

Abstract
Glioblastomas display variable phenotypes that include increased drug-resistance associated with enhanced migratory and anti-apoptotic characteristics. These shared characteristics contribute to failure of clinical treatment regimens. Identification of novel compounds that promote cell death and impair cellular motility is a logical strategy to develop more effective clinical protocols. We recently described the ability of the small molecule, KCC009, a tissue transglutaminase (TG2) inhibitor, to sensitize glioblastoma cells to chemotherapy. In the current study, we synthesized a series of related compounds that show variable ability to promote cell death and impair motility in glioblastomas, irrespective of their ability to inhibit TG2. Each compound has a 3-bromo-4,5-dihydroisoxazole component that presumably reacts with nucleophilic cysteine thiol residues in the active sites of proteins that have an affinity to the small molecule.
Our studies focused on the effects of the compound, ERW1227B. Treatment of glioblastoma cells with ERW1227B was associated with both down-regulation of the PI-3 kinase/Akt pathway, which enhanced cell death; as well as disruption of focal adhesive complexes and intracellular actin fibers, which impaired cellular mobility. Bioassays as well as time-lapse photography of glioblastoma cells treated with ERW1227B showed cell death and rapid loss of cellular motility. Mice studies with in vivo glioblastoma models demonstrated the ability of ERW1227B to sensitize tumor cells to cell death after treatment with either chemotherapy or radiation. The above findings identify ERW1227B as a potential novel therapeutic agent in the treatment of glioblastomas.
Keywords: Focal adhesive complexes, Glioblastomas, ERW1227B, Cell death, Drug-resistance, Cellular motility
Introduction
Prognosis for long-term survival in patients with glioblastomas remains poor, despite aggressive treatment with standard clinical strategies [1]. A major reason for failure to achieve effective clinical responses to treatment is attributed to resistance to radiation and chemotherapy protocols. Recent studies have identified multiple pro-survival molecular mechanisms that enhance resistance of glioblastoma cells to cytotoxic drug therapy. For example, changes in the activity of the DNA repair enzyme, O6-methylguanine-DNA methyltransferase (MGMT), in glioblastomas were correlated with methylation of the MGMT gene promoter. Decreased activity of MGMT was associated with an improved clinical response to temozolomide chemotherapy determined by increased survival in patients with glioblastomas [2]. Determination of MGMT activity in resected glioblastoma tissue allows one to better predict clinical response to temozolomide in individual patients. Unfortunately efforts to devise additional treatment strategies aimed at targets that decrease drug-resistance have been largely unsuccessful. Therefore it is important to identify additional novel compounds that inhibit drug-resistance in glioblastomas to allow development of new targeted clinical treatment strategies.
Recent studies have correlated the migratory phenotype and down-regulation of pro-apoptotic signals with resistance to cytotoxic drug therapy in glioma cells [3]. Tumor cells are associated with changes at the cell membrane important in regulating both cellular migration and resistance to apoptosis. Focal adhesive complexes are clusters of proteins organized at the cell membrane that facilitate the interaction of cells with the extracellular matrix (ECM). Focal adhesive complexes facilitate integrin-mediated cellular attachment by interacting with proteins in the ECM, e.g., fibronectin. The formation of focal adhesive complexes in cancer cells actively regulates “outside-in” pro-survival signals in malignant cells. Focal adhesive complexes are also important points of contact that promote the assembly and anchor intracellular cytoskeletal structures such as actin fibers that are important in cellular motility and invasion. Focal adhesive complexes at the cell membrane activate intracellular signaling pathways essential in promoting anti-apoptotic factors critical in drug-resistance. Direct downstream effectors of focal adhesive complexes include the phosphatidylinositol 3 kinase (PI-3 kinase)/Akt pathway, a key signaling cascade that has been linked with regulation of cell survival in glioblastomas. Increased activity of intratumoral phosphorylated Akt has also been associated with pro-survival signals and decreased response to treatment with radiation and chemotherapy in a number of systemic carcinomas including lung, breast, and prostate neoplasms. Inactivation of Akt and its downstream survival targets results in enhanced sensitivity to chemotherapy [4]. Up to 80% of glioblastomas express elevated levels of phosphorylated Akt [5]. Activation of the Akt pathway is related to progression of anaplastic astrocytomas to glioblastomas [6]. Elevated levels of phosphorylated Akt via the PI-3 kinase pathway have been specifically implicated in the pathogenesis of glioblastoma and resistance to chemotherapy [6,7]. Down regulation of the Akt pathway with an adenovirus containing MMAC/PTEN (Ad-MMAC) resulted in induction of anoikis in glioblastoma cells [8]. Inhibition of PI-3 kinase with LY204002 enhances cell death in glioblastoma cells with chemotherapy agents, particularly microtubule inhibitors [9].
In the course of our studies on the role of transglutaminase 2 (TG2) in glioblastoma growth and spreading [10], we synthesized a compound (ERW1227B) that lacked TG2 inhibitory activity but showed interesting activity in cell culture assays involving glioblastoma cells. We therefore investigated the activity of this compound, along with an analog ERW1095B, against glioblastoma cells in vitro. Our goal was to characterize the ability of these compounds to disrupt focal adhesive complexes, thereby perturbing the extracellular matrix and actin fibers, as well as to explore their effect on the activity of pro-survival proteins. Our data suggests that ERW1227B may be a useful agent in targeting focal adhesive complexes at the cell membrane and enhancing sensitivity to therapy in glioblastomas.
Material and Methods
Reagents and cell culture
N-N’-bis (2-chloroethyl)-N-nitrosourea (BCNU, carmustine), was purchased from Sigma (St. Louis, MO). KCC009 5-(N-benzloxycarbonyl-L-paratyrosinamidomethyl)-3-bromo-4′,5′-dihydroisoxazole was prepared as described earlier [11]. U87 cells were grown in cell culture as described previously [12]. After cells reached 70% confluence, groups were treated with vehicle (1% DMSO) or various small molecule agents.
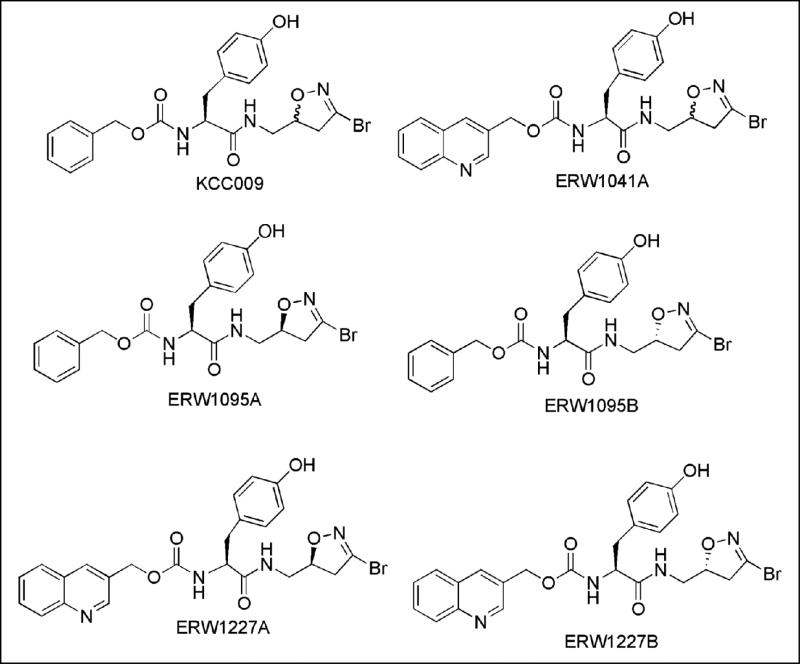
The structures of all compounds synthesized specifically for this study are shown in Table 1. Each compound has a 3-bromo-4,5-dihydroisoxazole electrophilic “warhead” that reacts with nucleophilic cysteine thiol residues in proteins that have an affinity to the small molecule. Their synthesis has been described earlier [11,13]. KCC009 and ERW1041A are diastereomeric mixtures that were prepared by coupling racemic 3-bromo-5-aminomethyl-4,5-dihydroisoxazole to the corresponding N-carbamoyl L-amino acid, and used in preliminary studies. ERW1095A, ERW1095B, ERW1227A and ERW1227B are enantiopure compounds; their preparation involved chiral resolution of the dihydroisoxazole prior to coupling with the N-protected amino acid moiety. Earlier studies [13] have established that ERW1095A and ERW1227A are inhibitors of human TG2, whereas ERW1095B and ERW1227B are not.
Table 1. Structures of synthetic small molecules used in this study.
KCC009 was prepared as a diastereomeric mixture of ERW1095A and ERW1095B, whereas ERW1041A is a diastereomeric mixture of ERW1227A and ERW1227B.
|
Crystal violet assay
U87 cells were plated at a density of 1×104 cells per well in 96-well microplate culture dishes. Cells were grown to approximately 50% confluence and assessed after treatment for 24 hours with 1% DMSO, 250 μM KCC009 and 250 μM ERW1041A. U87 cells were also assessed after treatment with 250 μM concentration of ERW1095A, ERW1095B, ERW1227A or ERW1227B for 24 hours. Each condition was examined in triplicate. Cells were fixed with 4% paraformaldehyde in PBS and stained with 1% crystal violet (Diagnostic Systems, Inc., NJ, USA) for 4 hours. The plates were washed with dH2O and allowed to air dry. The crystal violet was solubilized with 300 μl of 1% SDS per well. Absorbance was read at 570 nm with a microplate reader. Differences were assessed with a two-tailed Student's t-test for independent variables. Significance was determined with a p< 0.05.
Colorimetric (MTT) assay
Human glioblastoma cell lines U87, U118, U373, and murine glioblastoma cell line DBT, were grown to 80% confluence in 96 well plates and treated with vehicle-only, ERW1227B 125 μM, ERW1227B 250 μM and ERW1227B 500 μM. After 24 hours cells were evaluated in triplicate for viability with MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrasodium bromide) assay (Chemicon, CA). Results were determined with a Synergy HTTR plate-reader (BIO-TEK instrument Inc, VT) and KC4 software (BIO-TEK instrument Inc, VT) and differences were assessed with a two-tailed Student's t-test for independent variables.
Cell migration (scratch) assay
Human glioblastoma U87 cells were grown in 24 well- plates. The plates were scratched with a pipet tip separating the cells. Cells were maintained in a medium that contained 0.2% FBS . The cells were treated with vehicle-only, ERW1227B 125 μM, 250 μM for another 24 hours and assayed for migration of cells across the gap at two time points.
Time Lapse Video
U87 cell cultures were grown in Delta T dishes (0.17 mm) purchased from Fisher Scientific. Cell cultures were imaged for 24 hours at 30 minute intervals with an automated microscopy system [14]. Four microscopic fields were observed in each culture with the 20× (NA=0.4) objective of an inverted, wide-field differential interference contrast/epiflourescent microscope (Leica DMIRE2, Leica Microsystems, Wetzlar, Germany) as described in Kozel et al 2006 [15]. For each field and microscopy mode 3 images were taken in multiple focal planes, separated by 10 μm to avoid cells moving out of focus. The best-focused optical plane was selected from the acquired “z-stacks”. Image processing, including focal plane “collapsing,” field merging, and registering, was performed using a custom made program used in previous cell tracking studies [16]. The software automatically calculates most in focus block in X by X pixel blocks for each Z-plane and collapses them into a single image. Slight shifts in X position can also be corrected automatically. The photographs were processed with image analysis using the ImageJ (http://rsb.info.nih.gov/ij) functions and plug-ins.
In vitro immunofluorescent histology
Changes in focal adhesive complexes were evaluated with immunofluorescent histology. U87 cells were grown on cover slips. At various time points after treatment cells were fixed in 4% paraformaldehyde for 15 minutes, followed by 0.25% Triton for 5 minutes; and then placed in blocking solution (5% BSA, 1% donkey serum, and 1% horse serum) for one hour. Alternatively, cells stained for phosphorylated Akt (Cell Signalling, Beverly, MA, USA, 1:100) were permeabilized with cold methanol for 10 minutes on ice, instead of Triton for 5 minutes at room temperature. Primary antibodies were added to the same blocking solution in the following dilutions: mouse anti-human TG2 (1:50, NeoMarker Fremont, CA), mouse anti-human α5β1 integrin (1:100, Millipore, CA), mouse anti-human vinculin (1:200, Sigma, St. Louis, MO), Texas ReD-X Phalloidin (1:40, Molecular Probes, Eugene, Oregon). Primary antibodies were incubated at 4°C overnight. After washing, cells were incubated sequentially with anti-mice IgG conjugated with Cy2, anti-rabbit IgG conjugated with Cy3 (both from Chemicon), and Hoechst dye 33342 (Sigma, St. Louis, MO) for one hour. Slides were viewed with a fluorescent microscope (Nikon) and analyzed with Metamorph 6.2 image analysis software.
In vivo immunofluorescent histology
The incidence of apoptosis was determined with terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate biotin nick-end labeling (TUNEL) in tumors from the orthotopic DBT glioblastomas in mice. Specimens were fixed in 4% paraformaldehyde and sectioned in 5μm thick sections. The tissue was then stained with TUNEL. The assay labels nuclei that are undergoing DNA fragmentation characteristic of apoptosis. Pre-labeling and labeling were performed with a commercially available TUNEL kit, (In situ Death Detection Kit TMR Red, BD Biosciences Pharmingen, San Diego, CA, USA), in accordance with the manufacturer's instructions. Total nuclei were stained with Hoescht 33342 (Sigma, Saint Louis, MO, USA). Slides were viewed with a Nikon fluorescent microscope and photomicrographs were analyzed with Metamorph 6.2 image analysis software. Random images were assessed from twenty regions from each group, and the incidence of TUNEL positive cells was quantified from between 3000 and 4000 cells per specimen. Differences were assessed with a two-tailed Student's t-test for independent variables. Significance was determined with a p< 0.05.
Western blotting
Glioblastoma cells were grown in 100 mm dishes to approximately 70% confluence. Cells were washed with PBS and scraped in lysis buffer (50 mM Tris 150 mM NaCl, 1% NP-40, 0.25% Na-deoxycholate, 1 mM EDTA) with proteinase inhibitors (Roche Diagnostics, Germany). Protein levels were determined with the Bio-Rad Kit and equivalent amount of protein (15 μg per lane) was loaded on SDS-PAGE gels (Bio-Rad). Following electrophoresis, the proteins were transferred onto Immobilon-P membranes. The membranes were blocked with either 5% milk or 5% BSA in TBS with 0.05% Tween20; then blotted with primary antibody; followed by the HRP-labeled secondary antibody (Piscataway, NJ, USA). The reaction was developed with ECL Plus from Amersham (Piscataway, NJ, USA). Antibodies utilized for immunoblotting include rabbit anti-human phosphorylated Akt; rabbit total Akt; survivin; phosphorylated GSK-3β (Cell Signalling, Beverly, MA, USA); Bim (Stressgene Biotech, San Diego, CA); and tubulin antibody (Sigma, Saint Louis, MO, USA).
DBT glioblastoma orthotopic mouse models
In vivo research was performed in accordance with the Washington University Animal Studies Committee guidelines. Balb/C mice (20 grams), were purchased from Charles River Laboratories (Wilmington, MA, USA), and anesthetized with ketamine. Two in vivo glioblastoma mouse models were studied. The first was a subcutaneous tumor model. DBT glioblastoma cells, 1×106 in 50μl, were injected into the subcutaneous tissues of each flank. One week after tumor cell implantation, groups of mice (n=5, per group) were treated with intraperitoneal injections of vehicle-only; ERW1227B (25mg/kg); vehicle-only plus BCNU 5mg/kg; or ERW1227B (25mg/kg) plus BCNU (5mg/kg). The ERW1227B was given in 9 daily injections and BCNU was given 24 hours prior to sacrificing the mice. Tumors were removed and immediately frozen in −80°C for cutting, followed by TUNEL staining.
The second variation of the DBT model studied orthotopic intracranial glioblastoma tumors in mice treated with ERW1227B and radiation. Each animal subject was irradiated using a conformal small animal micro irradiator. The instrument consists of an Ir-192 brachytherapy source with a nominal source strength of 4.03 cGy m2/h used in a teletherapy configuration [17]. The irradiator operating parameters were tuned to deliver a dose of 2.5 Gy to the target tumor with a 5 mm diameter beam. Animal positioning was performed using a mouse bed with a stereotactic device specially designed to irradiate murine brains [18]. Verification of the animal positioning, dose delivery and beam location was performed with radiochromic films (Film Type EBT, International Specialty Products, Wayne, NJ). One week later groups of mice (n=5) were treated with intraperitoneal injections of vehicle-only; ERW1227B (50mg/kg); vehicle-only plus radiation 2.5 Gy alone; or ERW1227B (50mg/kg) plus radiation 2.5 Gy. The ERW1227B was given in 9 daily injections and radiation on day 3, 6, and day 9, twenty-four hours prior to sacrificing the mice. The tumors were collected and assessed for cell death with TUNEL as described above.
Results
Comparative efficacy of selected dihydroisoxazole inhibitors against cultured U87 glioblastoma cells
We prepared and characterized the potency of a number of analogs of KCC009 (chemical structure shown in Table 1), a small molecule dihydroisoxazole inhibitor of human tissue transglutaminase [13]. Motivated by the chemo-sensitizing activity of KCC009 against glioblastoma [10], we screened the apoptotic activity of a subset of these compounds against U87 glioblastoma cells. After 24 hours in culture, cells were treated with 250 μM of ERW1041A and KCC009, respectively. U87 cells treated with ERW1041A resulted in dramatic morphological changes characterized by rounding of the cell bodies, loss of processes, and detachment from the surface of the culture plate within 4 hours. In distinction to ERW1041A, cells treated with KCC009 showed only minimal morphological changes after 4 hours with a slight decrease in spreading on the surface of the culture plate. The cells maintained their typical morphology at 24 hours (Figure 1, A1-A3).
Figure 1.

A. Photomicrographs from U87 glioblastoma cells 24 hours after treatment with equivalent doses of KCC009 and ERW1041A. Cells were treated with (A1) vehicle-only; (A2) ERW1041A (250 μM); or (A3) KCC009 (250 μM). U87 cells treated with ERW1041A resulted in dramatic morphological changes characterized by rounding of the cell bodies, loss of neurites, and detachment from the surface of the culture plate. In contrast to ERW1041A, cells treated with an equal dose of KCC009 showed only minimal morphological changes after 24 hours with a slight decrease in spreading on the surface of the culture plate. B. Photomicrographs from U87 glioblastoma cells 4 hours after treatment with equivalent doses (250 μM) of enantiopure analogs of ERW1041A and KCC009. (B1) ERW1227B; (B2) ERW1227A; (B3) ERW1095B; or (B4) ERW1095A. U87 cells treated with ERW1227B resulted in dramatic morphological changes characterized by rounding of the cell bodies, loss of neurites, and detachment from the surface of the culture plate within 4 hours (B1). In contrast to ERW1227B, cells treated with ERW1227A maintained distinct cellular morphology, neuritic extensions, and attachment to the cell surface (B2). By 24 hours after treatment cells treated with the ERW1227B and the ERW1227A isomer demonstrated similar changes. Cells treated with ERW1095B showed rounding of the cell bodies and decreased neurites in contrast to those treated with ERW1095A that maintained normal cellular morphology (B3, B4).
(C). Comparison of U87 cell survival after treatment with between ERW1227B and ERW1095B. Twenty-four hours after treatment with either vehicle-only; ERW1227B 250 μM; or ERW1095B 250 μM; cells were assessed with the crystal violet assay for survival. Cells treated with ERW1227B showed a significantly decreased survival compared with either control or ERW1095B treated cells (P<0.05).
Two unexpected findings emerged from our preliminary screening efforts. First, a relatively poor correlation was observed between TG2 inhibitory potency and activity against glioblastoma cells. For example, analogs with >10-fold higher TG2 inhibitory activities than KCC009 were less active in U87 cell assays (data not shown). In contrast, ERW1041A (Table 1), an analog with less TG2 inhibitory activity comparable to that of KCC009, induced dramatic morphological changes in U87 cultures characterized by rounding of the cell bodies, loss of processes, and detachment from the surface of the plate within 4 hours. Similar morphological changes were only observed in U87 cells treated with KCC009 at higher concentrations (500 μM) (data not shown).
Second, the potency of KCC009 and ERW1041A was reflected in the activities of enantiopure analogs ERW1095B and ERW1227B (Table 1) respectively, whereas the TG2 inhibitors ERW1095A and ERW1227A were considerably less effective at causing cell detachment. For example, treatment of U87 cells with 250 μM ERW1227B for four hours induced rounding of the cell bodies and detachment from the culture surface and disruption of normal cell structures. In contrast, significantly less pronounced morphological changes were observed when cells were treated with 250 μM of the diastereomer ERW1227A up to 24 hours (Figure 1, B1-B2). It should be noted that at later time points cells treated with ERW1227A also showed progressive changes in morphology similar to those with ERW1227B (data not shown). There was also a similar, but less pronounced, effect on cell morphology with ERW1095B as compared to ERW1095A (Figure 1, B3-B4).
The higher potency of ERW1227B relative to ERW1095B after 24 hours of treatment with U87 cells was quantified in a crystal violet survival assay (Figure 1C). Consistent with the changes in morphology, there was a greater effect noted with ERW1227B compared with ERW1095B. There was significantly less survival in cells treated with ERW1227B (31%) vs ERW1095B (76%) (Student's t-test; p<0.05). Accordingly, subsequent experiments aimed at elucidating the biology of this new class of small molecule agents used ERW1227B as the benchmark compound.
ERW1227B results in the cell death in glioblastoma cell lines
The ability of ERW1227B to induce cell death in cultured glioblastoma cell lines was assessed in multiple glioblastoma cell lines. Cell viability was determined with a MTT assay after ERW1227B treatments at a concentration of 250 μM at 24 hours that resulted in cell death in U373 (43.3%), U118 (55.5%), U87 (54%) ,and DBT (67.7%) (Figure 2A).
Figure 2.

A. Survival curves of multiple cell lines to a dose-response ERW1227B in 24 hours. Cell death of U373, U118, U87 and DBT at 250 μM ERW1227B is 43.3%, 55.5%, 54% and 67.7% respectively. B. Scratching assay of U87 at 24 hours. U87 cells’ migration was nearly completely blocked by 250 μM ERW1227B. C. Time-lapse photography shows U87 cells treated with 250 μM ERW1227B results in loss of motility within 2 hours. The loss of motility correlated with disruption of actin fiber structure. (see time-lapse video in Supplemental Material).
ERW1227B impairs the motility of cells
To further investigate the ability of ERW1227B to impair the ability of glioblastoma cells to migrate, two additional experiments were performed. U87 cells were grown in culture to full confluence followed by treatment with either vehicle or with ERW1227B at doses of 125 μM, 250 μM. Figure 2B shows, the result of the scratch assay 24 hours after treatment. The cells treated with vehicle-only migrated and crossed the gap created by the scratch. The cells treated with ERW1227B 125 μM and 250 μM showed progressively impaired migration across the gap. Treatment with ERW227B also resulted in changes in morphology in the U87 cells (Figure 2B). The ability of ERW1227B to inhibit motility in U87 cells was dramatically demonstrated by time-lapse photography (Figure 2C, see videos in Supplemental Material). Images were processed as videos at thirty minute intervals for a period of 24 hours with phase-contrast photography. Untreated U87 cells were observed to maintain active motility throughout this time period. They were also noted to maintain normal morphology and to extend processes and move randomly in the cell dish. In contrast, the glioblastoma cells treated with ERW1227B showed typical changes in morphology with rounding of their cell bodies and progressive loss of motility identified within the first two hours.
Treatment of glioblastoma cells with ERW1227B results in disruption of focal adhesive complexes
Glioblastoma cells were evaluated with immunofluorescent histology after treatment with either vehicle-only or 250 μM ERW1227B. Four hours after treatment, the U87 cells were stained with antibodies to α5β1 integrin and phalloidin (which binds to actin) to assess the development of focal adhesive complexes. Control glioblastoma cells showed α5β1 integrins localized in small clusters on the cell membrane with anchored intracellular attachments of actin fibers. Identification of intracellular actin fibers that emanate from focal adhesive complexes via clusters of α5β1 integrin staining at the cell membrane confirms the nature of these structures (Figure 3A1-3A3). Within 4 hours, treatment of U87 cells with ERW1227B resulted in disruption of the normal cyto-architecture of focal adhesive complexes at the cell membrane. The majority of glioblastoma cells showed changes in staining of α5β1 integrin and actin characterized by enlarged dense clumps localized in the cell membrane with disruption of actin fiber formation (Figure 3B1-3B3). Alternatively, in a minority of cells, a visible loss of normal staining of small clusters of α5β1 integrins was associated with disrupted actin fiber formation at the cell membrane (data not shown).
Figure 3.

U87 cells grown in culture after treatment with vehicle-only (A) or with ERW1227B 250 μM (B). Cells were stained 4 hours after treatment for actin with phalloidin (A2, B2); with antibodies for α5β1 integrin (A1, B1); merged images (A3,B3). Control cells show staining of actin fibers co-localized with staining of clusters of α5β1 integrins along the cell membrane (A). ERW1227B treated cells show actin staining co-localized with dense clumps of α5β1 integrin and loss of staining of organized actin fibers (B). C. U87 cells grown in culture and treated with vehicle-only (C1) or 250 μM ERW1227B at 2 hours (C2) and 4 hours (C3). U87 cells treated with vehicle-only showed filamental actin after growing 24 hours on coverslips, while U87 cells treated with 250 μM ERW1227B demonstrated clustered actin and enlarged lamellipodium on the edge of cell membrane.
In addition, U87 cells were stained for filamentary actin (F-actin) in U87 cells after 2 hours and 4 hours following treatment with ERW1227B. In the vehicle-only group, F-actin filaments were anchored on the cell membranes that formed long strands crossing the cytoplasm within the cell bodies. U87 cell cultured overnight demonstrated a few filopodium on the cell surface (Figure 3C1). Within 2 hours after treatment with ERW1227B, F-actin filaments were dramatically disrupted and were no longer visible in the cytoplasm and the cell bodies had lost their processes. High density F-actin staining clusters were noted on the cell membrane (Figure 3C2). Four hours after treatment with ERW1227B, the majority of filamentary actin had disappeared with occasional foci of dense clumps of F-actin staining along the cell membrane (Figure 3C3). The findings were consistent with disruption of the focal adhesions on the cell membrane.
Dissociation of vinculin with focal adhesive complexes after treatment with ERW1227B
The ability of vinculin to interact with focal adhesive complexes along the inner surface of cell membranes was examined after treatment of U87 cells in culture. Within 30 minutes of plating, vinculin was expressed in the paranuclear cytoplasm and on occasion clustered along the cell membrane (Figure 4A). Two hours, and particularly four hours, after plating U87 cells, vinculin and actin extended to cell membranes in filopodia and lamellipodia. Both proteins are co-localized in focal adhesion complexes along the inner surface of cell membranes (Figure 4B-4C). Cells treated with ERW1227B for the same period, demonstrated dissociation of actin and vinculin (Figure 4D-4E). These findings are consistent with the observation that ERW1227B treatment results in disruption of focal adhesive complexes with loss of membrane associated F-actin cyto-architecture and decreased cellular motility.
Figure 4.

Vinculin growth in U87 cells with vehicle-only (A, B, C) and with 250 μM ERW1227B (D, E). Directly after plating U87 on coverslips for 30min (A), vinculin can be visualized at the cell edge and furthermore colocalized with F-actin at the cell surface at 2 hours (B). At four hours after plating, F-actin became filament and vinculin was located on the edge of cell surface and colocalized with F-actin (C). With 250 μM ERW1227B at 2 hours (D), vinculin was still on the cell surface but dislocated with F-actin. At 4 hours after treatment, most vinculin disappeared from the cell surface and transferred into the cytoplasm (E).
ERW1227B down regulates anti-apoptotic proteins and up regulates pro-apoptotic proteins
The formation of focal adhesive complexes has been associated with an increase in pro-survival signals mediated through the activation of the PI-3 kinase/Akt pathway. Increased levels of phosphorylated Akt have been correlated with malignancy and decreased sensitivity to chemotherapy in astrocytomas. U87 cells were grown in culture as described above. Groups of cells were treated with either vehicle-only or with 250 μM ERW1227B for various time points up to 24 hours. Cells were then stained with antibodies to α5β1 integrins to assess the development of focal adhesive complexes. Cells were co-stained with antibodies to phosphorylated Akt. There was co-localization of α5β1 integrin staining with phosphorylated Akt at the cell membrane (Figure 5A). Four hours after treatment with ERW1227B there was approximately 50% loss of phosphorylated Akt staining associated with focal adhesive complexes (Figure 5B). At later time points, both α5β1 integrin staining and phosphorylated Akt were lost at cell membrane (data not shown).
Figure 5.

ERW1227B resulted in anti-apoptosis protein decreased and pro- apoptosis protein increased in U87 cells. U87 cells grown in culture were treated with vehicle-only (A) or 250 μM ERW1227B (B) for four hours, and protein lysates following treatment with 250 μM ERW1227B were harvest over a 24 hour time course (C). Cells were stained with antibodies for α5β1 integrin (A1, B1); phosphorylated Akt (A2, B2); and merged images (A3, B3). Control cells showed co-localization of staining for α5β1 integrin and phosphorylated Akt at the cell membrane (A). Treatment with ERW1227B resulted in decreased staining of phosphorylated Akt at the cell membranes (B). Immunoblots (C) were done with antibodies against phosphorylated-Akt; total Akt; phosphorylated GSK-3β, survivin, and Bim; and tubulin (see text for discussion).
The effect of treatment of U87 cells with ERW1227B on the PI-3 kinase/Akt signaling pathway was also examined by Western blotting. Groups of U87 cells were treated with either vehicle-only or with 250 μM ERW1227B for various time points up to 24 hours. Cell lysates were collected at specified times and examined with Western blots. Treatment with ERW1227B resulted in decreased levels of phosphorylated Akt with no change in total Akt. Alterations in proteins downstream from phosphorylated Akt included decreased levels of the anti-apoptotic proteins, phosphorylated GSK-3β and survivin (Figure 5C). ERW1227B treatment also resulted in increased levels of the pro-apoptotic BH3-only protein, Bim (Figure 5C). The changes in levels of the above proteins reflect an increased sensitivity toward apoptosis.
The changes in motility and survival in the glioblastoma cells treated with ERW1227B corresponded with the changes noted in focal adhesive complex staining with loss of vinculin and F-actin association. Focal adhesive complexes are important in multiple cell functions including survival, migration and invasiveness. The ability of dihydroisoxazole-derived compounds such as ERW1227B to interfere with the formation and maintenance of focal adhesions at the cell membrane suggest their potential as a new class of therapeutic anti-neoplastic agents.
Ability of ERW1227B to sensitize glioblastomas to chemotherapy and radiation treatment
Four groups of mice were injected with DBT tumor cells via injection in the right and left flanks as previously described [12]. One week after the cells were implanted, the four groups of mice were treated with either vehicle, ERW1227B (25 mg/kg × 9 days), BCNU (5 mg/kg, injected one day prior to sacrifice), or a combination of ERW1227B (25 mg/kg × 9 days) and BCNU (5 mg/kg, injected one day prior to sacrifice). It should also be noted that this dosage of ERW1227B was well-tolerated in the experimental groups with no deaths or undesired effects. At the end of the study, the tumors were evaluated for the incidence of apoptosis determined with TUNEL staining in each of the groups. TUNEL positive cells were quantified, and the relative incidence of apoptotic cells in each group was calculated to be 3.5% (vehicle-only); 4.6% (ERW1227B-treated); 12.6% (BCNU-treated); and 17.5% (combination ERW1227B + BCNU-treated). The incidence of apoptosis was significantly increased in the combination ERW1227B + BCNU group compared with each of the other groups including control (p<0.002); KCC009-only (p<0.01); and BCNU-only (p<0.02) (Figure 6A). These findings demonstrate that ERW1227B promotes cell death in DBT glioblastoma cells treated with chemotherapy.
Figure 6.

(A) Incidence of apoptosis in DBT glioblastoma subcutaneous flank xenografts from mice treated with ERW1227B (25mg/kg) intraperitoneally daily for 9 days and with BCNU (5mg/kg) 24 hours prior to sacrifice. The incidence of apoptosis was determined with TUNEL staining. There was an increased incidence of apoptosis in the groups treated with ERW1227B either alone and ERW1227B combined with BCNU compared with vehicle-only (*). Differences were assessed with a two-tailed Student's t-test for independent variables. Significance was determined with a P< 0.05. (B) Incidence of apoptosis in an orthotopic intracranial glioblastoma model from mice treated with ERW1227B (50 mg/kg) intraperitoneally daily for 9 days and with radiation (2.5 Gy) at days 3, 6, and 9. The mice were sacrificed 24 hours after the last treatment. The incidence of apoptosis was determined with TUNEL staining. There was an increased incidence of apoptosis in the group treated with ERW1227B combined with radiation compared with each of the other groups (*). Differences were assessed with a two-tailed Student's t-test for independent variables. Significance was determined with a P< 0.05 in all comparisons.
In a second group of experiments, four groups of mice were treated with DBT tumor cells via injection in the right frontal lobe of the brain as previously described [10], One week after the cells were implanted, four groups of mice were treated with either vehicle, ERW1227B (50 mg/kg × 9 days), radiation (2.5 Gy at day 3, 6 and 9), or a combination of ERW1227B (50 mg/kg × 9 days) and radiation (2.5 Gy at day 3, 6 and 9). In each case the vehicle or ERW1227B was injected intraperitoneally four hours prior to the radiation to the whole brain. Twenty-four hours following the last treatment, the tumors were evaluated for the incidence of apoptosis determined with TUNEL staining in each of the groups. TUNEL positive cells were quantified, and the relative incidence of apoptotic cells in each group was calculated to be 1.2% (vehicle alone); 1.6% (ERW1227B alone); 4.5% (radiation alone); 8.9% (combination ERW1227B plus radiation). The incidence of apoptosis was significantly increased in the combination ERW1227B + radiation group compared with each of the other groups including control (p<0.002); ERW1227B (p<0.007); and radiation alone (p<0.03) (Figure 6B). These findings demonstrate that ERW1227B promotes cell death by sensitizing DBT glioblastoma cells to radiation treatment.
Discussion
Failure to achieve long-term survival in patients with glioblastomas has been attributed to increased resistance to apoptosis after treatment with radiation or chemotherapy. Glioblastomas have developed alterations in apoptotic regulatory proteins that increase their resistance to chemotherapy. In vitro studies have demonstrated that manipulation of pro- or anti- apoptotic proteins can regulate cell death in glioblastoma cells. Down-regulation of the anti-apoptotic proteins Bcl-2 and Bcl-xL with antisense oligonucleotides resulted in caspase-dependent cell death in glioblastoma cells in vitro [19]. Chemosensitive oligodendroglioma-derived cell lines treated with BCNU showed decreased expression of both Bcl-xL and Bcl-2 in association with cell death [20]. Up-regulation of the pro-apoptotic BH3-only protein, Bim, was associated with cell death in glioblastomas cells after treatment with lovastatin [21]. Conditions in vitro that favor increased levels of “pro-apoptotic” and decreased levels of “anti-apoptotic” proteins, are capable of promoting apoptosis in glioblastomas after chemotherapy treatment. ERW1227B treatment resulted in decreased levels of anti-apoptotic proteins, survivin and GSK-3β, while increasing levels of the proapoptotic protein, Bim. The finding of an increased incidence of apoptosis in glioblastoma cells in mice after systemic treatment with ERW1227B provides evidence that the in vitro observations are well correlated with in vivo studies. New agents that modulate these cell-survival signaling pathways are promising in the development of new therapies for the clinical treatment of glioblastomas.
Glioblastomas are infiltrative tumors characterized by cells that intermingle with normal CNS cells located outside the “core” tumor as demonstrated on as an enhancing lesion on MR imaging. The widespread dispersal of migratory glioblastoma cells contribute to failure of long-term control despite resection of the “core” tumor and standard radiation treatment to a 2 cm perimeter around the resection bed/residual tumor based on MR imaging. Chemotherapy is unsuccessful in effectively treating these migratory cells. Recent work has demonstrated that the migratory phenotype of glioma cells is associated with a shift in multiple regulatory apoptotic proteins that support resistance to cell death [3]. Migrating glioma cells have increased levels of phosphorylated Akt and downstream anti-apoptotic protein, phosphorylated GSK-3β. In vitro studies that compare migrating “peripheral” glioblastoma cells with migration-restricted “core” cells demonstrate elevated PI-3 kinase/Akt pathway activity [22]. The link between glioma cell migration and activation of the pro-survival PI-3 kinase/Akt pathway suggests that compounds that target both cell motility and the relative levels of pro- and anti- apoptotic protein levels may be effective in decreasing resistance to chemotherapy from “peripherally” located tumor cells. Our data show that ERW1227B was effective at inhibiting cell motility as demonstrated by time-lapse videos. The loss of motility correlates well with the finding that ERW1227B treatment resulted in disruption of actin fiber cyto-architecture. Down regulation of the PI-3 kinase pathway was assessed by measuring Akt activation. Levels of phosphorylated Akt were determined by Western blotting after treatment of glioblastoma cells in culture with ERW1227B. Immunohistochemical staining of phosphorylated Akt consistently demonstrated loss of co-localization of phosphorylated Akt with α5β1 integrins at the cell membrane after treatment with ERW1227B.
Integrin-mediated focal adhesive complexes provide a nexus for cell membrane/ECM membrane interaction and intracellular transmission of pro-survival signals. Tumor cells have developed specific mechanisms to promote survival that distinguish them from normal cells. They utilize multiple major existing pathways that cross-talk, increase resistance to cell death, and permit tumor cells to escape monotherapy directed at one pathway. Directing targets at major crossroads of survival pathways that are modulated in glioblastomas are more likely to be successful. One potential target present in tumor cells is focal adhesive complexes that coordinate extra-cellular and intra-cellular survival signals at the cell membrane. Focal adhesive complexes include integrins, adapter proteins, and tyrosine kinase receptors that initiate intracellular survival pathways [23]. Integrin signaling has been associated with regulation of cell death in cancer cells including αvβ3 integrin in melanoma cells [24, 25]; and α5β1 integrins in MCF-7 breast carcinoma cells. β1 integrin-mediated inhibition of drug-induced apoptosis in breast cancer cells was linked with activation of the PI-3 kinase/Akt pathway. Thus, targeting α5β1 integrin-mediated pathways at the cell membranes by interfering with function of focal adhesive complexes provides a logical target to enhance cell death in glioblastomas.
During tumorigenesis, neoplastic cells typically remodel their ECM distinct from surrounding normal cells. The remodeled stroma interacts with focal adhesive complexes on tumor cell membranes that provide survival-promoting advantages. Alterations in the ECM in malignant neoplasms generally occur by either proteolytic degradation of pre-existing proteins, or de novo synthesis of proteins unique to tumor cells. The latter category of proteins includes fibronectin, collagen, laminin, and tenascin [26]. Malignant tumor cells generally do not maintain normal cellular adhesion. Disruption of adhesion between normal cells results in a programmed cell death known as anoikis [27]. Malignant tumor cells, however, have developed mechanisms that allow them to escape anoikis. One such ECM protein, fibronectin, plays a key role in promoting tumor cell survival via interactions with cell surface integrins [28, 29]. Fibronectin binds to α5β1 integrin through a consensus Arg-Gly-Asp (RGD) binding motif within its Type III domain. The significance of this interaction in adhesion-mediated survival has been characterized using synthetic agents such as cyclic RGD peptides that competitively bind to the RGD-binding site and function to enhance cellular apoptosis [30, 31]. The ability of ERW1227B to disrupt focal adhesive complexes proves that it is a potential drug capable of inhibiting cellular motility and blocking the pro-survival signals downstream of the focal adhesive complexes.
The recruitment of α5β1 integrins and fibronectin promote the formation of focal adhesive complexes at the cell membrane. Clustering of α5β1 integrins promote the assembly of intracellular proteins at the cytoplasmic α5β1 integrin tail. The tail serves as a scaffold, which includes FAK, integrin-linked kinase, and activation of Src-family tyrosine kinases. The FAK-Src complex activates the pro-survival PI-3 kinase/Akt pathway that enhances resistance to chemotherapy in tumor cells [32]. Paxillan connects integrins to actin that activates RhoA and assembly of actin stress fibers. These structures are important in cell adhesion, spreading and migration, and survival. Impairment of focal adhesive complex formation is important in regulating anoikis via integrin-driven cell death signaling as well as cell-adhesion mediated drug resistance [33, 34].
Vinculin is an ubiquitously expressed actin-binding protein that is frequently used as a marker for both cell-cell and cell-extracellular matrix adhesion-type junctions. The actin-binding protein is localized on the cytoplasmic side of the integrin-mediated cell-extracellular junctions. It is a key protein and has an important function in cell adhesion, cell spreading, focal adhesive complex stability, cell migration and resistance to apoptosis [35]. A well-known function of vinculin supports the regulation of actin dynamics at the cell membrane. The ability of vinculin to interact with focal adhesive complexes along the inner surface of cell membranes allows it to promote actin polymerization at discrete sites on the plasma membrane and also at specific intracellular membrane compartments. Polymerization of actin is critical in the regulation of cellular motility.
The precise molecular target of ERW1227B remains unknown. Treatment of glioblastoma cells in vitro with ERW1227B was associated with impairment of cellular motility associated with loss of FN linearization in the ECM, focal adhesion formation at the cell membrane, dissociation of F-actin to vinculin, and disruption of intracellular actin fiber cyto-architecture. In addition ERW1227B treatment resulted in down-regulation of anti-apoptotic intracellular signals including phosphorylated Akt, survivin, and GSK-3β kinase, and increased levels of the pro-apoptotic protein, Bim. These changes are consistent with in vivo studies that identified increased incidence of apoptosis after treatment of tumor-harboring mice with ERW1227B plus either BCNU chemotherapy or radiation. These findings suggest the capability of ERW1227B as a novel agent targeting glioblastoma cells with disruption of key proteins important in migratory and anti-apoptotic phenotypes.
Supplementary Material
Acknowledgments
This work is supported by grants from ABC2 Foundation (K.M.R.), the Stanford Center for Children's Brain Tumors (C.K.), and support from Alvine Pharmaceuticals. K.M.R. and C.K. have a financial interest with Alvine Pharmaceuticals. We thank Ms. Bethany Kassebaum for her help with the radiation studies; and Mr. Cameron Ball for his assistance in analyzing the time-lapse imaging data. Time-lapse imaging (TiLa) code was created by the Computational Imaging Group at the University of Kansas Medical Center, under the direction of Drs. C. Little, B. Rongish, and A. Czirok. Dr. Czirok devised the original code for image acquisition and processing, which has been further developed and modified by Alan Petersen, Michael Filla, and Dr. Evan Zamir. A current version of this open source code is available from the Computational Imaging Group upon request (clittle@kumc.edu).
References
- 1.Castro MG, Cowen R, Williamson IK, et al. Current and future strategies for the treatment of malignant brain tumors. Pharmacol Ther. 2003;98:71–108. doi: 10.1016/s0163-7258(03)00014-7. [DOI] [PubMed] [Google Scholar]
- 2.Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 3.Mariani L, Beaudry C, McDonough WS, et al. Glioma cell motility is associated with reduced transcription of proapoptotic and proliferation genes: a cDNA microarray analysis. J Neurooncol. 2001;53:161–76. doi: 10.1023/a:1012253317934. [DOI] [PubMed] [Google Scholar]
- 4.McCormick F. Cancer: survival pathways meet their end. Nature. 2004;428:267–9. doi: 10.1038/428267a. [DOI] [PubMed] [Google Scholar]
- 5.Haas-Kogan D, Shalev N, Wong M, et al. Protein kinase B (PKB/Akt) activity is elevated in glioblastoma cells due to mutation of the tumor suppressor PTEN/MMAC. Curr Biol. 1998;8:1195–8. doi: 10.1016/s0960-9822(07)00493-9. [DOI] [PubMed] [Google Scholar]
- 6.Sonoda Y, Ozawa T, Aldape KD, et al. Akt pathway activation converts anaplastic astrocytoma to glioblastoma multiforme in a human astrocyte model of glioma. Cancer Res. 2001;61:6674–8. [PubMed] [Google Scholar]
- 7.Li B, Chang CM, Yuan M, et al. Resistance to small molecule inhibitors of epidermal growth factor receptor in malignant gliomas. Cancer Res. 2003;63:7443–50. [PubMed] [Google Scholar]
- 8.Davies MA, Lu Y, Sano T, et al. Adenoviral transgene expression of MMAC/PTEN in human glioma cells inhibits Akt activation and induces anoikis. Cancer Res. 1998;58:5285–90. [PubMed] [Google Scholar]
- 9.Shingu T, Yamada K, Hara N, et al. Synergistic augmentation of antimicrotubule agent-induced cytotoxicity by a phosphoinositide 3-kinase inhibitor in human malignant glioma cells. Cancer Res. 2003;63:4044–7. [PubMed] [Google Scholar]
- 10.Yuan L, Siegel M, Choi K, et al. Transglutaminase 2 inhibitor, KCC009, disrupts fibronectin assembly in the extracellular matrix and sensitizes orthotopic glioblastomas to chemotherapy. Oncogene. 2007;26:2563–73. doi: 10.1038/sj.onc.1210048. [DOI] [PubMed] [Google Scholar]
- 11.Choi K, Siegel M, Piper JL, et al. Chemistry and biology of dihydroisoxazole derivatives: selective inhibitors of human transglutaminase 2. Chem Biol. 2005;12:469–75. doi: 10.1016/j.chembiol.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 12.Yuan L, Choi K, Khosla C, et al. Tissue transglutaminase 2 inhibition promotes cell death and chemosensitivity in glioblastomas. Mol Cancer Ther. 2005;4:1293–302. doi: 10.1158/1535-7163.MCT-04-0328. [DOI] [PubMed] [Google Scholar]
- 13.Watts RE, Siegel M, Khosla C. Structure-activity relationship analysis of the selective inhibition of transglutaminase 2 by dihydroisoxazoles. J Med Chem. 2006;49:7493–501. doi: 10.1021/jm060839a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Czirók A, Rupp PA, Rongish BJ, et al. Multi-field 3D scanning light microscopy of early embryogenesis. J Microsc. 2002;206:209–17. doi: 10.1046/j.1365-2818.2002.01032.x. [DOI] [PubMed] [Google Scholar]
- 15.Kozel BA, Rongish BJ, Czirok A, et al. Elastic fiber formation: a dynamic view of extracellular matrix assembly using timer reporters. J Cell Physiol. 2006;207:87–96. doi: 10.1002/jcp.20546. [DOI] [PubMed] [Google Scholar]
- 16.Rupp PA, András Czirók A, Little CD. αvβ3 integrin-dependent endothelial cell dynamics in vivo. Development. 131:2887–97. doi: 10.1242/dev.01160. [DOI] [PubMed] [Google Scholar]
- 17.Stojadinovic S, Low DA, Hope AJ, et al. MicroRT-small animal conformal irradiator. Med Phys. 2007;34:4706–16. doi: 10.1118/1.2799887. [DOI] [PubMed] [Google Scholar]
- 18.Kiehl EL, Stojadinovic S, Malinowski KT, et al. Feasibility of small animal cranial irradiation with the microRT system. Med Phys. 2008;35:4735–43. doi: 10.1118/1.2977762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiang Z, Zheng X, Rich KM. Down-regulation of Bcl-2 and Bcl-xL expression with bispecific antisense treatment in glioblastoma cell lines induce cell death. J Neurochem. 2003;84:273–81. doi: 10.1046/j.1471-4159.2003.01522.x. [DOI] [PubMed] [Google Scholar]
- 20.Lytle RA, Jiang Z, Zheng X, et al. BCNU down-regulates anti-apoptotic proteins bcl-xL and Bcl-2 in association with cell death in oligodendroglioma-derived cells. J Neurooncol. 2004;68:233–41. doi: 10.1023/b:neon.0000033382.40601.5a. [DOI] [PubMed] [Google Scholar]
- 21.Jiang Z, Zheng X, Lytle RA, Higashikubo R, et al. Lovastatin-induced up-regulation of the BH3-only protein, Bim, and cell death in glioblastoma cells. J Neurochem. 2004;89:168–78. doi: 10.1111/j.1471-4159.2004.02319.x. [DOI] [PubMed] [Google Scholar]
- 22.Joy AM, Beaudry CE, Tran NL, et al. Migrating glioma cells activate the PI3-K pathway and display decreased susceptibility to apoptosis. J Cell Sci. 2003;1:4409–17. doi: 10.1242/jcs.00712. [DOI] [PubMed] [Google Scholar]
- 23.Burridge K, Fath K, Kelly T, et al. Nuckolls G, Turner C. Focal adhesions: transmembrane junctions between the extracellular matrix and the cytoskeleton. Annu Rev Cell Biol. 1988;4:487–525. doi: 10.1146/annurev.cb.04.110188.002415. [DOI] [PubMed] [Google Scholar]
- 24.Montgomery AM, Reisfeld RA, Cheresh DA. Integrin alpha v beta 3 rescues melanoma cells from apoptosis in three-dimensional dermal collagen. Proc Natl Acad Sci U S A. 91:8856–60. doi: 10.1073/pnas.91.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Petitclerc E, Strömblad S, von Schalscha TL, et al. Integrin alpha(v)beta3 promotes M21 melanoma growth in human skin by regulating tumor cell survival. Cancer Res. 1999;59:2724–30. [PubMed] [Google Scholar]
- 26.Pupa SM, Ménard S, Forti S, et al. New insights into the role of extracellular matrix during tumor onset and progression. Cell Physiol. 2002;192:259–67. doi: 10.1002/jcp.10142. [DOI] [PubMed] [Google Scholar]
- 27.Frisch SM, Francis H. Disruption of epithelial cell-matrix interactions induces apoptosis. J Cell Biol. 1994;124:619–26. doi: 10.1083/jcb.124.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Danen EH, Yamada KM. Fibronectin, integrins, and growth control. J Cell Physiol. 2001;189:1–13. doi: 10.1002/jcp.1137. [DOI] [PubMed] [Google Scholar]
- 29.Wierzbicka-Patynowski I, Schwarzbauer JE. The ins and outs of fibronectin matrix assembly. J Cell Sci. 2003;15:3269–76. doi: 10.1242/jcs.00670. [DOI] [PubMed] [Google Scholar]
- 30.Taga T, Suzuki A, Gonzalez-Gomez I, et al. Alpha v-integrin antagonist EMD 121974 induces apoptosis in brain tumor cells growing on vitronectin and tenascin. Int J Cancer. 2002;98:690–697. doi: 10.1002/ijc.10265. [DOI] [PubMed] [Google Scholar]
- 31.Buckley CD, Pilling D, Henriquez NV, et al. RGD peptides induce apoptosis by direct caspase-3 activation. Nature. 1999;397:534–9. doi: 10.1038/17409. [DOI] [PubMed] [Google Scholar]
- 32.Deschesnes RG, Patenaude A, Rousseau JL, et al. Microtubuledestabilizing agents induce focal adhesion structure disorganization and anoikis in cancer cells. J Pharmacol Exp Ther. 2007;320:853–64. doi: 10.1124/jpet.106.110957. [DOI] [PubMed] [Google Scholar]
- 33.Vachon PH, Harnois C, Grenier A, et al. Differentiation state-selective roles of p38 isoforms in human intestinal epithelial cell anoikis. Gastroenterology. 2002;123:1980–91. doi: 10.1053/gast.2002.37072. [DOI] [PubMed] [Google Scholar]
- 34.Harnois C, Demers MJ, Bouchard V, et al. Human intestinal epithelial crypt cell survival and death: Complex modulations of Bcl-2 homologs by Fak, PI3-K/Akt-1, MEK/Erk, and p38 signaling pathways. J Cell Physiol. 2004;198:209–22. doi: 10.1002/jcp.10399. [DOI] [PubMed] [Google Scholar]
- 35.Ziegler WH, Liddington RC, Critchley DR. The structure and regulation of vinculin. Trends in Cell Biol. 2006;16:453–460. doi: 10.1016/j.tcb.2006.07.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.