


Abstract
Cell cycle checkpoints induced by DNA damage play an integral role in preservation of genomic stability by allowing cells to limit the propagation of deleterious mutations. The retinoblastoma tumor suppressor (RB) is crucial for the maintenance of the DNA damage checkpoint function because it elicits cell cycle arrest in response to a variety of genotoxic stresses. Although sporadic loss of RB is characteristic of most cancers and results in the bypass of the DNA damage checkpoint, the consequence of RB loss upon chemotherapeutic responsiveness has been largely uninvestigated. Here, we employed a conditional knockout approach to ablate RB in adult fibroblasts. This system enabled us to examine the DNA damage response of adult cells following acute RB deletion. Using this system, we demonstrated that loss of RB disrupted the DNA damage checkpoint elicited by either cisplatin or camptothecin exposure. Strikingly, this bypass was not associated with enhanced repair, but rather the accumulation of phosphorylated H2AX (γH2AX) foci, which indicate DNA double-strand breaks. The formation of γH2AX foci was due to ongoing replication following chemotherapeutic treatment in the RB-deficient cells. Additionally, peak γH2AX accumulation occurred in S-phase cells undergoing DNA replication in the presence of damage, and these γH2AX foci co-localized with replication foci. These results demonstrate that acute RB loss abrogates DNA damage-induced cell cycle arrest to induce γH2AX foci formation. Thus, secondary genetic lesions induced by RB loss have implications for the chemotherapeutic response and the development of genetic instability.
INTRODUCTION
DNA damage checkpoints are elicited to ensure the maintenance of genomic stability following genotoxic insult (1,2). First observed in unicellular eukaryotes, it has subsequently become clear that highly conserved checkpoint pathways are present in virtually all organisms (3). It is believed that these signaling pathways lead to transient inhibition of cell cycle progression thereby ensuring repair of damaged DNA prior to DNA replication and cellular division. Alternatively, severe damage may lead to permanent cell cycle withdrawal or apoptosis to prevent the proliferation of cells harboring irreparable genetic lesions. As such, loss of critical checkpoint pathways represents a means for cells to accumulate deleterious mutations and is implicated in tumorigenesis (4–6).
Several tumor suppressor proteins play key roles in the maintenance of appropriate DNA damage response. For example, the p53 tumor suppressor was observed to accumulate in cells treated with UV or ionizing radiation and is required for G1/S cell cycle inhibition (7). Cells mutant for p53 are defective in delaying cell cycle progression in response to DNA damage (8). Similarly, ataxia telangiectasia mutated (ATM) activity is stimulated immediately following recognition of ionizing radiation-induced DNA damage. ATM-deficient cells have been found to undergo radioresistant DNA synthesis, erroneous entry into mitosis, and to exhibit extensive loss of the G1/S cell cycle checkpoint (9–11). In both the case of ATM and p53, loss of checkpoint function following DNA damage is associated with genomic instability and tumor predisposition.
While the retinoblastoma tumor suppressor (RB) is a central regulator of cell cycle progression, its role in checkpoint processes and genomic stability is less well understood. Initially, described as the gene mutated in retinoblastoma, it has subsequently become clear that RB is inactivated at high frequency in human cancers (5,12–17). RB functions to inhibit cell cycle progression by assembling repressor complexes that inhibit the expression of genes required for progression through S-phase (18–20). Following mitogenic signaling, RB is phosphorylated through the combined activities of CDK4- and CDK2-associated complexes (21–25). These phosphorylation events disrupt RB-mediated transcriptional repression and enable progression through the cell cycle (26–29). It has recently been shown that DNA damage acts through RB to inhibit cell cycle progression (20). Specifically, it was observed that DNA damage signals to prevent RB phosphorylation, thus activating RB-signaling pathways (30). Cells deficient in RB exhibit checkpoint deregulation in the presence of DNA-damaging agents, however, the extent and consequence of this genetic damage has not been specifically investigated.
A high proportion of tumors that are treated with DNA-damaging chemotherapeutic agents happen to be RB deficient (31–33). As such, understanding the interaction between RB loss and the chemotherapeutic response to these drugs is important for the improvement of existing therapeutics and the development of novel treatments. In principal, RB loss could have various effects on the processing of DNA lesions induced by chemotherapeutics and several non-exclusive possibilities exist. First, it has recently been demonstrated that RB negatively regulates the expression of a wide range of DNA repair factors (34–36); thus, in RB-deficient cells these lesions may be readily repaired due to the elevated levels of repair factors. Secondly, loss of RB in cells could deregulate cell cycle checkpoints permitting the propagation of deleterious mutations as is postulated to promote tumor progression. Thirdly, RB loss and subsequent checkpoint bypass could compromise viability in a manner analogous to that observed in checkpoint-deficient yeast, wherein a failure to elicit checkpoints leads to cell cycle catastrophe and death (37–40). In order to probe these responses in RB mutant cells, here we investigated the development of double-strand breaks (DSBs) as a secondary event from cisplatin (CDDP) or camptothecin (CPT) treatment.
MATERIALS AND METHODS
Isolation of primary RbloxP/loxP murine adult fibroblasts
Mice of the RbloxP/loxP genotype of mixed 129/FVBN background (41), at least 5 weeks of age, were killed by CO2 anesthetization, followed by cervical dislocation. Fibroblasts were isolated from the peritoneal fascia by excision and mincing of the peritoneum, followed by dissociation by constant agitation for 40 min at 37°C in 0.2 mg/ml collagenase (Type I, Sigma) supplemented with 100 U DNase I (Roche). The dissociated tissue was washed with PBS and subsequently incubated for 20 min at 37°C in 0.25% trypsin (Gibco) with constant agitation. After washing twice, the isolated cells were plated in tissue culture dishes.
Cell culture, recombinant adenoviral infections
RbloxP/loxP murine adult fibroblasts (MAFs) were subcultured in DMEM containing 10% FBS supplemented with 100 U/ml penicillin/streptomycin and 2 mM l-glutamine at 37°C in air containing 5% CO2. Primary cells used in this study were between passages 3 and 5. Replication-defective recombinant adenovirus expressing green fluorescent protein (Ad-GFP) or GFP in addition to Cre recombinase (Ad-GFP-Cre) were obtained from G. Leone (Department of Molecular Genetics, Ohio State University). The conditional RB knockout in primary RbloxP/loxP MAFs was attained by infecting cells with adenovirus at approximately 2 × 107 virus particles per dish to achieve an infection efficiency of 90–95% as determined by GFP immunofluoresence. Cells were cultured for at least 4 days post-adenoviral infection before use. The same passage number and length of time post-infection of the Ad-GFP and Ad-GFP-Cre infected MAFs were used in all experiments.
Immunoblotting
Cells infected with Ad-GFP or Ad-GFP-Cre were harvested by trypsinization and lysed in RIPA buffer and equal amounts of protein were resolved by SDS–PAGE. Specific proteins were detected by standard immunoblotting procedures using the following primary antibodies: PCNA (pc10) (Santa Cruz, 1:5000 dilution), cyclin E (HE12), cyclin A (C19), β-tubulin (D10) and anti-RB (G3-245; Becton Dickinson, 1:1000 dilution).
RT–PCR analysis of recombination
RT–PCR analysis was performed to verify adenoviral-Cre-mediated recombination in primary MAFs. Total RNA was extracted using Trizol (Gibco) and cDNA was synthesized from 1 µg of RNA with the SuperScript RT–PCR system (Gibco) according to the manufacturer’s protocol. cDNAs were amplified using PCR and the following primers: sense, 5′-CTGGCCAGGCTTGAGTTTGAAG-3′; and antisense, 5′-CAGTAGATAACGCACTGCTG-3′. PCR conditions consisted of initial denaturation for 2 min at 94°C, followed by 30 cycles of 30 s at 94°C, 30 s at 51°C and 1 min at 72°C, followed by a final extension for 5 min at 72°C. Ten microliters of PCR product was run on a 2% agarose gel and visualized by ethidium bromide staining.
DNA damage, bromodeoxyuridine (BrdU) labeling, γH2AX immunofluorescence
Primary MAFs infected with Ad-GFP or Ad-GFP-Cre were seeded on coverslips in six-well dishes and allowed to attach. Cells were then incubated with chemotherapeutic drugs at the indicated concentrations for 16 h. Drugs were then removed by washing cells for 3 × 5 min with tissue culture media and labeled with BrdU (Amersham Pharmacia Biotech) to detect DNA synthesis. Cells were labeled for 8 h, then washed, fixed in 3.7% formaldehyde, and processed to detect BrdU incorporation as previously described (42). CPT was purchased from Sigma while clinical grade cis-diamminedichloroplatinum II (CDDP) was from Bristol Oncology.
γH2AX staining was performed as described by the manu facturer using anti-phospho-H2AX ser 139 mouse primary antibody (Upstate Biotechnology).
BrdU and γH2AX co-localization studies were performed by BrdU labeling cells for 20 min, followed by methanol fixation. Cells were stained for γH2AX as described above using Alexa 488 secondary 1:100 (Molecular Probes). Cells were then fixed in 3.7% formaldehyde for 20 min, followed by 0.5% NP-40 for 5 min and washed with water. HCl (1.5 N) was used to depurinate the cell nuclei for 30 min, followed by washing for 3 × 5 min with water, permeabilization with 0.3% Triton X-100 for 20 min, and BrdU staining as above, using 1:100 rat anti-BrdU primary antibody (McAb). Donkey anti-rat IgG rhodamine-red X-conjugated secondary antibody (Jackson Immunoresearch) was used at a 1:100 dilution and counterstained with DAPI (Sigma).
Flow cytometry for γH2AX
Cells (5 × 105) were harvested and fixed in 70% ethanol. On the day of analysis, cells were prepared and analyzed as previously described (43). The cell cycle distribution was determined using Mod-Fit software.
RESULTS
Efficient deletion of RB from adult cells
During the genesis of human cancer, RB is acutely lost in adult cells. As most prior studies have used mouse embryo fibroblasts (MEFs) harboring loss of RB throughout development or tumor lines which have been extensively cultured, a specific role for RB loss has been difficult to ascertain. To eliminate these complications, we utilized a novel system involving mice harboring a conditional Rb allele (RbloxP/loxP mice) in which loxP sites flank Rb exon 3. Through adenoviral expression of Cre recombinase, acute RB loss in cells that contain a genetically stable primary cell background have recently been reported (41).
To examine the role of RB in adult cells, we employed MAFs infected with recombinant adenoviruses expressing both GFP and Cre recombinase (Ad-GFP-Cre) or GFP alone (Ad-GFP) as a control. Efficient infection was demonstrated under previously described conditions such that >90% Ad-GFP-Cre infected cells expressed high levels of GFP fluorescence 16–24 h post-infection (data not shown). To confirm Cre-mediated recombination, primers in exon 2 and exon 4 of the Rb gene were utilized as described in Materials and Methods. RNA was prepared from uninfected MAFs (0 h) or those infected with Ad-GFP-Cre at 48 h post-infection. Analysis of the Rb transcript using RT–PCR showed the accumulation of the Δexon3-PCR product in the infected cells relative to the control, confirming efficient recombination at this locus (Fig. 1A). Western blotting with anti-RB monoclonal antibody revealed that the Cre-mediated recombination resulted in acute downregulation of RB protein in Ad-GFP-Cre infected MAFs 5 days post-infection (Fig. 1B).
Figure 1.
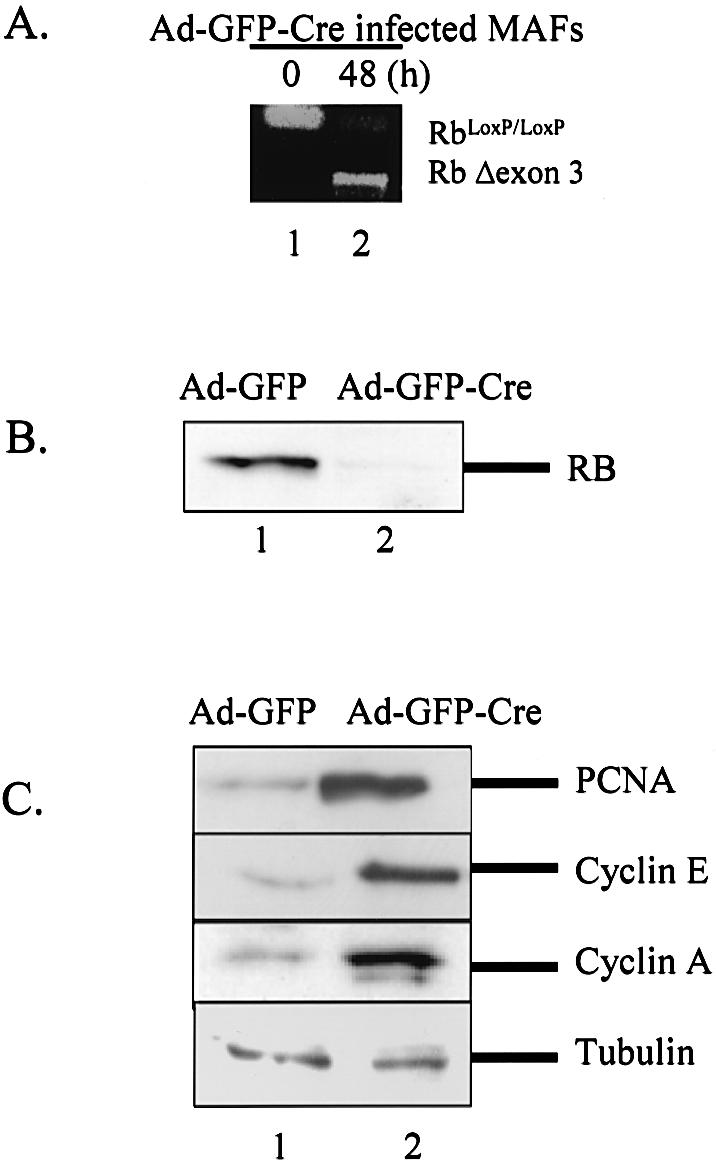
Infection of primary RbloxP/loxP MAFs with adenoviral Cre yields acute downregulation of RB protein and deregulation of RB repression targets. (A) Asynchronously proliferating primary RbloxP/loxP MAFs were infected with Ad-GFP or Ad-GFP-Cre adenovirus. RNA was isolated at 0 and 48 h post-infection and RT–PCR was performed employing primers specific for regions flanking the loxP sites in the murine RB gene. Recombination at the floxed RB locus is evident by the appearance of the smaller transcript post-infection. (B) Asynchronously proliferating primary MAFs were infected with Ad-GFP or Ad-GFP-Cre and harvested in RIPA buffer. Protein concentrations were normalized by Bio-Rad DC assay. Equal amounts of protein were separated by electrophoresis and immunoblotted with anti-RB monoclonal antibody. (C) Asynchronously proliferating MAFs were infected with adenovirus, harvested, and protein concentrations were quantified as in (B). The effect of acute RB loss upon downstream targets was determined by immunoblotting for PCNA, cyclin E and cyclin A. Lysates were immunoblotted for β-tubulin to confirm equal loading.
To determine the biochemical effect of acute RB loss, we evaluated the expression of downstream targets in the RB/E2F signaling axis (30,35,44). MAFs infected with either Ad-GFP or Ad-GFP-Cre were harvested 5 days post-infection and levels of known RB target proteins were analyzed by immunoblotting. Relative to control (lane 1), the Ad-GFP-Cre infected MAFs exhibited increased levels of proteins downstream of RB signaling including, PCNA, cyclin E and cyclin A (Fig. 1C). No changes were detected in tubulin protein levels, which served as a loading control. Thus, RB can be conditionally ablated in primary adult cells and leads to specific target gene deregulation.
RB loss in primary adult fibroblasts impairs the cell cycle response to cisplatin and camptothecin damage
In order to evaluate the role of RB in the DNA damage response of adult fibroblasts, asynchronously proliferating Ad-GFP or Ad-GFP-Cre infected MAFs were treated with various concentrations of CDDP for 16 h. Initially, we used an anti-platinum (Pt) antibody that detects Pt-DNA adducts to determine that both Ad-GFP and Ad-GFP-Cre infected MAFs treated with CDDP produced similar amounts of Pt-DNA adducts based on equal staining intensities (Fig. 2A, left) (45). To quantify the amounts of anti-Pt staining present in these MAFs, the average pixel intensity was compared using Metamorph software and displayed graphically to confirm that there is no significant difference in the levels of initial Pt-adduct damage (Fig. 2A, right). In parallel experiments, cells were treated with CDDP for 16 h, washed of drug, and then processed in one of the three following ways: (i) pulsed with BrdU for 8 h and subsequently fixed, (ii) allowed to proliferate to 40 h before BrdU labeling and fixing, (iii) allowed to proliferate to 64 h before BrdU labeling and fixing (Fig. 2B). The replicative fraction of treated cells was determined with respect to untreated control cells by detecting BrdU incorporation. MAFs containing functional RB exhibited a dose-dependent cell cycle inhibition, whereas cells lacking RB exhibited significantly reduced levels of arrest at each time point (Fig. 2C). Analogous RB-mediated dose-dependent checkpoint results were also observed when treating infected MAFs with CPT (Fig. 2D). Together, these data indicate that RB plays an instrumental role in the cell cycle response to DNA damage, in that acute loss of RB in adult cells uncouples this checkpoint.
Figure 2.


RB loss in primary adult fibroblasts impairs replication in response to DNA damage. (A) (Left) Immunofluorescence using an anti-Pt antibody was performed on Ad-GFP and Ad-GFP-Cre infected RbloxP/loxP MAFs in order to confirm that the cells incorporated similar amounts of Pt-DNA adducts after 16 h of CDDP treatment. (Right) The average pixel intensities from the anti-Pt staining shown in the left panel are represented graphically to verify similar Pt-DNA adduct incorporation after CDDP treatment. (B) The following studies incorporate the experimental format as outlined, such that asynchronously proliferating primary RbloxP/loxP MAFs infected with Ad-GFP or Ad-GFP-Cre were treated with 0, 4 and 8 µM CDDP for 16 h. The drug was washed away and a subset of the cells were allowed to proliferate to 40 and 64 h. (C) CDDP-treated cells from (B) were washed of drug and pulsed with BrdU for 8 h. The proliferative fraction of treated cells was determined with respect to an untreated control through immunofluorescence. (D) The adenovirus infected primary cells from (B) were treated with 0, 5 and 10 µM CPT for 16 h and the proliferative fraction of cells was determined by BrdU incorporation to show an RB-mediated role in the dose-dependent cell cycle response to DNA damage.
Acute RB knockout influences the accumulation of DNA double-strand breaks
Since neither CDDP nor CPT act directly to elicit DNA DSBs (46,47), we studied the impact of RB loss upon the accumulation of these lesions. H2AX, a submember of the histone H2A family is rapidly phosphorylated at its C-terminus (γH2AX) in response to DSBs, and serves as an efficient measure of DSB accumulation (46–48). Ad-GFP and Ad-GFP-Cre infected primary MAFs were treated with CDDP for 16 h and examined for γH2AX focus formation using immunofluorescence. Capturing images of these drug-treated cells at equal exposures revealed that the number and intensities of γH2AX foci increased with CDDP treatment in both types of infected MAFs (Fig. 3A). Interestingly, cells lacking RB, in fact, have a significantly increased quantity and intensity of γH2AX foci as compared with the Ad-GFP infected MAFs after 16 h of treatment (Fig. 3A, top). Analogous results were evident 48 h post-treatment (T = 64 h) (Fig. 3A, bottom). In order to quantify the observed intensity differences between the γH2AX foci in Ad-GFP- and Ad-GFP-Cre infected MAFs, cells were imaged by confocal microscopy and analyzed for signal intensity using Metamorph software (Fig. 3B). These data show that the cells containing functional RB accumulate lower levels of DSBs that increase in proportion to CDDP concentrations immediately after and 48 h post-treatment (T = 64 h) as compared with MAFs lacking RB. RB-deficient MAFs exhibit a much higher intensity level of γH2AX staining that increases with CDDP dose such that the staining level appeared to saturate at the 4 µM dose (Fig. 3B). Cells containing wild-type RB treated for 16 h with CDDP, washed free of drug, and maintained in medium to 64 h contained similar levels of staining as compared with the cells stained after 16 h of treatment. We confirmed these results by evaluating the percentage of cells in the populations staining positive for γH2AX (Fig. 3C). Similar results were evident after CPT treatment for 16 h (Fig. 3D). These data demonstrate the significant differences in amount and intensity of nuclear γH2AX foci present in cells with and without RB and attests to the importance of RB in preventing the accumulation of DSBs in response to chemotherapeutic treatment.
Figure 3.


Acute RB loss influences the accumulation of DNA DSBs. (A) (Top) Asynchronously proliferating primary RbloxP/loxP MAFs infected with Ad-GFP or Ad-GFP-Cre were treated with 0, 4 and 8 µM CDDP for 16 h. MAFs were analyzed for γH2AX foci formation by immunofluorescence using an anti-γH2AX monoclonal antibody. (Bottom) A subset of the CDDP treated cells from (A) was washed of drug and allowed to proliferate to 64 h. The γH2AX positive foci were analyzed as above to show the effect of RB on the levels of DNA DSBs at longer periods after chemotherapeutic treatment. (B) The difference in γH2AX staining intensity between the Ad-GFP and Ad-GFP-Cre infected MAFs treated with 0, 4 and 8 µM CDDP for 16 h from (A) was further quantified using Metamorph software to analyze average pixel intensities of each stained nucleus. These data are represented graphically as the relative increase in γH2AX intensity. (C) In order to quantify the effect of RB loss upon levels of DNA damage, the Ad-GFP and Ad-GFP-Cre infected MAFs from (A) were treated with 0, 1, 4 and 8 µM CDDP for 16 h and either fixed or maintained in media to 64 h. These cells were then scored as being positive or negative for γH2AX foci and the data were represented graphically. (D) Ad-GFP or Ad-GFP-Cre infected MAFs were treated with 0, 5 and 10 µM CPT for 16 h and immunofluorescence using an anti-γH2AX monoclonal antibody is performed. As in (B), the cells were scored for the presence of γH2AX-positive foci and the data were represented graphically.
Replication-dependent DNA double-strand break accumulation
There are two possible explanations for the observed increase in accumulation of DNA damage in response to chemotherapeutic drugs in RB mutant cells. First, loss of RB may contribute to DSBs due to downstream target deregulation that promotes breaks (e.g. enhanced repair or nuclease expression). Secondly, replication across the primary lesion promotes additional DSBs. We were able to address these possibilities by uncoupling RB loss from checkpoint deregulation. As shown in Figure 4A, serum starvation for 3 days inhibits both RB-proficient and -deficient cells from undergoing replication. Under these conditions no significant difference in the percentage of γH2AX-positive foci per cell was evident between the Ad-GFP and Ad-GFP-Cre infected MAFs following 16 h of CDDP treatment (Fig. 4B). These data indicate that most of the DSB damage acquired by the RB-deficient cells occurred during cell cycle progression, as is demonstrated by comparing Figure 3C with Figure 4B.
Figure 4.


Replication-dependent DSB accumulation. (A) Primary RbloxP/loxP MAFs infected with Ad-GFP or Ad-GFP-Cre were serum starved for 3 days and treated with 0, 4 and 8 µM CDDP for 16 h. The cells were washed of drug, labeled with BrdU for 8 h to confirm that none of the cells were able to enter S-phase. (B) CDDP-treated MAFs from (A) were analyzed for γH2AX foci formation by immunofluorescence using an anti-γH2AX monoclonal antibody. (C) Asynchronously proliferating primary RbloxP/loxP MAFs infected with Ad-GFP-Cre were treated with 0 and 4 µM CDDP for 16 h. The cells were harvested and processed for flow cytometry by staining with γH2AX–FITC and propidium iodide. These data were analyzed using ModFit software.
To specifically consider the possibility that DSB damage is replication dependent, flow cytometric analysis was utilized to examine cell cycle phase-specific accumulation of γH2AX-positive foci. Asynchronous Ad-GFP-Cre infected MAFs were analyzed following 16 h of CDDP treatment. By separating cells stained for γH2AX and DNA content, peak γH2AX staining was, in fact, specifically observed in cells with an S-phase DNA content (Fig. 4C). This profile is consistent with the idea that continued replication in the CDDP-treated RB-deficient cells drives the accumulation of γH2AX.
To directly associate γH2AX with ongoing replication, co-labeling experiments were performed where DNA replication and γH2AX foci were coordinately analyzed. Initially, as a control to validate the specificity of the antibodies utilized, we demonstrated that the γH2AX background staining observed in untreated cells did not co-localize with BrdU incorporation (Fig. 5A). Ad-GFP and Ad-GFP-Cre infected MAFs treated with CDDP for 16 h, were washed of drug and pulsed with BrdU for 20 min, at which time they were fixed. Immunofluoresence was used to observe sites of BrdU incorporation and γH2AX foci localization in untreated control and CDDP-treated cells. No co-localization was evident in cells containing functional RB due to lower accumulation of γH2AX foci and a functional checkpoint yielding inhibition of BrdU incorporation following 16 h of 4 µM CDDP treatment. Conversely, in RB-deficient cells with the same CDDP exposure, γH2AX foci co-localized with the sites of ongoing BrdU incorporation (Fig. 5B). The BrdU- and γH2AX-stained cells were analyzed in greater detail using morphometric analysis to identify individual foci overlap throughout the entire nucleus (Fig. 5C). As a control for background staining, the pixel intensity thresholds for both the red and green channels in these 8-bit images were held constant at 133 and 50, respectively. Sites of BrdU incorporation were found to co-localize with ∼7% of the γH2AX background stained sites in untreated Ad-GFP infected MAFs (data not shown). However, in Ad-GFP-Cre infected MAFs that were treated with 4 µM CDDP for 16 h, sites of threshold BrdU incorporation staining were found to co-localize with 69 ± 8% of threshold γH2AX foci staining present in the nuclei. These data demonstrate that sites of ongoing replication and DSBs co-localize in RB-deficient adult cells following DNA damage.
Figure 5.



BrdU and γH2AX foci co-localization. (A) Immunofluorescence analysis of BrdU and γH2AX foci co-labeling in untreated primary RbloxP/loxP MAFs infected with Ad-GFP or Ad-GFP-Cre. (B) Cells from (A) were treated for 16 h with 4 µM CDDP and immunofluorescence was used to analyze BrdU and γH2AX foci localization. (C) Co-labeled Ad-GFP-Cre infected cells treated with CDDP from (B) were imaged by confocal micoroscopy and examined using morphometric analysis. As a control for background staining, the pixel intensity thresholds for both the red (BrdU) and green (γH2AX) channels in these 8-bit images were held constant at 133 and 50, respectively. Throughout the entire nucleus, sites of BrdU incorporation were found to co-localize with 69 ± 8% of γH2AX foci (n = 5).
DISCUSSION
Since RB is functionally inactivated in the majority of human cancers, understanding the role of RB in modifying the cellular response to chemotherapeutics is critical for the development of efficacious therapeutics. We and others have shown that chronic RB loss in MEFs is sufficient to inactivate the DNA damage checkpoint and yield deregulated growth (20,42). Here, we observed that primary MAFs with functional RB were able to successfully inhibit replication following treatment with CDDP, which forms Pt-DNA adducts, and with CPT, which is an inhibitor of topoisomerase I. However, acute RB loss in these same primary cells was sufficient to impair cell cycle arrest after exposure to both chemotherapeutic agents. These results indicate that RB is also required to mediate cell cycle checkpoints in adult cells, consistent with what has been observed in MEFs (42,48).
It has become increasingly clear that DNA damage functions to elicit cell cycle checkpoints via signaling pathways. One of the critical determinants of the signaling from DNA DSBs is the rapid phosphorylation of histone H2AX at the site of the lesion (γH2AX) (49). In this study, we utilize antibodies specific for γH2AX to probe for the induction of DSBs (as described below). However, the data from this study indicate that loss of RB does not compromise the signaling to phosphorylate H2AX. These results are consistent with the observation that RB function after DNA damage signaling demonstrates delayed kinetics relative to the rapid phosphorylation of H2AX by ATM that can occur within minutes of a formed DSB (54). As such, these results position RB downstream from the signaling pathways, giving rise to γH2AX.
Among the most damaging lesions caused by chemotherapeutic drugs are DSBs. Several potent anti-cancer agents induce this lesion as the primary form of damage (e.g. ionizing radiation or doxorubicin). However, a number of chemotherapeutics do not generate DSBs as the primary lesion. For example, CDDP or alkylating agents form DNA adducts that do not spontaneously give rise to DSBs. Similarly, CPT and other topoisomerase I inhibitors cause only single-strand breaks. However, the processing of these types of lesions can lead to the formation of DSBs (50–54). Here, we specifically focused on determining the role of RB loss on the development of DSBs following damage with CDDP and CPT. Presumably, the loss of RB could have diverse effects on the processing of these forms of damage. First, it has been found that RB regulates the expression of a large number of repair factors that are involved in the repair of adducts and DSBs (e.g. RAD51, BRCA1, FEN1, RPA, MSH2) (34–36). The upregulation of such factors could actually limit the amount of damage and therefore lead to less DSBs. Alternatively, loss of RB and the subsequent failure to halt DNA replication (30) could facilitate replication across the lesion and give rise to DSBs (50–53). Analysis of these lesions, arising both after CPT and CDDP damage, indicated that loss of RB does in fact lead to additional DSBs (Fig. 3). This occurred on a per cell basis with low dose CDDP and CPT (data not shown) damage capable of eliciting significantly more breaks in RB-deficient cells. Additionally, this was valid for the entire population of cultured cells, wherein RB-deficient cells acquired increased amounts of γH2AX foci at a given concentration of DNA-damaging agent. This effect could be explained by the ability of the cells lacking RB to replicate despite the presence of DNA damage as we observed through BrdU incorporation assays, so that failure to inhibit replication leads to secondary lesions.
Because functional inactivation of the RB pathway is characteristic of 90% of all tumors, understanding the relationship between chemotherapeutic response and the genetic status of a tumor has high clinical relevance and is crucial for the development of effective treatments. Our data suggest that CDDP and CPT may be particularly effective in the treatment of RB-deficient neoplasms because DSBs represent a difficult lesion to repair and can fuel the subsequent cell death elicited by the agents. Loss of RB synergizes with the drug to make it more effective in inducing damage. This observation may indicate why certain tumors characterized by loss of RB have good response rates to chemotherapeutics and why RB-deficient cells are generally more sensitive to cell death by such agents (42,55,56). However, the generation of additional genetic lesions through the loss of RB (in this case DSBs) can also have a deleterious consequence, as such lesions could fuel tumor progression. Such an idea is not without precedent as it has been reported that 30% of heritable retinoblastoma patients present with secondary primary nonocular neoplasms outside of the field of their initial radiation therapy by age 40 (57).
Our results provide a framework for elucidating the consequence of RB loss on chemotherapeutic response. We demonstrate that acute RB loss abrogates the DNA damage checkpoint in adult cells. These cells acquired increased amounts of DSBs as compared with RB-proficient cells following CDDP or CPT treatment. Lastly, the accumulation of DSBs in RB-deficient cells was shown to be replication dependent. Thus, loss of RB gives rise to secondary genetic lesions that will impact the response to therapeutic agents.
Acknowledgments
ACKNOWLEDGEMENTS
We are grateful to Dr Karen Knudsen and Dr Peter Stambrook for their helpful comments on the manuscript and all members of the Knudsen laboratories for insightful discussions. We thank Sandy Schwemberger and Dr George Babcock for providing their expert assistance with the flow cytometry. This work was supported by ACS grant RSG-01-254-01-CCG to E.S.K. E.E.B. is supported by National Institute of Environmental Health Sciences Training Grant 5T32 ES 07250-16. C.N.M. is supported by National Cancer Institute Training Grant T32 CA 59268. T.J. is an investigator of Howard Hughes Medical Institute.
REFERENCES
- 1.Hartwell L.H. and Weinert,T.A. (1989) Checkpoints: controls that ensure the order of cell cycle events. Science, 246, 629–634. [DOI] [PubMed] [Google Scholar]
- 2.Nyberg K.A., Michelson,R.J., Putnam,C.W. and Weinert,T.A. (2002) Toward maintaining the genome: DNA damage and replication checkpoints. Annu. Rev. Genet., 36, 617–656. [DOI] [PubMed] [Google Scholar]
- 3.Elledge S.J. (1996) Cell cycle checkpoints: preventing an identity crisis. Science, 274, 1664–1672. [DOI] [PubMed] [Google Scholar]
- 4.Bartek J. and Lukas,J. (2001) Mammalian G1- and S-phase checkpoints in response to DNA damage. Curr. Opin. Cell Biol., 13, 738–747. [DOI] [PubMed] [Google Scholar]
- 5.Bartkova J., Lukas,J. and Bartek,J. (1997) Aberrations of the G1- and G1/S-regulating genes in human cancer. Prog. Cell Cycle Res., 3, 211–220. [DOI] [PubMed] [Google Scholar]
- 6.Flatt P.M. and Pietenpol,J.A. (2000) Mechanisms of cell-cycle checkpoints: at the crossroads of carcinogenesis and drug discovery. Drug Metab. Rev., 32, 283–305. [DOI] [PubMed] [Google Scholar]
- 7.Kastan M.B., Zhan,Q., el-Deiry,W.S., Carrier,F., Jacks,T., Walsh,W.V., Plunkett,B.S., Vogelstein,B. and Fornace,A.J.,Jr (1992) A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell, 71, 587–597. [DOI] [PubMed] [Google Scholar]
- 8.Lu X. and Lane,D.P. (1993) Differential induction of transcriptionally active p53 following UV or ionizing radiation: defects in chromosome instability syndromes? Cell, 75, 765–778. [DOI] [PubMed] [Google Scholar]
- 9.Savitsky K., Bar-Shira,A., Gilad,S., Rotman,G., Ziv,Y., Vanagaite,L., Tagle,D.A., Smith,S., Uziel,T., Sfez,S. et al. (1995) A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science, 268, 1749–1753. [DOI] [PubMed] [Google Scholar]
- 10.Canman C.E., Lim,D.S., Cimprich,K.A., Taya,Y., Tamai,K., Sakaguchi,K., Appella,E., Kastan,M.B. and Siliciano,J.D. (1998) Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science, 281, 1677–1679. [DOI] [PubMed] [Google Scholar]
- 11.Lavin M.F. and Shiloh,Y. (1997) The genetic defect in ataxia-telangiectasia. Annu. Rev. Immunol., 15, 177–202. [DOI] [PubMed] [Google Scholar]
- 12.Wang J.Y., Knudsen,E.S. and Welch,P.J. (1994) The retinoblastoma tumor suppressor protein. Adv. Cancer Res., 64, 25–85. [DOI] [PubMed] [Google Scholar]
- 13.Weinberg R.A. (1995) The retinoblastoma protein and cell cycle control. Cell, 81, 323–330. [DOI] [PubMed] [Google Scholar]
- 14.Sherr C.J. (1996) Cancer cell cycles. Science, 274, 1672–1677. [DOI] [PubMed] [Google Scholar]
- 15.Reed S.I. (1997) Control of the G1/S transition. Cancer Surv., 29, 7–23. [PubMed] [Google Scholar]
- 16.Bartek J., Bartkova,J. and Lukas,J. (1997) The retinoblastoma protein pathway in cell cycle control and cancer. Exp. Cell Res., 237, 1–6. [DOI] [PubMed] [Google Scholar]
- 17.Kaelin W.G. Jr (1997) Recent insights into the functions of the retinoblastoma susceptibility gene product. Cancer Invest., 15, 243–254. [DOI] [PubMed] [Google Scholar]
- 18.Harbour J.W. and Dean,D.C. (2000) The Rb/E2F pathway: expanding roles and emerging paradigms. Genes Dev., 14, 2393–2409. [DOI] [PubMed] [Google Scholar]
- 19.Dyson N. (1998) The regulation of E2F by pRB-family proteins. Genes Dev., 12, 2245–2262. [DOI] [PubMed] [Google Scholar]
- 20.Harrington E.A., Bruce,J.L., Harlow,E. and Dyson,N. (1998) pRB plays an essential role in cell cycle arrest induced by DNA damage. Proc. Natl Acad. Sci. USA, 95, 11945–11950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brugarolas J., Chandrasekaran,C., Gordon,J.I., Beach,D., Jacks,T. and Hannon,G.J. (1995) Radiation-induced cell cycle arrest compromised by p21 deficiency. Nature, 377, 552–557. [DOI] [PubMed] [Google Scholar]
- 22.Lundberg A.S. and Weinberg,R.A. (1998) Functional inactivation of the retinoblastoma protein requires sequential modification by at least two distinct cyclin-cdk complexes. Mol. Cell. Biol., 18, 753–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mittnacht S. (1998) Control of pRB phosphorylation. Curr. Opin. Genet. Dev., 8, 21–27. [DOI] [PubMed] [Google Scholar]
- 24.Zarkowska T. and Mittnacht,S. (1997) Differential phosphorylation of the retinoblastoma protein by G1/S cyclin-dependent kinases. J. Biol. Chem., 272, 12738–12746. [DOI] [PubMed] [Google Scholar]
- 25.Resnitzky D., Hengst,L. and Reed,S.I. (1995) Cyclin A-associated kinase activity is rate limiting for entrance into S phase and is negatively regulated in G1 by p27Kip1. Mol. Cell. Biol., 15, 4347–4352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bremner R., Cohen,B.L., Sopta,M., Hamel,P.A., Ingles,C.J., Gallie,B.L. and Phillips,R.A. (1995) Direct transcriptional repression by pRB and its reversal by specific cyclins. Mol. Cell. Biol., 15, 3256–3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weintraub S.J., Prater,C.A. and Dean,D.C. (1992) Retinoblastoma protein switches the E2F site from positive to negative element. Nature, 358, 259–261. [DOI] [PubMed] [Google Scholar]
- 28.Sellers W.R., Rodgers,J.W. and Kaelin,W.G.,Jr (1995) A potent transrepression domain in the retinoblastoma protein induces a cell cycle arrest when bound to E2F sites. Proc. Natl Acad. Sci. USA, 92, 11544–11548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weintraub S.J., Chow,K.N., Luo,R.X., Zhang,S.H., He,S. and Dean,D.C. (1995) Mechanism of active transcriptional repression by the retinoblastoma protein. Nature, 375, 812–815. [DOI] [PubMed] [Google Scholar]
- 30.DeGregori J., Kowalik,T. and Nevins,J.R. (1995) Cellular targets for activation by the E2F1 transcription factor include DNA synthesis- and G1/S-regulatory genes. Mol. Cell. Biol., 15, 4215–4224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bornfeld N., Schuler,A., Bechrakis,N., Henze,G. and Havers,W. (1997) Preliminary results of primary chemotherapy in retinoblastoma. Klin. Padiatr., 209, 216–221. [DOI] [PubMed] [Google Scholar]
- 32.Zheng L. and Lee,W.H. (2002) Retinoblastoma tumor suppressor and genome stability. Adv. Cancer Res., 85, 13–50. [DOI] [PubMed] [Google Scholar]
- 33.Yamasaki L. (2003) Role of the RB tumor suppressor in cancer. Cancer Treat. Res., 115, 209–239. [DOI] [PubMed] [Google Scholar]
- 34.Polager S., Kalma,Y., Berkovich,E. and Ginsberg,D. (2002) E2Fs up-regulate expression of genes involved in DNA replication, DNA repair and mitosis. Oncogene, 21, 437–446. [DOI] [PubMed] [Google Scholar]
- 35.Markey M.P., Angus,S.P., Strobeck,M.W., Williams,S.L., Gunawardena,R.W., Aronow,B.J. and Knudsen,E.S. (2002) Unbiased analysis of RB-mediated transcriptional repression identifies novel targets and distinctions from E2F action. Cancer Res., 62, 6587–6597. [PubMed] [Google Scholar]
- 36.Ishida S., Huang,E., Zuzan,H., Spang,R., Leone,G., West,M. and Nevins,J.R. (2001) Role for E2F in control of both DNA replication and mitotic functions as revealed from DNA microarray analysis. Mol. Cell. Biol., 21, 4684–4699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tercero J.A. and Diffley,J.F. (2001) Regulation of DNA replication fork progression through damaged DNA by the Mec1/Rad53 checkpoint. Nature, 412, 553–557. [DOI] [PubMed] [Google Scholar]
- 38.Takada S., Kelkar,A. and Theurkauf,W.E. (2003) Drosophila checkpoint kinase 2 couples centrosome function and spindle assembly to genomic integrity. Cell, 113, 87–99. [DOI] [PubMed] [Google Scholar]
- 39.Andreassen P.R., Lacroix,F.B., Lohez,O.D. and Margolis,R.L. (2001) Neither p21WAF1 nor 14–3-3sigma prevents G2 progression to mitotic catastrophe in human colon carcinoma cells after DNA damage, but p21WAF1 induces stable G1 arrest in resulting tetraploid cells. Cancer Res., 61, 7660–7668. [PubMed] [Google Scholar]
- 40.Arroyo M.P. and Wang,T.S. (1998) Mutant PCNA alleles are associated with cdc phenotypes and sensitivity to DNA damage in fission yeast. Mol. Gen. Genet., 257, 505–518. [DOI] [PubMed] [Google Scholar]
- 41.Sage J., Miller,A.L., Perez-Mancera,P.A., Wysocki,J.M. and Jacks,T. (2003) Acute mutation of retinoblastoma gene function is sufficient for cell cycle re-entry. Nature, 424, 223–228. [DOI] [PubMed] [Google Scholar]
- 42.Knudsen K.E., Booth,D., Naderi,S., Sever-Chroneos,Z., Fribourg,A.F., Hunton,I.C., Feramisco,J.R., Wang,J.Y. and Knudsen,E.S. (2000) RB-dependent S-phase response to DNA damage. Mol. Cell. Biol., 20, 7751–7763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.MacPhail S.H., Banath,J.P., Yu,Y., Chu,E. and Olive,P.L. (2003) Cell cycle-dependent expression of phosphorylated histone H2AX: reduced expression in unirradiated but not X-irradiated G1-phase cells. Radiat. Res., 159, 759–767. [DOI] [PubMed] [Google Scholar]
- 44.Sever-Chroneos Z., Angus,S.P., Fribourg,A.F., Wan,H., Todorov,I., Knudsen,K.E. and Knudsen,E.S. (2001) Retinoblastoma tumor suppressor protein signals through inhibition of cyclin-dependent kinase 2 activity to disrupt PCNA function in S phase. Mol. Cell. Biol., 21, 4032–4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ghazal-Aswad S., Tilby,M.J., Lind,M., Baily,N., Sinha,D.P., Calvert,A.H. and Newell,D.R. (1999) Pharmacokinetically guided dose escalation of carboplatin in epithelial ovarian cancer: effect on drug-plasma AUC and peripheral blood drug-DNA adduct levels. Ann. Oncol., 10, 329–334. [DOI] [PubMed] [Google Scholar]
- 46.Fuertesa M.A., Castillab,J., Alonsoa,C. and Perez,J.M. (2003) Cisplatin biochemical mechanism of action: from cytotoxicity to induction of cell death through interconnections between apoptotic and necrotic pathways. Curr. Med. Chem., 10, 257–266. [DOI] [PubMed] [Google Scholar]
- 47.Streltsov S.A. (2002) Action models for the antitumor drug camptothecin: formation of alkali-labile complex with DNA and inhibition of human DNA topoisomerase I. J. Biomol. Struct. Dyn., 20, 447–454. [DOI] [PubMed] [Google Scholar]
- 48.Lan Z., Sever-Chroneos,Z., Strobeck,M.W., Park,C.H., Baskaran,R., Edelmann,W., Leone,G. and Knudsen,E.S. (2002) DNA damage invokes mismatch repair-dependent cyclin D1 attenuation and retinoblastoma signaling pathways to inhibit CDK2. J. Biol. Chem., 277, 8372–8381. [DOI] [PubMed] [Google Scholar]
- 49.Rogakou E.P., Pilch,D.R., Orr,A.H., Ivanova,V.S. and Bonner,W.M. (1998) DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem., 273, 5858–5868. [DOI] [PubMed] [Google Scholar]
- 50.Vaisman A., Lim,S.E., Patrick,S.M., Copeland,W.C., Hinkle,D.C., Turchi,J.J. and Chaney,S.G. (1999) Effect of DNA polymerases and high mobility group protein 1 on the carrier ligand specificity for translesion synthesis past platinum-DNA adducts. Biochemistry, 38, 11026–11039. [DOI] [PubMed] [Google Scholar]
- 51.Vaisman A. and Chaney,S.G. (2000) The efficiency and fidelity of translesion synthesis past cisplatin and oxaliplatin GpG adducts by human DNA polymerase beta. J. Biol. Chem., 275, 13017–13025. [DOI] [PubMed] [Google Scholar]
- 52.Vaisman A., Masutani,C., Hanaoka,F. and Chaney,S.G. (2000) Efficient translesion replication past oxaliplatin and cisplatin GpG adducts by human DNA polymerase eta. Biochemistry, 39, 4575–4580. [DOI] [PubMed] [Google Scholar]
- 53.Strumberg D., Pilon,A.A., Smith,M., Hickey,R., Malkas,L. and Pommier,Y. (2000) Conversion of topoisomerase I cleavage complexes on the leading strand of ribosomal DNA into 5′-phosphorylated DNA double-strand breaks by replication runoff. Mol. Cell. Biol., 20, 3977–3987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Furuta T., Takemura,H., Liao,Z.Y., Aune,G.J., Redon,C., Sedelnikova,O.A., Pilch,D.R., Rogakou,E.P., Celeste,A., Chen,H.T. et al. (2003) Phosphorylation of histone H2AX and activation of Mre11, Rad50 and Nbs1 in response to replication-dependent DNA double-strand breaks induced by mammalian DNA topoisomerase I cleavage complexes. J. Biol. Chem., 278, 20303–20312. [DOI] [PubMed] [Google Scholar]
- 55.Samuelson A.V. and Lowe,S.W. (1997) Selective induction of p53 and chemosensitivity in RB-deficient cells by E1A mutants unable to bind the RB-related proteins. Proc. Natl Acad. Sci. USA, 94, 12094–12099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nahle Z., Polakoff,J., Davuluri,R.V., McCurrach,M.E., Jacobson,M.D., Narita,M., Zhang,M.Q., Lazebnik,Y., Bar-Sagi,D. and Lowe,S.W. (2002) Direct coupling of the cell cycle and cell death machinery by E2F. Nature Cell. Biol., 4, 859–864. [DOI] [PubMed] [Google Scholar]
- 57.Mohney B.G., Robertson,D.M., Schomberg,P.J. and Hodge,D.O. (1998) Second nonocular tumors in survivors of heritable retinoblastoma and prior radiation therapy. Am. J. Ophthalmol., 126, 269–277. [DOI] [PubMed] [Google Scholar]