observations of alloimmune responses to transfused RBCs from newly generated transgenic donors expressing a human RBC antigen. These donors have RBC specific expression of the KEL glycoprotein34, which makes up a blood group of antigens in humans. The KEL glycoprotein in these animals has intact expression of multiple epitopes found in humans, including Jsb, Kpb, and KEL2 (also known as Cellano)34. KEL2 RBCs transfused into C57BL/6 animals, which lack the human KEL antigen altogether, result in robust, dose titratable, and boostable anti-KEL glycoprotein IgM and IgG responses, which are further enhanced by recipient inflammation with the double stranded RNA poly (I:C).
“Responder” rates of KEL alloimmunization approach 100% in this model, presumably due in part to the degree of donor/recipient RBC antigenic differences. The model somewhat resembles that of D in humans, in that donors may express the D antigen which recipients lack altogether. It should be noted that murine KEL is approximately 75% orthologous to human KEL at the amino acid level35, and thus the human KEL glycoprotein is not altogether foreign to the murine recipients. Given that very few humans actually lack the KEL glycoprotein, additional transgenic animals were generated to test the impact on RBC alloimmunogenicity of a single amino acid polymorphism between donor and recipient. Both KEL2 and KEL1 animals have RBC specific expression of the human KEL glycoprotein, with a single amino acid polymorphism difference. Unlike the high responder rates observed when a relatively large antigenic barrier (KEL2 into wild type C57BL/6) was crossed, no antibody responses to date have been detected with KEL2 RBCs transfused into KEL1 recipients or vice versa. It is unclear if KEL1 or KEL2 mice are capable of presenting the polymorphic peptide between KEL1 and KEL2 in their MHC II; as C57BL/6 animals have a single MHC II molecule, this is a distinct possibility. Ongoing studies are continuing to investigate KEL1/KEL2 responses, including MHC diversity analyses.
In addition to studies of immunity induction, this KEL model allows for studies of the fate of transfused RBCs. Human studies utilizing chromium labeling are somewhat limited to blunt evaluations of clearance curves. The strategy of labeling RBCs with a lipophilic dye, as previously described by our group, allows for in depth flow cytometric evaluation of the transfused cells. A slow but reproducible pattern of KEL2 specific RBC clearance was noted following an initial transfusion, with more rapid clearance and a pro-inflammatory serum cytokine response observed in previously alloimmunized recipients with higher titer alloantibodies. The degree of intravascular versus extravascular hemolysis cannot be evaluated from the current studies, as extensive blood or urine chemistry analyses were not completed. However, there was no appreciable “by-stander” hemolysis of the recipient’s own RBCs, as hematocrit values were noted to be similar in alloimmunized and non-alloimmunized recipients of KEL2 RBCs (data not shown). Clearance patterns correlated with detectable serum and RBC bound anti-KEL Igs, with no clearance of antigen negative C57BL/6 RBCs noted. However, in no experiment were all transfused KEL2 RBCs cleared by day 28. These findings suggest that KEL2 RBCs remaining in circulation may be acquiring a survival advantage of sorts. We have previously reported RBC populations resistant to hemolysis in the hGPA system36. In addition, KEL antigens are known to have the potential to undergo weakened antigenicity during incompatibility in human settings45,46; ongoing studies are investigating clearance as well as clearance resistance mechanisms in our model.
Limitations to the KEL into C57BL/6 model and experiments described must be considered. One such consideration is that this model resembles that of D in humans more so than that of KEL1/KEL2, in that the human KEL antigen is present on donor RBCs but lacking on recipient RBCs. This is, perhaps, more of a deviation from what is “human” than a true scientific limitation of the model; findings from this described model will likely have applicability to some but not all antigens. Other considerations include the potential confounders involved in utilizing lipophilic dyes to track transfused RBCs; despite careful titration and control studies, their impact on study results cannot definitively be ruled out. Another consideration is that poly (I:C) is just one type of recipient inflammation, and other toll like receptor agonists43 or authentic infections may have differential effects on alloimmunization outcomes. A last consideration is that these studies were completed utilizing KEL2 and KEL1 animals which have approximately 1200 and 800 copies of KEL glycoprotein per RBC34. Additional KEL transgenic animals have since been created with lower and higher RBC antigen copy numbers, with ongoing studies investigating the impact of copy number and dose responses on alloimmunogenicity and clinical consequences.
In sum, the KEL model described herein offers a reductionist system in which to better understand both the induction of KEL specific RBC alloantibodies as well as their clinical consequences. A number of questions remain unanswered, and much work remains to be completed. However, this model, with a boostable, clinically significant alloantibody response, will serve as a platform for the investigation of immunomodulatory strategies to prevent or mitigate RBC alloantibody formation as well as subsequent sequelae. An improvement in the understanding and treatment of RBC alloimmunization, in a bench to bedside and back manner, remains a necessity in improving transfusion safety in men, women, and children alike.
Acknowledgments
This work was supported in part by funding from the National Institutes of health to JEH (K08 HL092959, R21 HL115696) and to JCZ (P01 HL086773).
Footnotes
All authors performed the laboratory work and contributed to the writing and proofreading of this manuscript.
The authors declare they have no conflicts of interest related to this work; the transgenic animals were generated in part by funding from Immucor to JCZ.
References
- 1.FDA. Fatalities Reported to the FDA Following Blood Collection. 2012 http://wwwfdagov/BiologicsBloodVaccines/SafetyAvailability.
- 2.Moise KJ, Jr, Argoti PS. Management and prevention of red cell alloimmunization in pregnancy: a systematic review. Obstet Gynecol. 2012;120(5):1132–1139. doi: 10.1097/aog.0b013e31826d7dc1. [DOI] [PubMed] [Google Scholar]
- 3.Reid ME, Mohandas N. Red blood cell blood group antigens: structure and function. Seminars in Hematology. 2004;41(2):93–117. doi: 10.1053/j.seminhematol.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 4.Reid ME, Lomas-Francis C. The Blood Group Antigen Facts Book. 2. Amsterdam: Elsevier Academic Press; 2004. [Google Scholar]
- 5.Brinc D, Denomme GA, Lazarus AH. Mechanisms of anti-D action in the prevention of hemolytic disease of the fetus and newborn: what can we learn from rodent models? Curr Opin Hematol. 2009;16(6):488–496. doi: 10.1097/MOH.0b013e32833199ed. [DOI] [PubMed] [Google Scholar]
- 6.Kumpel BM. On the immunologic basis of Rh immune globulin (anti-D) prophylaxis. Transfusion. 2006;46(9):1652–1656. doi: 10.1111/j.1537-2995.2006.00924_1.x. [DOI] [PubMed] [Google Scholar]
- 7.Kumpel BM. Monoclonal anti-D development programme. Transpl Immunol. 2002;10(2–3):199–204. doi: 10.1016/s0966-3274(02)00066-7. [DOI] [PubMed] [Google Scholar]
- 8.Kumpel BM. Efficacy of RhD monoclonal antibodies in clinical trials as replacement therapy for prophylactic anti-D immunoglobulin: more questions than answers. Vox Sang. 2007;93(2):99–111. doi: 10.1111/j.1423-0410.2007.00945.x. [DOI] [PubMed] [Google Scholar]
- 9.Amos DB, Zumpft M, Armstrong P. H-5.A AND H-6.A, TWO MOUSE ISOANTIGENS ON RED CELLS AND TISSUES DETECTED SEROLOGICALLY. Transplantation. 1963;1:270–283. doi: 10.1097/00007890-196301030-00002. [DOI] [PubMed] [Google Scholar]
- 10.Azen EA, Davisson MT, Cherry M, Taylor BA. Prp (proline-rich protein) genes linked to markers Es-12 (esterase-12), Ea-10 (erythrocyte alloantigen), and loci on distal mouse chromosome 6. Genomics. 1989;5(3):415–422. doi: 10.1016/0888-7543(89)90004-9. [DOI] [PubMed] [Google Scholar]
- 11.Hod EA, Zimring JC, Spitalnik SL. Lessons learned from mouse models of hemolytic transfusion reactions. Curr Opin Hematol. 2008;15(6):601–605. doi: 10.1097/MOH.0b013e328311f40a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zimring JC, Hendrickson JE. The role of inflammation in alloimmunization to antigens on transfused red blood cells. Current Opinion in Hematology. 2008;15(6):631–635. doi: 10.1097/MOH.0b013e328313695e. [DOI] [PubMed] [Google Scholar]
- 13.Hendrickson JE, Chadwick TE, Roback JD, Hillyer CD, Zimring JC. Inflammation enhances consumption and presentation of transfused RBC antigens by dendritic cells. Blood. 2007;110(7):2736–2743. doi: 10.1182/blood-2007-03-083105. [DOI] [PubMed] [Google Scholar]
- 14.Auffray I, Marfatia S, de Jong K, et al. Glycophorin A dimerization and band 3 interaction during erythroid membrane biogenesis: in vivo studies in human glycophorin A transgenic mice. Blood. 2001;97(9):2872–2878. doi: 10.1182/blood.v97.9.2872. [DOI] [PubMed] [Google Scholar]
- 15.Smith NH, Hod EA, Spitalnik SL, Zimring JC, Hendrickson JE. Transfusion in the absence of inflammation induces antigen-specific tolerance to murine RBCs. Blood. 2012;119(6):1566–1569. doi: 10.1182/blood-2011-09-382655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Campbell-Lee SA, Liu J, Velliquette RW, et al. The production of red blood cell alloantibodies in mice transfused with blood from transgenic Fyb-expressing mice. Transfusion. 2006;46(10):1682–1688. doi: 10.1111/j.1537-2995.2006.00966.x. [DOI] [PubMed] [Google Scholar]
- 17.Desmarets M, Cadwell CM, Peterson KR, Neades R, Zimring JC. Minor histocompatibility antigens on transfused leukoreduced units of red blood cells induce bone marrow transplant rejection in a mouse model. Blood. 2009;114(11):2315–2322. doi: 10.1182/blood-2009-04-214387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hendrickson JE, Hod EA, Spitalnik SL, Hillyer CD, Zimring JC. Storage of murine red blood cells enhances alloantibody responses to an erythroid-specific model antigen. Transfusion. 2010;50(3):642–648. doi: 10.1111/j.1537-2995.2009.02481.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zimring JC, Hair GA, Chadwick TE, et al. Nonhemolytic antibody-induced loss of erythrocyte surface antigen. Blood. 2005;106(3):1105–1112. doi: 10.1182/blood-2005-03-1040. [DOI] [PubMed] [Google Scholar]
- 20.Bao W, Yu J, Heck S, Yazdanbakhsh K. Regulatory T-cell status in red cell alloimmunized responder and nonresponder mice. Blood. 2009;113(22):5624–5627. doi: 10.1182/blood-2008-12-193748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schirmer DA, Song SC, Baliff JP, et al. Mouse models of IgG- and IgM-mediated hemolysis. Blood. 2007;109(7):3099–3107. doi: 10.1182/blood-2006-08-040139. [DOI] [PubMed] [Google Scholar]
- 22.Hod EA, Arinsburg SA, Francis RO, Hendrickson JE, Zimring JC, Spitalnik SL. Use of mouse models to study the mechanisms and consequences of RBC clearance. Vox Sang. 2010;99(2):99–111. doi: 10.1111/j.1423-0410.2010.01327.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hod EA, Cadwell CM, Liepkalns JS, et al. Cytokine storm in a mouse model of IgG-mediated hemolytic transfusion reactions. Blood. 2008;112(3):891–894. doi: 10.1182/blood-2008-01-132092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liepkalns JS, Hod EA, Stowell SR, Cadwell CM, Spitalnik SL, Zimring JC. Biphasic clearance of incompatible red blood cells through a novel mechanism requiring neither complement nor Fcgamma receptors in a murine model. Transfusion. 2012;52(12):2631–2645. doi: 10.1111/j.1537-2995.2012.03647.x. [DOI] [PubMed] [Google Scholar]
- 25.Coombs RR, Mourant AE, Race RR. In-vivo isosensitisation of red cells in babies with haemolytic disease. Lancet. 1946;1(6391):264–266. doi: 10.1016/s0140-6736(46)91925-3. [DOI] [PubMed] [Google Scholar]
- 26.Ottensooser F, Mellone O, Blancalana A. Fatal transfusion reaction due to the Kell factor. Blood. 1953;8(11):1029–1033. [PubMed] [Google Scholar]
- 27.Westhoff CM, Reid ME. Review: the Kell, Duffy, and Kidd blood group systems. Immunohematology. 2004;20(1):37–49. [PubMed] [Google Scholar]
- 28.Bowman JM, Pollock JM, Manning FA, Harman CR, Menticoglou S. Maternal Kell blood group alloimmunization. Obstet Gynecol. 1992;79(2):239–244. [PubMed] [Google Scholar]
- 29.Eder AF. Update on HDFN: new information on long-standing controversies. Immunohematology. 2006;22(4):188–195. [PubMed] [Google Scholar]
- 30.Moise KJ. Fetal anemia due to non-Rhesus-D red-cell alloimmunization. Semin Fetal Neonatal Med. 2008;13(4):207–214. doi: 10.1016/j.siny.2008.02.007. [DOI] [PubMed] [Google Scholar]
- 31.Rimon E, Peltz R, Gamzu R, et al. Management of Kell isoimmunization--evaluation of a Doppler-guided approach. Ultrasound Obstet Gynecol. 2006;28(6):814–820. doi: 10.1002/uog.2837. [DOI] [PubMed] [Google Scholar]
- 32.Vaughan JI, Manning M, Warwick RM, Letsky EA, Murray NA, Roberts IA. Inhibition of erythroid progenitor cells by anti-Kell antibodies in fetal alloimmune anemia. N Engl J Med. 1998;338(12):798–803. doi: 10.1056/NEJM199803193381204. [DOI] [PubMed] [Google Scholar]
- 33.Vaughan JI, Warwick R, Welch CR, Letsky EA. Anti-Kell in pregnancy. Br J Obstet Gynaecol. 1991;98(9):944–945. doi: 10.1111/j.1471-0528.1991.tb13522.x. [DOI] [PubMed] [Google Scholar]
- 34.Smith NH, Henry KL, Cadwell CM, et al. Generation of transgenic mice with antithetical KEL1 and KEL2 human blood group antigens on red blood cells. Transfusion. 2012 doi: 10.1111/j.1537-2995.2012.03641.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee S, Russo DC, Pu J, Ho M, Redman CM. The mouse Kell blood group gene (Kel): cDNA sequence, genomic organization, expression, and enzymatic function. Immunogenetics. 2000;52(1–2):53–62. doi: 10.1007/s002510000251. [DOI] [PubMed] [Google Scholar]
- 36.Liepkalns JS, Cadwell CM, Stowell SR, Hod EA, Spitalnik SL, Zimring JC. Resistance of a subset of red blood cells to clearance by antibodies in a mouse model of incompatible transfusion. Transfusion. 2012 doi: 10.1111/j.1537-2995.2012.03910.x. [DOI] [PubMed] [Google Scholar]
- 37.Zimring JC, Cadwell CM, Chadwick TE, et al. Nonhemolytic antigen loss from red blood cells requires cooperative binding of multiple antibodies recognizing different epitopes. Blood. 2007;110(6):2201–2208. doi: 10.1182/blood-2007-04-083097. [DOI] [PubMed] [Google Scholar]
- 38.Branch DR, Muensch HA, Sy Siok Hian AL, Petz LD. Disulfide bonds are a requirement for Kell and Cartwright (Yta) blood group antigen integrity. Br J Haematol. 1983;54(4):573–578. doi: 10.1111/j.1365-2141.1983.tb02136.x. [DOI] [PubMed] [Google Scholar]
- 39.Tormey CA, Fisk J, Stack G. Red blood cell alloantibody frequency, specificity, and properties in a population of male military veterans. Transfusion. 2008;48(10):2069–2076. doi: 10.1111/j.1537-2995.2008.01815.x. [DOI] [PubMed] [Google Scholar]
- 40.Tormey CA, Stack G. The persistence and evanescence of blood group alloantibodies in men. Transfusion. 2009;49(3):505–512. doi: 10.1111/j.1537-2995.2008.02014.x. [DOI] [PubMed] [Google Scholar]
- 41.King KE, Shirey RS, Lankiewicz MW, Young-Ramsaran J, Ness PM. Delayed hemolytic transfusion reactions in sickle cell disease: simultaneous destruction of recipients’ red cells.[see comment] Transfusion. 1997;37(4):376–381. doi: 10.1046/j.1537-2995.1997.37497265337.x. [DOI] [PubMed] [Google Scholar]
- 42.Matzinger P. The danger model: a renewed sense of self. Science. 2002;296(5566):301–305. doi: 10.1126/science.1071059. [DOI] [PubMed] [Google Scholar]
- 43.Hendrickson J, Roback JD, Hillyer CD, Easley KA, Zimring JC. Discrete toll like receptor agonists have differential effects on alloimmunization to red blood cells. Transfusion. 2008;(48):1869–1877. doi: 10.1111/j.1537-2995.2008.01801.x. [DOI] [PubMed] [Google Scholar]
- 44.Hendrickson JE, Desmarets M, Deshpande SS, et al. Recipient inflammation affects the frequency and magnitude of immunization to transfused red blood cells. Transfusion. 2006;46(9):1526–1536. doi: 10.1111/j.1537-2995.2006.00946.x. [DOI] [PubMed] [Google Scholar]
- 45.Beck ML, Marsh WL, Pierce SR, DiNapoli J, Oyen R, Nichols ME. Auto anti-Kpb associated with weakened antigenicity in the Kell blood group system: a second example. Transfusion. 1979;19(2):197–202. doi: 10.1046/j.1537-2995.1979.19279160294.x. [DOI] [PubMed] [Google Scholar]
- 46.Bosco A, Xenocostas A, Kinney J, Cadwell CM, Zimring JC. An autoanti-Kp b immunoglobulin M that simulates antigen suppression. Transfusion. 2009;49(4):750–756. doi: 10.1111/j.1537-2995.2008.02045.x. [DOI] [PubMed] [Google Scholar]