

Abstract
Resistance-nodulation-cell division (RND) superfamily efflux systems are responsible for the active transport of toxic compounds from the Gram-negative bacterial cell. These pumps typically assemble as tripartite complexes, spanning the inner and outer membranes of the cell envelope. In Escherichia coli, the CusC(F)BA complex, which exports copper(I) and silver(I) and mediates resistance to these two metal ions, is the only known RND transporter with a specificity for heavy metals. We have determined the crystal structures of both the inner membrane pump CusA and membrane fusion protein CusB, as well as the adaptor-transporter CusBA complex formed by these two efflux proteins. In addition, the crystal structures of the outer membrane channel CusC and the periplasmic metallochaperone CusF have been resolved. Based on these structures, the entire assembled model of the tripartite efflux system has been developed, and this efflux complex should be in the form of CusC3-CusB6-CusA3. It has been shown that CusA utilizes methionine clusters to bind and export Cu(I) and Ag(I). This pump is likely to undergo a conformational change, and utilize a relay network of methionine clusters as well as conserved charged residues to extrude the metal ions from the bacterial cell.
Introduction
Membrane transport proteins provide molecule-specific pathways in and out of the cell. In addition to the transport of nutrients in through the cell membrane, these proteins are responsible for pumping toxic compounds out of the cell. In Gram-negative bacteria, such as Escherichia coli, the latter activity is dominated by transporters belonging to the resistance-nodulation-cell division (RND) superfamily (Tseng et al. 1999), a class of efflux pumps which is found ubiquitously in bacteria, archaea and eukaryotes. These RND efflux pumps span both the inner and outer membranes of the Gram-negative bacterial cell envelope through tripartite protein complexes. These complexes consist of an inner-membrane, substrate-binding transporter (or pump), a periplasmic membrane fusion protein (or adaptor), and an outer membrane-anchored channel. The assembled tripartite efflux complex is responsible for removing toxic compounds out of the cell, and mediating bacterial resistance to these noxious chemicals (Fig. 1).
Figure 1.

Model of the CusCBA tripartite efflux pump. The three components of this HME-RND system form a CusCBA efflux complex, which spans both the inner and outer membranes of E. coli to export Cu+ and Ag+ directly out of the cell (blue, CusA; green, CusB; red, CusC). The periplasmic chaperone CusF (magenta) is involved in delivering the metal ions to the CusCBA complex. Heavy-metal efflux is driven by the proton-motive force.
In E. Coli, there are seven such transport complexes. The efflux pumps AcrB (Zgurskaya and Nikaido 1999; Nishino and Yamaguchi 2001; Murakami et al. 2002; Yu et al. 2003; Yu et al. 2005; Murakami et al. 2006; Seeger et al. 2006; Su et al. 2006; Sennhauser et al. 2007; Das et al. 2007; Törnroth- Horsefield et al. 2007; Nakashima et al. 2011), AcrD (Rosenberg et al. 2000; Nishino and Yamaguchi 2001; Aires and Nikaido 2005), AcrF (Ma et al. 1993; Nishino and Yamaguchi 2001; Lau and Zgurskaya 2005), MdtB (Nishino and Yamaguchi 2001; Baranova and Nikaido 2002; Nagakubo et al. 2002), MdtC (Nishino and Yamaguchi 2001; Baranova and Nikaido 2002; Nagakubo et al. 2002), and YhiV (Nishino and Yamaguchi 2001; Kobayashi et al. 2001; Bohnert et al. 2007) all belong to the hydrophobic and amphiphilic efflux RND (HAE-RND) subfamily. The HAE-RND proteins typically confer multi-drug resistance to the cell, with AcrB showing the widest substrate-binding specificity. In addition to these six HAE-RND multi-drug efflux pumps, E. coli possesses the CusA efflux pump, which belongs to the heavy-metal efflux RND (HME-RND) sub-family, and confers specific resistance to Cu(I) and Ag(I) ions (Franke et al. 2001; Franke et al. 2003).
CusA is the transporter protein that operates in conjunction with the membrane fusion protein CusB and outer channel protein CusC to form the tripartite efflux pump CusCBA. Additionally, the Cus system includes a small, periplasmic, metal-binding protein CusF, a component that has no analog in the HAE-RND complexes. Crystal structures of individual components, including CusA (Long et al. 2010), CusB (Su et al. 2009), CusC (Kulathila et al. 2011), and CusF (Loftin et al. 2005; Loftin et al. 2007; Xue et al. 2008), of the CusC(F)BA system have been determined. In addition, the x-ray structure of the CusBA adaptor-transporter efflux complex formed by the CusA and CusB proteins has also been resolved in 2011 (Su et al. 2011).
Here we summarize the known structural information for the CusC(F)BA system. Based on these structural data, we put forward our opinion on the mechanism for the binding and extrusion of Cu(I) and Ag(I) by the CusC(F)BA efflux system.
The CusB Membrane Fusion Protein
Structure of CusB
The mature protein of CusB consists of 379 amino acids (residues 29 through 407). The crystal structure (pdb code: 3H9I) comprising 78% of the protein (residues 89- 385) was determined to a resolution of 3.40 Å (Su et al. 2009). The crystal structure reveals that the CusB adaptor is an elongated molecule of ~120 Å long and ~40 Å wide (Fig. 2a). Each molecule of CusB can be divided into four distinct domains, the first three domains consisting of mostly β-strands. However, the fourth domain forms an all α-helical secondary structure, featuring with a three-helical bundle.
Figure 2.

Crystal structure of the CusB membrane fusion protein. (a) The structure can be divided into four distinct domains. Domain 1 is formed by the N and C-termini and is located above the inner membrane. The loops between domains 2 and 3 appear to form an effective hinge to allow the molecule to shift from an open conformation to a more compact structure. Domain 4 is folded into an anti-parallel, three-helix bundle, which is thought to be located near the outer membrane. (b) This figure depicts a comparison of the two conformations of CusB observed in the crystal. Superimposition of domains 3 + 4 of the two crystal structures of CusB displays an overall shift of the -strands of domain 1 of CusB by ~23°.
The first β-domain (domain 1) is formed by the N and C-terminal ends of the polypeptide (residues 89–102 and 324–385). It is composed of six β-strands, with the N-terminal end forming one of the β-strands and the C-terminus contributing the other five strands. The second β-domain (domain 2) is formed by residues 105–115 and 243–320. Again, the N-terminal residues form one of the β-strands that is incorporated into this domain. The C-terminal residues contribute a β-strand, an α-helix and four anti-parallel β-sheets. Domain 3 consists of residues 121–154 and 207–239, with a majority of these residues folded into eight β-strands. Domain 4 of CusB forms an all-helical domain, which is folded into an anti-parallel, three-helix bundle, and creates a ~27 Å long helix-turn-helix-turn-helix secondary structure. This structural feature, not found in other known membrane fusion proteins, highlights the uniqueness of the CusB adaptor.
Two distinct conformations of CusB were captured in the single crystal, which suggests that the protein is quite flexible. Presumably, these structures represent two transient states of the membrane fusion protein. The two conformations are closely related, and a small hinge motion can be attributed to the difference (Fig. 2b). Superimposition of these two molecules gives an overall rmsd of 2.6 Å calculated over the Cα atoms. Comparison of these two structures reveals that one of the molecules of CusB adopts a more open conformation, while the other molecule exhibits a relatively compact form of the structure. Thus, these two molecules may correspond to open and closed states of this membrane fusion protein.
Structure of CusB-Cu(I) and CusB-Ag(I)
The crystal structure of CusB was also determined for apo-crystals soaked in metal ions (Su et al. 2009). Two strong peaks at the copper edge of the CusB-Cu(I) molecule were observed, indicating the presence of two possible Cu(I) binding sites. The first Cu(I) binding site is located in the first domain formed by the N and C-termini of the protein, located near the bottom of the elongated CusB molecule. Coordinating with the bound Cu(I) ion at this site are M324, F358 and R368. The second Cu(I) binding site is located close to the center of the three-helix bundle in domain 4. Cu(I) in this location is bound by M190, W158 and Q162.
For the CusB-Ag(I) complex crystal, we have found one anomalous difference Fourier peak corresponding to the potential Ag(I) binding site. It appears that the location of this Ag(I) binding site is the same as that of first binding site for Cu(I). Although these binding sites are found to coordinate Cu(I) and Ag(I) ions, it has later been found that these sites in CusB are not physiological relevant (Kim et al. 2011).
Based on experimental results from extended x-ray absorption fine structure (EXAFS) and site-specific mutagenesis (Bagai et al. 2007), it has been proposed that residues M49, M64 and M66 of CusB form the three-coordinate binding site for Cu(I) and Ag(I). These residues are located at the N-terminal end of CusB, a region that is intrinsically disordered and for which structural information is not available. Although these methionine residues were excluded in the structural model, they could form an ideal binding site for Cu(I) and Ag(I). According to the crystal structure of CusB, this proposed metal binding site may be located right above the inner membrane, adjacent to the periplasmic domain of the CusA efflux pump. It is possible that this proposed metal binding site might directly interact with the inner membrane protein. In this case, CusA may capture the metal that is released from CusB through this proposed metal binding site. In addition, if the α-helices at domain 4 of CusB interact with the outer membrane channel CusC, the CusB membrane fusion protein may be directly involved in delivering the bound metal ions to CusC and eventually exporting the metal ions out of the cell.
Cross-linking and mass spectrometry
To determine how CusB interacts with the CusA efflux pump and the relative orientation of CusB in the efflux complex, we cross-linked the purified CusA and CusB proteins using the lysine-lysine cross-linker disuccinimidyl suberate (DSS). The resulting product was digested with trypsin and examined using LC-MS/MS. Analysis of the mass spectral data suggests that the lysine residue of the polypeptide β (IDPTQTQNLGVKTATVTR) originating from the N-terminal residues (84–101) of CusB directly interacts with the lysine residue of peptide α (SGKHDLADLR) which belongs to the periplasmic domain (residues 148–157) of the CusA efflux pump. This result, together with the crystal structures, suggests that domain 1, formed by the N and C-terminal ends of CusB, should interact with the periplasmic domain of the CusA transporter (Su et al. 2009).
The CusA Heavy-Metal Efflux Pump
Structure of CusA
The crystal structure of the inner-membrane CusA (pdb code: 3K07) was determined to 3.52 Å with 98% of the 1,047 residues (residues 5–504 and 516–1040) included in the final model (Long et al. 2010) (Fig. 3). According to our model, CusA forms a homotrimer. Each subunit contains 12 transmembrane helices (TM1–TM12) and a large periplasmic domain formed by two loops between TM1 and TM2, and between TM7 and TM8. The two helices TM2 and TM8 protrude into the periplasm and form a large periplasmic domain of CusA. In addition, TM4, TM5, TM10, and TM11 extend into the cytoplasm to form a small cytoplasmic domain of the pump (Fig. 3a).
Figure 3.

Crystal structures of the CusA efflux pump. (a) Ribbon diagram of the CusA homotrimer viewed in the membrane plane. Each molecule is labeled with a different color: molecule 1 is green, molecule 2 is pink, and molecule 3 is cyan. Each subunit of CusA consists of 12 transmembrane helices (TM1-TM12) and a large periplasmic domain formed by two periplasmic loops between TM1 and TM2, and TM7 and TM8, respectively. (B) Ribbon diagram of the CusA homotrimer subdomains as viewed from the top. The six subdomains are PN1 (red), PN2 (blue), PC1 (yellow), PC2 (teal), DN (green) and DC (pink).
The periplasmic domain of CusA can be divided into six sub-domains (PN1, PN2, PC1, PC2, DN, and DC) (Fig. 3b). In the trimer, sub-domains PN1, PN2, PC1 and PC2 form the pore domain, and PN1 forms the central pore, which stabilizes the trimeric organization. Sub-domains DN and DC, contribute to the docking domain, presumably interacting with the CusC channel. The trimeric CusA structure suggests that subdomains PN2, PC1 and PC2 are located at the outermost core of the periplasmic domain, facing the periplasm. PC1 and PC2 also form an external cleft, and this cleft is closed in the apo-CusA crystal structure. A structure formed by one α-helix (residues 690–706) and three β-sheets (residues 681–687, 711–716 and 821–827) in PC2 tilts into the cleft to close the opening. Intriguingly, residues 665–675, located at the bottom of the cleft, form a horizontal-positioned α-helix. This unique structural feature is later found to govern the specificity of CusA. The α-helix orients horizontally and roughly divides the transmembrane and periplasmic domains into two regions.
Structure of CusA-Cu(I) and CusA-Ag(I)
We have also determined the crystal structure of CusA in the presence of Cu(I) (pdb code: 3KSS) and Ag(I) (pdb code: 3KSO) (Long et al. 2010). Anomalous peaks at the copper-edge wavelength showed that binding of these two monovalent metal ions is coordinated by three proximal methionines, M573, M623 and M672, located at the bottom of the periplasmic cleft. Binding of the metal ion is shown to induce a conformational change in the periplasmic as well as the transmembrane domains. The most dramatic change shifts the entire PC2 sub-domain, and this shift can be interpreted as a 30° swing of PC2 that opens the cleft between PC1 and PC2, with G721 and P810 forming the hinge (Fig. 4). In addition, the C-terminal end of the horizontal helix in the cleft is found to tilt upward by 21°, coordinating M573, M623 and M672 to form a three-methionine metal binding site.
Figure 4.

Metal bound structure of CusA. (a) Superposition of a monomer of apo-CusA (red) onto a monomer of Cu(I) bound-CusA (green). The bound Cu(I) is pink. The arrow represents a major swing of the PC2 sub-domain initiated by Cu(I) binding. (b) The periplasmic cleft between PC1 and PC2 forms a metal binding site. The bound copper in the CusA-Cu(I) structure is in red. M573, M623 and M672 forming a metal binding site at the periplasmic cleft are shown in stick form (green).
It appears that binding of Cu(I) or Ag(I) also triggers significant conformational changes in the other transmembrane helices of the pump. Based on these ion-bound CusA structures, the horizontal helix in the cleft directly interacts with the N-terminal end of TM8. Coupled with the movement of the horizontal helix, TM8 also shifts in position to a more vertical orientation while retaining its -helical structure. Overall, the N-terminal end of TM8 is found to shift away from the core by 10 Å after metal binding. The movement of TM8 may relate to transmembrane signaling, which could initiate proton translocation across the membrane. In addition to the movement of TM8, all other transmembrane helices except TM2 shift horizontally by as much as 4 Å. Further, TM1, TM3 and TM6 readjust in an approximately 3 Å upward shift with respect to the inner membrane surface. The net result is that all three of these transmembrane helices move towards the periplasm by one turn. Coupled with the above conformational changes, PN1 also shifts upward by 3 Å. This moves the central pore helix upward by one turn upon metal binding.
In vivo metal susceptibility assay
The trimeric structure of CusA shows that each protomer forms a channel spanning the entire transmembrane region up to the bottom of the periplasmic funnel. This channel includes the four methionine-pairs (M410–M501, M403–M486, M391–M1009, and M271–M755) as well as the three-methionine binding site (M573, M623 and M672). Taken together these five methionine clusters are likely to form a relay network facilitating metal transport. To test this, we prepared an E. coli knockout strain lacking both cueO and cusA (Long et al. 2010). M573, M623 and M672, comprising the three-methionine transmembrane binding site, were mutated into isoleucines (M573I, M623I and M672I). These three mutants were expressed in BL21(DE3)ΔcueOΔcusA and tested for their ability to confer Ag(I) and Cu(I) resistance. Our studies show that the CusA mutants, M573I, M623I and M672I, are unable to relieve the copper or silver sensitivity of strain BL21(DE3)ΔcueOΔcusA. Further mutants M391I, M410I, M486I and M755I were prepared to disrupt the methionine pairs formed by the pump. Expression of these four mutants in BL21(DE3)ΔcueOΔcusA also showed a significant decrease in the levels of Ag(I)/Cu(I) tolerance. Together, these results strongly support the importance of these methionine pairs/clusters for the methionine-residue ion relay channel.
In vitro metal transport assay
We reconstituted the purified CusA pump into liposomes containing the fluorescent indicator Phen Green SK. A stopped-flow transport assay was then employed to determine whether these proteoliposomes can capture silver ions from the extravesicular medium. Surprisingly, the experiment suggested that the CusA pump is able to transport Ag+ from the cytosol (Long et al. 2010). To determine the importance of the methionine pairs and clusters for metal transport in the relay network, this assay was repeated for the CusA mutants M391I, M486I, M573I, M623I, M672I and M755I. As expected, these mutant transporters did not take up Ag+ from the extravesicular medium of the proteoliposomes (Long et al., 2010). The collective experiments provide direct compelling evidence that CusA is capable of taking up Ag+ from the cytoplasm through the methionine-relay network of the pump.
The CusBA Adaptor-Transporter Efflux Complex
Structure of CusBA
We have determined the crystal structure of the CusBA complex (pdb code: 3NE5) to a resolution of 2.90 Å with 93% of the residues included in the final protein (Su et al. 2011). The structure reveals that the periplasmic domain of CusA interacts specifically with two adaptor molecules of CusB (molecules 1 and 2), which are tilted at an angle of ~50° with respect to the membrane surface (Fig. 5). Therefore, the trimeric CusA pump is found to contact six CusB adaptor molecules, which form a hexameric channel right above the periplasmic domain of CusA. The structure of the CusB hexamer mimics an inverted funnel. Domain 1 and the lower half of domain 2 of CusB create a cap-like structure, whereas domains 3, 4, and the upper half of domain 2 form the central channel of the funnel. The inner surface of the cap fits closely on top of the periplasmic domain of the CusA trimer. The channel formed above the cap of the adaptor is ~62 Å in length with an average internal diameter of ~37 Å. Thus, the interior of the channel gives rise to a large elongated cavity with a volume of ~65,000 Å3. The lower half of the channel is primarily created by β-barrels, whereas the upper half is an entirely α-helical tunnel. The diameter of the channel is gradually constricted and then dilates as it approaches the outer membrane. The narrowest section of the central channel is located at residue D232 of each CusB protomer. The widest section of the channel appears to form at the top edge, with an inner diameter of ~56 Å. The inner surface of the channel is predominantly negatively charged, as indicated by the electrostatic surface distribution, which suggests that the interior surface of the channel may have the capacity to bind to positively charged metal ions.
Figure 5.

Crystal structures of the CusBA adaptor-transporter efflux complex. The full structure includes the hexameric CusB adaptor as well as the trimeric CusA transporter. Each CusA and CusB molecule is indicated using a different color.
CusA and CusB interactions
Molecule 1 of CusB establishes close contact with the upper regions of PN2 and PC1 and the DN subdomain of CusA, and interacts primarily through charge–charge interactions. The residues K95, D386, E388 and R397 of molecule 1 form four salt bridges, with D155, R771, R777 and E584 of CusA, respectively (Su et al. 2011). In addition, T89, the backbone oxygen of N91, and R292 of molecule 1 form hydrogen bonds with K594, R147 and the backbone oxygen of Q198 of CusA, respectively. Molecule 2 of CusB, however, bridges the upper regions of PC1 and PC2, and the DC subdomain by charge–dipole and dipole–dipole interactions. Specifically, Q108, S109, S253 and N312 of molecule 2 form hydrogen bonds with Q785, Q194, D800 and Q198 of CusA, respectively (Su et al. 2011). In addition, the backbone oxygens of L92 and T335 of molecule 2 contribute two additional hydrogen bonds with the side chains of K591 and T808 of CusA.
Domains 1–3 of CusB are involved in binding molecules 1 and 2 of CusB. Residues E118, Y119, R186, E252 and R292 of molecule 1 form hydrogen bonds with T139, D142, T206, N312 and N113 of molecule 2, respectively. In the hexameric structure, residues N113, N228 and N312 of molecule 1 pair with R292, the backbone oxygen of A126, and E252 of molecule 6 of CusB, respectively, to form three hydrogen bonds. In addition, D142 of molecule 1 of CusB forms two hydrogen bonds with Y119 and R297 of molecule 6 (Su et al. 2011).
Isothermal titration calorimetry for CusA and CusB binding
We used isothermal titration calorimetry (ITC) to determine the binding affinity between CusA and CusB. Interestingly, the molecular ratio for this binding reaction based on ITC is one CusA monomer for every two CusB protomers (Su et al. 2011). This result is indeed confirmed by the crystal structure where each protomer of CusA binds two molecules of CusB.
The CusBA-Cu(I) Complexes
Structures of CusBA-Cu(I)
In the absence of the CusB adaptor, two distinct structures of CusA were obtained by x-ray crystallography (Long et al. 2010). These structures most likely capture two different conformational states of the pump in the transport cycle. The apo-CusA conformation should represent the “resting” state where the external periplasmic cleft is closed. However, the Cu(I) and Ag(I)-bound CusA structures should correspond to the “binding” state where the periplasmic cleft is open. Upon metal ion transport, the pump must go through other transient states to actively remove the metal ions. Recently, the co-crystal structures of the CusBA-Cu(I) complexes have been determined. These structures have allowed us to capture the conformation of different intermediates transitioning between the “binding” and “resting” states in the transport cycle.
CusBA-Cu(I) was crystallized in three distinct conformations, each containing one CusA and two CusB molecules (pdb codes: 3T56, 3T51 and 3T53) (Su et al. 2012). These conformations were designated as “pre-extrusion 1”, “pre-extrusion 2” and “extrusion” states (Fig. 6). The conformation of the “pre-extrusion 1” state appears to be similar to but is also different from the “binding” structure, where the periplasmic cleft is open. However, the conformation of the “pre-extrusion 2” state seems to be closer to but is distinct from the “resting” structure of the pump. In this conformation, the periplasmic cleft is closed. In the “pre-extrusion 1” state, a single bound Cu(I) is found to coordinate residues M573, M623 and M672, which forms the distinct three-methionine ion binding site inside the cleft of the periplasmic domain of CusA. The nearby conserved charged residue E625 is also involved in the binding (Su et al. 2012). In comparison with the structure of the “binding” state, the horizontal helix, formed by residues 665–675, inside the cleft of the structure of “pre-extrusion 1” state is lower in position (Fig. 7). The change can be interpreted as a 3° downward tilting motion of the C-terminal end of the horizontal helix in this structure when compared with the CusA-Cu(I) form. This motion also shifts M672 away from M573 and M623, seemingly to disassemble the three-sulfur coordination site and to release the bound Cu(I) ion from this site. In contrast to the “binding” and “resting” structures, no continuous channel is found in each CusA protomer of “pre-extrusion 1” state.
Figure 6.

Structures of the CusBA-Cu(I) efflux complexes. (a) Ribbon diagram of the structure of form Ia (blue) viewed in the membrane plane. The bound copper is shown as a purple sphere. This structure represents the “pre-extrusion 1” state of the pump. (b) Ribbon diagram of the structure of form Ib (yellow) viewed in the membrane plane. The bound copper is shown as a red sphere. This structure represents the “pre-extrusion 2” state of the pump. (c) Ribbon diagram of the structure of form II (green) viewed in the membrane plane. The bound copper is shown as a red sphere. This structure represents the “pre-extrusion 2” state of the pump. (d) Ribbon diagram of the structure of form III (magenta) viewed in the membrane plane. The bound copper is shown as a blue sphere. This structure represents the “extrusion” state of the pump. For clarity, only the periplasmic domain (subdomains PN2, PC1 and PC2) and part of the transmembrane region of CusA are shown in (a)–(d). The molecules of CusB in each complex are not included.
Figure 7.

Different conformational states of CusBA-Cu(I). (a) Superimposition of subdomain PC2 and TM8 of each state of the pump. The arrow indicates a 30° swing of PC2 upon conformational transition from the “pre-extrusion 1” (cyan) to “pre-extrusion 2” (green) and “extrusion” (magenta) states. The structures of forms Ia, II and III are used to represent the “pre-extrusion 1”, “pre-extrusion 2” and “extrusion” states, respectively. (b) Superimposition of subdomain PC1 and the horizontal helix of each state of the pump. The figure illustrates the change in conformations of the PC1 helix (residues 582–589) and the flexible loop (residues 609–626) upon CusB binding. For clarity, only the short C-terminal helix (residues 391–400) of molecule 1 of CusB (red) is included. The change in conformation of the horizontal helix at different states of the pump is also shown in this superimposition (apo-CusBA, gray; CusA-Cu(I), orange; form Ia, cyan; form II, green; form III, magenta). (c) Superimposition of the transmembrane helices 5 (TM5) and 6 (TM6) of each state of the pump. The figure illustrates the change in positions of TM5 and TM6 (residues 447–495) within the transport cycle (apo-CusBA, gray; CusA-Cu(I), orange; form Ia, cyan; form II, green; form III, magenta). As both forms Ib and II are in the same state, the conformation of form Ib is not included in (a)–(c) for clarity.
In the “pre-extrusion 2” state, the channel formed by the methionine relay network of each protomer of CusA has been closed up (Su et al. 2012). The only cavity that can be identified is the space nearby the copper signal. This copper signal is found in the familiar three-methionine binding site formed by M573, M623 and M672. However, its binding mode is quite distinct from that found in the “binding” state. First, the C-terminal end of the horizontal helix (residues 665–675) significantly tilts downward by ~10° in this “pre-extrusion 2” state in comparison with the “binding” form. This motion shifts the position of M672 away from M573 and M623, probably weakening the binding for Cu(I) within the methionine triad. Secondly, coupled with this movement, the side chain of a conserved anionic charged residue E625, located deep inside the periplasmic cleft, is found to flip towards the bound Cu(I) and interact with this ion by electrostatic interaction. In addition, a cluster of conserved charged residues, including R83, E567, D617, E625, R669 and K678, is found nearby this copper signal. Interestingly, these conserved charged residues line along the metal-ion transport channel formed by the methionine relay network, suggesting that these charged residues may be important for the transporter’s functioning (Su et al. 2012).
In addition to the above movements, the transmembrane helices 5 and 6 (residues 447–495) are found to shift downward by 5 Å with respect to the membrane plane in both “pre-extrusion 1” and “pre-extrusion 2” states, mimicking the change in position of the horizontal helix, when compared with the conformation of the “binding” state (Fig. 7).
The conformation of the “extrusion” state is nearly identical to that of apo-CusBA, and one bound Cu(I) is found to coordinate M573, M623, M672 and E625. The conformation of CusA in this complex structure is also very similar to that of apo-CusA at the “resting” state. However, binding of the CusB adaptor triggers a subtler but significant conformational change at the upper portion of subdomain PC1 of the CusA transporter. It is observed that a short C-terminal helix (residues 391–400) of molecule 1 of CusB directly interacts with and pushes a helix (residues 582–589) located at the upper half of PC1 of CusA (Fig. 7). The consequence is that the N-terminal end of this PC1 helix is found to tilt downwards by 8° in the CusBA form in comparison with the apo-CusA structure. This tilting motion in turn further pushes a loop (residues 609–626) located right below the PC1 helix downward. On the basis of these conformational changes, we propose that CusB may be involved in tuning the width of the channel formed by the methionine metal-ion relay network to its optimal level through the above mechanism.
An empirical measurement defined as the C -C distance between E622 and M812 was used to identify the degree of opening of the channel. In view with the structures, these C -C distances are 13 Å, 12 Å, 10 Å, 10 A and 11 A for the “resting”, “binding”, “pre-extrusion 1”, “pre-extrusion 2” and “extrusion” states, respectively. Thus, the channel is fully open the “resting” form, and subsequently contracts and expands between the “binding” and “extrusion” states (Su et al. 2012).
In vivo metal susceptibility assay
The structures of CusBA–Cu(I) reveal that conserved charged residues (R83, E567, D617, E625, R669, and K678) line the walls of the methionine relay tunnel in the periplasmic domain of CusA. To test whether this conserved charged residues play an important role in elevating the level of copper resistance, these residues were mutated into alanines (R83A, E567A, D617A, E625A, R669A, and K678A) or aspartate (E625D), and the corresponding mutant transporters were expressed in BL21(DE3)ΔcueOΔcusA (Su et al. 2012). The ability of these mutant transporters to confer copper resistance was then tested in vivo using a metal susceptibility assay. The results indicate that none of these CusA mutants were able to relieve the copper sensitivity of strain BL21(DE3)ΔcueOΔcusA, suggesting that these residues are critical for the function of the pump.
In vitro metal transport assay
To investigate whether the charged residues are essential for metal ion transport, we reconstituted the purified R83A, E567A, D617A, E625A, R669A, and K678A mutant proteins into liposomes containing the fluorescent indicator Phen Green SK. A similar stopped-flow transport assay was used to determine whether these proteoliposomes can capture Ag(I) from the extravesicular medium. When Ag(I) was added to the extravesicular medium, no quenching of the fluorescence signal was detected, suggesting that these mutant transporters are incapable of taking up Ag(I).
The stopped-flow assay was also used to study the transport activity of the reconstituted CusBA proteoliposomes (Su et al. 2012). The results suggest that the CusBA proteoliposomes are more active than those liposomes containing CusA only for metal transport as indicated by the position of the 50% attenuation of the fluorescent signal. No transport activity has been observed in liposomes containing CusB only, indicating that these CusB protoliposomes cannot uptake silver ions. These collective experiments provide compelling evidence that these conserved charged residues have primary roles in the function of the pump.
The CusC Channel
Structure of CusC
The crystal structure of CusC was determined to a resolution of 2.3 Å (pdb code: 3PIK), with ~98% of the 440 residues included in the final structure (Kulathila et al. 2011). This structure reveals that trimeric CusC forms a 130 Å long barrel, with each monomer contributing 4 β-strands to the 12-stranded outer membrane β-barrel, and 4 α-helices to the periplasmic α-barrel (Fig. 8a). The interior of this trimeric channel forms a large cylindrical cavity with a volume of ~28,000 Å3. As in the hexamic CusB channel, the exterior of the trimeric CusC channel is relatively uncharged, while the interior is predominantly electronegative. Thus, the interior of the CusC channel may have the capacity to bind positively charged ligands. In contrast, the surface of the periplasmic end of the α-barrel, which is likely to interact with CusA, is relatively hydrophobic. The channel is closed on this end by van der Waals interactions between the L403 residues of the three monomers. An adjacent ring of negatively charged residues (D407) provides an additional layer of closure on the periplasmic side. On the contrary, the transmembrane domain of this channel is widely open. Based on the crystal structure, CusC is mostly likely triacylated at the N-terminal cysteine residue to promote anchoring at the outer membrane. The structure indicates that the CusC channel does not possess any obvious structural features that would make it specific for metal ions, suggesting that the narrow specificity of the CusC(F)BA efflux system is provided by other components of the complex.
Figure 8.

Crystal structures of CusC and CusF. (a) Ribbon diagram of the CusC homotrimer (pdb code: 3PIK) (Kulathila et al. 2011) viewed in the membrane plane. Each molecule is labeled with a different color (green, pink, and cyan). (b) Ribbon diagram of Cu(I)-bound CusF (pdb code: 2VB2) (Xue et al. 2008). H36, M47, M49 and W44 forming a metal binding site are shown in stick form. The bound Cu(I) is in red.
The CusF Metallochaperone
Structure of CusF
The crystal structure of CusF (pdb code: 1ZEQ) was determined to a resolution of 1.5 Å, with ~79% of the residues (6–88) included in the final model (Loftin et al. 2005). CusF takes the shape of a small β-barrel formed by five strands (β1-β5). These five strands are arranged in two three-stranded antiparallel β-sheets, with both sheets sharing the first β-strand. Thus, strand 1 is composed of β1, β2, and β3, and strand 2 is associated with β4, β5, and β1. The barrel is closed by interactions between β3 and β5 in a parallel arrangement.
To determine which residues are involved in Cu(I) binding, an NMR spectrum of CusF was collected with and without Cu(I) (Loftin et al. 2005). Based on the experimental data and homologous sequence alignment, the most likely candidates for coordinating with the bound Cu(I) in the protein are H36, M47 and M48. These residues are found primarily in β2, in β3, and at the C-terminal end of β4.
The crystal structures of Cu(I) and Ag(I)-bound CusF indeed depict that the bound metal ions are tetragonally coordinated with these three residues (H36, M47 and M48) as well as W44 (Xue et al. 2008; Loftin et al. 2007). Overall, the crystal structures of the ion-bound forms are very similar to the structure of apo-CusF (Fig. 8b). Thus, the -barrel conformation of the metallochaperone CusF is retained and can accommodate the metal ion with only minimal local changes.
Recent NMR spectroscopy and chemical cross-linking coupled with mass spectrometry revealed that the N-terminal region of CusB most likely interacts with CusF in the vicinity of the metal-binding surface (Mealman et al. 2012). This result is in good agreement with previous study that the bound Ag+ ion in CusF can be directly transferred to the CusB fusion protein, and that the N-terminal end of CusB should contain a specific metal binding site (Bagai et al. 2008). Presumably, the specific interaction between CusF and CusB is critical for a direct transfer of metal ion, an important step to occur before ion extrusion through the tripartite efflux complex. Quantum-mechanical/molecular mechanical (QC/MM) calculations suggested that the protein’s internal motions should play an essential role in transferring the bound metal ion from the metallochaperone CusF to membrane fusion protein CusB (Chakravorty et al. 2011). The interaction between CusF and CusB may mark the initial step for metal ion extrusion by the CusCBA tripartite efflux complex.
Conclusion
The metal ions can enter the CusA pump via the periplasmic cleft as well as the cytoplasm. Presumably, the methionine pairs/clusters are responsible for metal ion transport. In the transmembrane regions, these methionine pairs are ~15 Å apart. Thus, it is possible that a direct transfer of metal ions from one methionine pairs to the other contributes to deliver the ions through the inner membrane. It is believed that CusA predominately carries out metal ion efflux through the periplasm (Kim et al. 2011). Thus, the major path for taking up metals should be across the cleft in the periplasmic space.
We propose that the CusA transporter utilizes methionine pairs/clusters to export Cu(I) and Ag(I). Presumably, the periplasmic cleft of CusA remains closed when there is no Cu(I)/Ag(I). In the presence of Cu(I) or Ag(I) ions, the periplasmic cleft likely opens. Metal ions could enter the three-methionine binding-site, formed by M573, M623 and M672, inside the cleft between sub-domains PN2 and PC1 on the periplasmic portion of CusA or via the methionine pairs within the transmembrane domain of the pump. It has been demonstrated that the chaperone protein CusF can directly transfer its bound Cu(I) to the CusB membrane fusion protein (Bagai et al. 2008). Thus, it is likely that CusF is responsible for delivering metal ions to the CusCBA tripartite efflux system within the periplasm. There is a chance that the initial step of metal transport through the periplasmic cleft of the CusA pump may involve a direct transfer from CusF to the previously proposed three-methionine metal binding site (Bagai et al. 2007) at the long N-terminal tail of CusB. Based on the co-crystal structure of CusBA, each flexible tail of CusB is located near the PN2/PC1 cleft of the CusA pump. Thus, the second step could deliver the bound metal ion from CusB to the three-methionine cluster (M573, M623 and M672) inside the periplasmic cleft of CusA (Long et al. 2010). The bound metal ion could then be released to the nearest methionine pair (M271-M755) located directly above the three-methionine metal binding site, from which the ion could be released into the central funnel of CusA and eventually extruded via the outer membrane channel CusC. Genetic data and transport assay have shown that the conserved charged residues lining the methionine relay channel are essential for the transporter’s functioning (Su et al. 2012). Therefore, it is possible that these conserved charged amino acids are responsible for delivering the metal ions from one residue to another in the methionine relay channel.
A series of crystal structures have been determined for the CusBA complex in the presence of Cus(I). These structures have depicted the sequential conformational changes of the pump. During the transport cycle, the CusBA efflux complex must go through different transient states, including the “resting”, “binding”, “pre-extrusion 1”, “pre-extrusion 2” and “extrusion” conformations, to complete the transport cycle (Fig. 9). Thus, in coordination with the motion of the periplasmic cleft, the transmembrane portion of the methionine relay channel should also be synchronized to open and close to accommodate for the export of metal ions.
Figure 9.
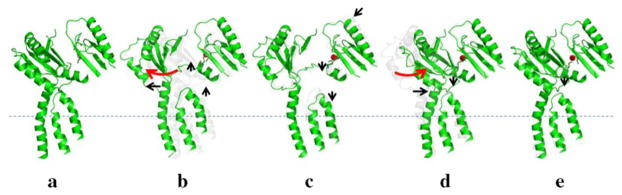
Sequential conformational transition of the CusBA pump. This figure depicts stepwise motions of CusA in the adaptor-transporter complex transitioning between the “resting” and “extrusion” states (a, “resting” state; b, “binding” state; c, “pre-extrusion 1” state; d, “pre-extrusion 2” state; e, “extrusion” state). For clarity, only subdomains PC1 and PC2 in the periplasmic domain and transmembrane helices 5, 6 and 8 in the transmembrane region of CusA are shown. The bound copper is in red sphere. The red arrows indicate the change in conformation of the protein within the transport cycle.
It is not yet known whether the interior of the hexameric CusB channel forms part of the extrusion pathway for Cu(I) and Ag(I). Based on the crystal structures of CusBA (Su et al. 2011) and CusC (Kulathila et al. 2011), the interior surface of both the hexameric CusB and trimeric CusC channels are predominantly negatively charged. However, the interior surface of CusC is more electronegative. Thus, it is likely that the last step of transport of cationic Cu(I) and Ag(I) is facilitated by the negative potential gradient created by the CusB and CusC channels. During metal-ion extrusion, a large conformational change of CusC may occur to open this outer membrane channel, similar to the cases of TolC (Vaccaro et al. 2008) and OprM (Phan et al. 2010). Exactly what this tripartite efflux system’s mechanism is must await confirmation by elucidation of additional crystal structures of the CusCBA tripartite complex.
Acknowledgments
This work is supported by an NIH Grant R01GM086431 (E.W.Y.).
References
- Aires JR, Nikaido H. Aminoglycosides are Captured from Both Periplasm and Cytoplasm by the AcrD Multidrug Efflux Transporter of Escherichia coli. J Bacteriol. 2005;187:1923–1929. doi: 10.1128/JB.187.6.1923-1929.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagai I, Liu W, Rensing C, Blackburn NJ, McEvoy MM. Substrate-Linked Conformational Change in the Periplasmic Component of a Cu(I)/Ag(I) Efflux System. J Biol Chem. 2007;282:35695–35702. doi: 10.1074/jbc.M703937200. [DOI] [PubMed] [Google Scholar]
- Bagai I, Rensing C, Blackburn NJ, McEvoy MM. Direct Metal Transfer Between Periplasmic Proteins Identifies a Bacterial Copper Chaperone. Biochemistry. 2008;47:11408–11414. doi: 10.1021/bi801638m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranova N, Nikaido H. The BaeSR Two-Component Regulatory System Activates Transcription of yegMNOB (mdtABCD) Transporter Gene Cluster in Escherichia coli and increases its Resistance to Novobiocin and Deoxycholate. J Bacteriol. 2002;184:4168–4176. doi: 10.1128/JB.184.15.4168-4176.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnert JA, Schuster S, Fähnrich E, Trittler R, Kern WV. Altered Spectrum of Multidrug Resistance Associated with a Single Point Mutation in the Escherichia coli RND-type MDR Efflux Pump YhiV (MdtF) J Antimicrob Chemother. 2007;6:1216–1222. doi: 10.1093/jac/dkl426. [DOI] [PubMed] [Google Scholar]
- Chakravorty DK, Wang B, Ucisik MN, Merz KM., Jr Insight into the Cation- Interaction at the Metal Binding Site of the Copper Metallochaperone CusF. J Am Chem Soc. 2011;133:19330–19333. doi: 10.1021/ja208662z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das D, Xu QS, Lee JY, Ankoudinova I, Huang C, Lou Y, Degiovanni A, Kim R, Kim SH. Crystal Structure of the Multidrug Efflux Transporter AcrB at 3.1 Å Resolution Reveals the N-terminal Region with Conserved Amino Acids. J Struct Biol. 2007;158:494–502. doi: 10.1016/j.jsb.2006.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke S, Grass G, Nies DH. The Product of the ybdE Gene of the Escherichia coli Chromosome is involved in Detoxification of Silver Ions. Microbiol. 2001;147:965–972. doi: 10.1099/00221287-147-4-965. [DOI] [PubMed] [Google Scholar]
- Franke S, Grass G, Rensing C, Nies DH. Molecular Analysis of the Copper-Transporting Efflux System CusCFBA of Escherichia coli. J Bacteriol. 2003;185:3804–3812. doi: 10.1128/JB.185.13.3804-3812.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EH, Nies DH, McEvoy MM, Rensing C. Switch or Funnel: How RND-Type Transport Systems Control Periplasmic Metal Homeostasis. J Bacteriol. 2011;193:2381–2387. doi: 10.1128/JB.01323-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi A, Hirakawa H, Hirata T, Nishino K, Yamaguchi A. Growth Phase-Dependent Expression of Drug Exporters in Escherichia coli and its Contribution to Drug Tolerance. J Bacteriol. 2001;16:5693–5703. doi: 10.1128/JB.00217-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulathila R, Kulathila R, Indic M, van den Berg B. Crystal Structure of Escherichia coli CusC, the Outer Membrane Component of a Heavy Metal Efflux Pump. PLoS One. 2011;6:e15610. doi: 10.1371/journal.pone.0015610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau SY, Zgurskaya HI. Cell Division Defects in Escherichia coli Deficient in the Multidrug Efflux Transporter AcrEF-TolC. J Bacteriol. 2005;187:7815–7825. doi: 10.1128/JB.187.22.7815-7825.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loftin IR, Franke S, Roberts SA, Weischsel A, Héroux A, Montfort WR, Rensing C, McEvoy MM. A Novel Copper-Binding Fold for the Periplasmic Copper Resistance Protein CusF. Biochemistry. 2005;44:10533–10540. doi: 10.1021/bi050827b. [DOI] [PubMed] [Google Scholar]
- Loftin IR, Franke S, Blackburn NJ, McEvoy MM. Unusual Cu(I)/Ag(I) Coordination of Escherichia coli CusF as Revealed by Atomic Resolution Crystallography and X-ray Absorption Spectroscopy. Prot Sci. 2007;16:2287–2293. doi: 10.1110/ps.073021307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long F, Su CC, Zimmermann MT, Boyken SE, Rajashankar KR, Jernigan RL, Yu EW. Crystal Structures of the CusA Efflux Pump Suggest Methionine-Mediated Metal Transport. Nature. 2010;467:484–488. doi: 10.1038/nature09395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma D, Cook DN, Alberti M, Pon NG, Nikaido H, Hearst E. Molecular Cloning of acrA and acrE Genes of Escherichia coli. J Bacteriol. 1993;175:6299– 6313. doi: 10.1128/jb.175.19.6299-6313.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mealman TD, Bagai I, Singh P, Goodlett DR, Rensing C, Zhou H, Wysocki VH, McEvoy MM. Biochemistry. 2012;50:2559–2566. doi: 10.1021/bi102012j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami S, Nakashima R, Yamashita E, Yamaguchi A. Crystal Structure of Bacterial Multidrug Efflux Transporter AcrB. Nature. 2002;419:587–593. doi: 10.1038/nature01050. [DOI] [PubMed] [Google Scholar]
- Murakami S, Nakashima R, Yamashita E, Matsumoto T, Yamaguchi A. Crystal Structures of a Multidrug Transporter Reveal a Functionally Rotating Mechanism. Nature. 2006;443:173–179. doi: 10.1038/nature05076. [DOI] [PubMed] [Google Scholar]
- Nakashima R, Sakurai K, Yamasaki S, Nishino K, Yamaguchi A. Structures of the multidrug exporter AcrB reveal aproximal multisite drug-binding pocket. Nature. 2011;480:565–569. doi: 10.1038/nature10641. [DOI] [PubMed] [Google Scholar]
- Nagakubo S, Nishino K, Hirata T, Yamaguchi A. The Putative Response Regulator BaeR Stimulates Multidrug Resistance of Escherichia coli via a Novel Multidrug Exporter System, MdtABC. J Bacteriol. 2002;184:4161–4167. doi: 10.1128/JB.184.15.4161-4167.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishino K, Yamaguchi A. Analysis of a Complete Library of Putative Drug Transporter Genes in Escherichia coli. J Bacteriol. 2001;183:5803–5812. doi: 10.1128/JB.183.20.5803-5812.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan G, Benabdelhak, Lascombe MB, Benas P, Rety S, Picard M, Ducruix A, Etchebest C, Broutin I. Structural and Dynamical Insights into the Opening Mechanism of P. aeruginosa OprM Channel. Structure. 2010;18:507–517. doi: 10.1016/j.str.2010.01.018. [DOI] [PubMed] [Google Scholar]
- Rosenberg EY, Ma D, Nikaido H. AcrD of Escherichia coli is an Aminoglycoside Efflux Pump. J Bacteriol. 2000;182:1754–1756. doi: 10.1128/jb.182.6.1754-1756.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeger MA, Schiefner A, Eicher T, Verrey F, Dietrichs K, Pos KM. Structural Asymmetry of AcrB Trimer Suggests a Peristaltic Pump Mechanism. Science. 2006;313:1295–1298. doi: 10.1126/science.1131542. [DOI] [PubMed] [Google Scholar]
- Sennhauser G, Amstutz P, Briand C, Storchengegger O, Grütter MG. Drug Export Pathway of Multidrug Exporter AcrB Revealed by DARPin Inhibitors. PLoS Biol. 2007;5:e7. doi: 10.1371/journal.pbio.0050007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su CC, Li M, Gu R, Takatsuka Y, McDermott G, Nikaido H, Yu EW. Conformation of the AcrB Multidrug Efflux Pump in Mutants of the Putative Proton Relay Pathway. J Bacteriol. 2006;188:7290–7296. doi: 10.1128/JB.00684-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su CC, Yang F, Long F, Reyon D, Routh MD, Kuo DW, Mokhtari AK, Van Ornam JD, Rabe KL, Hoy JA, Lee YJ, Rajashankar KR, Yu EW. Crystal Structure of the Membrane Fusion Protein CusB from Escherichia coli. J Mol Biol. 2009;393:342–355. doi: 10.1016/j.jmb.2009.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su CC, Long F, Zimmermann MT, Rajashankar KR, Jernigan RL, Yu EW. Crystal Structure of the CusBA Heavy-Metal Efflux Complex of Escherichia coli. Nature. 2011;470:558–562. doi: 10.1038/nature09743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su CC, Long F, Lei HT, Bolla JR, Do SV, Rajashankar KR, Yu EW. Charged Amino Acids (R83, E567, D617, E625, R669, and K678) of CusA Are Required for Metal Ion Transport in the Cus Efflux System. J Mol Biol. 2012;422:429–441. doi: 10.1016/j.jmb.2012.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Törnroth-Horsefield S, Gourdon P, Horsefield R, Brive L, Yamamoto N, Mori H, Snijder A, Neutze R. Crystal Structure of AcrB in Complex with a Single Transmembrane Subunit Reveals another Twist. Structure. 2007;15:1663–1673. doi: 10.1016/j.str.2007.09.023. [DOI] [PubMed] [Google Scholar]
- Tseng TT, Gratwick KS, Kollman J, Park D, Nies DH, Goffeau A, Saier MH., Jr The RND Permease Superfamily: an Ancient, Ubiquitous and Diverse Family that includes Human Disease and Development Protein. J Mol Microbiol Biotechnol. 1999;1:107–125. [PubMed] [Google Scholar]
- Vaccaro L, Scott KA, Sansom MSP. Gating at Both Ends and Breathing in the Middle: Conformational Dynamics of TolC. Biophys J. 2008;95:5681–5691. doi: 10.1529/biophysj.108.136028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Davis AV, Balakrishnan G, Stasser JP, Staehlin BM, Focia P, Spiro TG, Penner-Hahn JE, O’Halloran TV. Cu(I) Recognition via Cation-π and Methionine Interactions in CusF. Nature Chem Biol. 2008;4:107–109. doi: 10.1038/nchembio.2007.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu EW, McDermott G, Zgruskaya HI, Nikaido H, Koshland DE., Jr Structural Basis of Multiple Drug Binding Capacity of the AcrB Multidrug Efflux Pump. Science. 2003;300:976–980. doi: 10.1126/science.1083137. [DOI] [PubMed] [Google Scholar]
- Yu EW, Aires JR, McDermott G, Nikaido H. A Periplasmic Drug-Binding Site of the AcrB Multidrug Efflux Pump: a Crystallographic and Site-Directed Mutagenesis Study. J Bacteriol. 2005;187:6804–6815. doi: 10.1128/JB.187.19.6804-6815.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zgurskaya H, Nikaido H. Bypassing the Periplasm: Reconstitution of the AcrAB Multidrug Efflux Pump of Escherichia coli. Proc Natl Acad Sci USA. 1999;96:7190–7195. doi: 10.1073/pnas.96.13.7190. [DOI] [PMC free article] [PubMed] [Google Scholar]