


Abstract
Family D DNA polymerase (PolD) is a new type of DNA polymerase possessing polymerization and 3′–5′ exonuclease activities. Here we report the characterization of the nuclease activity of PolD from Pyrococcus horikoshii. By site-directed mutagenesis, we verified that the putative Mre11-like nuclease domain in the small subunit (DP1), predicted according to computer analysis and structure inference reported previously, is the catalytic domain. We show that D363, H365 and H454 are the essential residues, while D407, N453, H500, H563 and H565 are critical residues for the activity. We provide experimental evidence demonstrating that manganese, rather than magnesium, is the preferable metal ion for the nuclease activity of PolD. We also show that DP1 alone is insufficient to perform full catalysis, which additionally requires the formation of the PolD complex and manganese ion. We found that a 21 amino acid, subunit-interacting peptide of the sequence from cysteine cluster II of the large subunit (DP2) stimulates the exonuclease activity of DP1 and the internal deletion mutants of PolD lacking the 21-aa sequence. This indicates that the putative zinc finger motif of the cysteine cluster II is deeply involved in the nucleolytic catalysis.
INTRODUCTION
A number of DNA polymerases possess 3′–5′ exonuclease activity in addition to their DNA polymerization activity (1–4). The 3′–5′ exonuclease activity potentially enhances the DNA synthesis fidelity, termed exonucleolytic proofreading (1). The exonuclease activity is performed either by an exonuclease domain covalently connected with the DNA polymerase domain, such as in DNA polymerase I of Escherichia coli (5), yeast mitochondrial DNA polymerase (6) and numerous family B DNA polymerases including eukaryotic DNA polymerases delta and epsilon (1,7–10); or by an associated subunit, such as in E.coli and Salmonella typhimurium DNA polymerase III holoenzymes (2,11). The analysis of the amino acid sequences suggested that most of the polymerase-related 3′–5′ exonucleases share conserved motifs, Exo I, Exo II and Exo III, with carboxyl residues composing the catalytic center (12,13). Structures of a number of DNA polymerases possessing an exonuclease domain or associated exonuclease subunit have been solved (14–21), showing similar exonuclease topology among family A, B and C DNA polymerases. A two-metal-ion mechanism has been proposed to explain the exonucleolytic process (21–24).
Family D DNA polymerase (PolD) is a new DNA polymerase extensively existing in the Euryarchaeota subdomain of Archaea (25). The enzyme has strong 3′–5′ exonuclease activity in addition to its strong and processive DNA polymerization activity (26–29). Based on the nature of the enzymatic activities, the gene organization in Pyrococcus furiosus, P.horikoshii and P.abyssi, and the ability to interact with multiple proteins related to DNA replication, recombination and repair (30,31), PolD was suggested to be the replicase in Euryarchaeota. By site-directed mutagenesis, the C-terminal of the large subunit (DP2) has been identified as the catalytic domain for DNA polymerization (28). Through analysis of various recombinant terminal truncated proteins of the PolD complex at the small subunit (DP1) or DP2, the regions putatively involved in the subunit interaction (1255–1332 of DP2, 201–260 and 599–622 of DP1 in P.horikoshii), oligomerization (1–300 of DP2), and regulation of the exonuclease activity (1–200 of DP1) have been found (32). However, many aspects of the properties of PolD remain to be uncovered, for example, the catalytic domain and catalytic residues for the 3′–5′ exonuclease activity, as investigated in the present study, have not been identified.
Although PolD does not contain common motifs of the other DNA polymerase families for the catalytic activities (25), genome sequence and protein structure analysis indicates that the C-terminal half of DP1 is potentially the catalytic domain for the 3′–5′ exonuclease of PolD (33–37). A comprehensive computer analysis of genome sequences revealed that DP1 shares conserved motifs of a phosphoesterase super-family including archaeal DP1s, the second subunits of eukaryote DNA polymerases alpha, delta and epsilon, Mre11 nuclease subfamily, kidney bean purple acid phosphatase and human calcineurin (33). The motifs usually contain several putative metal chelating residues, predicted according to the three-dimensional structures of kidney bean purple acid phosphatase (34), human calcineurin (35,36) and the catalytic domain of Mre11Pfu, a nuclease from P.furiosus (37). In the structure of Mre11Pfu, two manganese ions were shown to be octahedrally coordinated by seven conserved residues of the phosphoesterase motifs and by a bridging water molecule (37). Two additional residues within these motifs were also shown to putatively help the products leaving in the catalysis. It appears that the C-terminal domain of DP1 resembles the nuclease domain of Mre11, however, there has been no experimental evidence to confirm that the conserved residues are indeed involved in the catalysis in Mre11Pfu and PolD. The roles of these residues have not been defined.
The alignment of DP1 with the Mre11 nuclease subfamily is intriguing. In eukaryotes, Mre11, Rad50 and Nbs1 form a complex with multiple functions including dsDNA break repair, generation and processing of meiotic dsDNA breaks, DNA replication and maintenance of the telomere length (38–40). Mre11 has manganese-dependent 3′–5′ exonuclease and ssDNA endonuclease activities, Rad50 has an ATPase activity, while the Mre11–Rad50 complex has a helicase activity. The complex is evolutionarily conserved across bacteria (42), archaea (43,44), bacteriophage T4 (41), as well as eukaryotes (38–41). The structural and catalytic similarities between DP1 and Mre11 nuclease, if proved, may suggest an evolutionary conserved link between DNA replication, recombination and repair in Archaea and Eukaryota.
In the present study, we report the construction and analysis of various alanine mutants of PolD from P.horikoshii (PolDPho), at residues in the C-terminal of DP1, presumably involved in the nuclease catalysis. We verified experimentally that PolDPho uses a Mre11-like nuclease domain for the exonuclease activity. We further show that three residues, D363, H365 and H454 in DP1 from P.horikoshii (DP1Pho) are essential residues, while the other five predicted residues D407, N453, H500, H563 and H565 also play important roles. We demonstrated that DP1Pho alone has weak exonuclease activity which could be stimulated by a short peptide from the C-terminal of DP2 from P.horikoshii (DP2Pho) in the presence of manganese. We found that PolDPho prefers manganese for the 3′–5′ nuclease activity and possesses manganese-dependent ssDNA endonuclease activity, whose catalytic center is probably the same as that of the exonuclease.
MATERIALS AND METHODS
Chemicals and bacterial strains
The pET15b vector and ultra competent E.coli XL2-Blue MRF’ cells were purchased from Stratagene (La Jolla, CA). The pGEMEX-1 vector was purchased from Promega (Madison, WI). Escherichia coli strain BL21-CondonPlus (DE3)-RIL-competent cells were obtained from Stratagene (La Jolla, CA). Deep Vent DNA polymerase was from New England Biolabs (Beverly, MA). Restriction enzymes were purchased from Takara Shuzo (Otsu, Shiga, Japan) and Promega (Madison, WI). The DNA ligation kit was from Takara Shuzo (Otsu, Shiga, Japan), and was used according to the manufacturer’s recommendation. Protease inhibitor mixture tablets (EDTA-free) were purchased from Roche Molecular Biochemicals (Mannheim, Germany). The FAM-labeled oligonucleotide was provided by Sawady–Qiagen (Tokyo, Japan). The synthetic polypeptides were provided by Hokkaido System Science Co. (Sapporo, Japan).
Construction of vectors for the expression of site-directed PolDPho mutants, DP1Pho, DP2Pho and the internal deletion mutants SL(DEL1289–1398) and SL(DEL1299–1308)
Site-directed alanine mutants at residues in the DP1Pho of PolDPho complex were constructed using overlap PCR methods (45) and the co-expression vector pET15b/SL (28,32). The upper fragment was amplified using the vector-derived primer pET-SphI (5′-CAAGGAATGGTGCATGC AAGGAGATGGC-3′) (32) and a site-specific downstream primer (Table 1). The lower fragment was amplified using a site-specific upstream primer (Table 1) and the primer pGEM-rbs (28,32). After amplification by PCR, the full-length fragment was digested with SphI and SacII and cloned back into the pET15b/SL vector digested with SphI and SacII (32). To construct a vector for DP2Pho, the co-expression vector pET15b/SL was used. The in-frame NdeI site at the C-terminal of DP2Pho was silenced by site-directed mutagenesis (28). The modified plasmid was digested with NdeI and self-ligated, giving rise to a vector pET15b/L containing the DP2Pho gene only. DNA sequencing was performed to confirm that no spurious mutations had been introduced during PCR. The construction of expression vectors for DP1Pho (pET15b/S) and internal deletion proteins, SL(DEL1289–1298) and SL(DEL1299–1308) [pET15b/SL(DEL1289–1298) and pET15b/SL(DEL1299–1308)] respectively, has been described elsewhere (28,32).
Table 1. List of the site-directed mutants and the site-specific oligonucleotides used in this study.
| Mutanta | Sequenceb |
|---|---|
| D363A | 5′-CGCCGTTTTGACGAGTGCTATCCACGTTGGAAGCAAGG-3′ |
| H365A | 5′-CGCCGTTTTGACGAGTGATATCGCCGTTGGAAGCAAGG-3′ |
| D407A | 5′-GGTATTTAATTATAGCAGGGGCTGTTGTCGATGGCATCGGAATTTATCCTGGCC-3′ |
| D410A | 5′-GGTATTTAATTATAGCAGGGGATGTTGTCGCTGGCATCGGAATTTATCCTGGCC-3′ |
| D421A | 5′-CCTGGCCAGTATTCTGCCCTAATAATTCCCGATATC-3′ |
| N453A | 5′-GATCTTCATAGGCCCCGGTGCTCATGATGCTGCAAGGC-3′ |
| N454A | 5′-CTTCATAGGCCCCGGTAATGCTGATGCTGCAAGGCCC-3′ |
| N479A | 5′-GCCCCTGTATAAGTTGAAGGCTACTGTGATAATCAGCAACCC-3′ |
| H492A | 5′-CCAGCGGTCATAAGGCTTGCTGGTAGGGATTTTTTGATAG-3′ |
| H500A | 5′-GGGATTTTTTGATAGCCGCTGGAAGGGGAATAGAGGATG-3′ |
| E553A | 5′-GAGGATCTATTGGTAATAGCTGAAGTTCCGGATCTAGTTC-3′ |
| D557A | 5′-GTAATAGAGGAAGTTCCGGCTCTAGTTCAGATGGGACATG-3′ |
| H563A | 5′-GATCTAGTTCAGATGGGAGCTGTGCATGTTTACGATACTGCGG-3′ |
| H565A | 5′-GATCTAGTTCAGATGGGACATGTGGCTGTTTACGATACTGCGG-3′ |
aThe mutants in bold indicate residues putatively involved in metal binding; the mutants in italics indicate residues probably involved in catalytic assistance and the rest of the mutations are residues conserved among DP1 species.
bOnly forward sequences are shown. Codes underlined indicate the sites of mutations.
Expression and purification of the enzymes
The co-expression vectors for the protein complexes of the wild-type PolDPho, the internal deletion mutants and site-directed PolDPho mutants (mutations in DP1Pho of the complexes), and vectors for single subunit DP1Pho and DP2Pho, were transformed into host E.coli BL21-CodonPlus (DE3)-RIL. The transformed cells were grown in 2× YT medium containing ampicillin (100 mg/l) at 37°C. When the A600 reached 0.6–1.0, isopropylthio-β-d-galactoside (2 mM) was added to induce the expression of the genes. After being cultured for 4 h at 37°C, the cells were harvested by centrifugation, re-suspended in 50 mM Tris–HCl pH 8.0 containing the protease inhibitor, and disrupted on ice by sonication. The disrupted cells were heated at 85°C for 30 min and centrifuged at 27 000 g for 20 min to remove the cell debris and denatured proteins. The expression of DP1Pho and DP2Pho was checked by SDS–PAGE performed on a 10–15% gradient gel using the Phast system (Amersham Biosciences). The protein bands were visualized by staining with Coomassie Brilliant Blue R-250. The supernatant was dialyzed against buffer A (50 mM Tris–HCl pH 8.0) then buffer B (50 mM Tris–HCl pH 7.0). The dialysate was loaded onto a Hitrap Q column (Amersham Biosciences) which was pre-equilibrated using the FPLC system (Amersham Biosciences). The column was developed with a linear gradient of 0–1000 mM NaCl. Fractions containing the PolDPho were dialysed against buffer A to remove the salt. For further purification, the samples were loaded onto a nickel column (Novagen). After a wash with a buffer containing 20 mM imidazole, the enzymes were eluted with the elution buffer containing 150 mM imidazole. For purification by gel filtration, the samples were dialysed with 50 mM Tris–HCl buffer pH 8.0, 200 mM NaCl, concentrated using a microfilter (Microcon YM-10, Millipore Corp, Beddord, MA), and loaded onto a Hiload Superdex-200 (16/60) column (Amersham Biosciences) equilibrated with 50 mM Tris–HCl buffer, 200 mM NaCl pH 8.0. The fraction size was 0.5 ml and the flow rate was 0.5 ml/min. The peak fractions were collected and concentrated. The protein content was determined with the protein assay dye reagent (Bio-Rad Lab., CA) using bovine serum albumin as the standard protein.
Preparation of the substrates and the assay of primer extension activity
The primer extension ability was assayed using a substrate of 84mer oligomer whose sequences were taken from M13mp18 ssDNA (5′-TCCTCTAGAGTCGACCTGCAG GCATGCAAGCTTGGCACTGGCCGTCCTTTTACAACG TCGTGACTGGGAAAACCCTGGCGTTAC-3′, from positions 6254 to 6337) annealed with a 25 mer (5′-GTAACG CCAGGGTTTTCCCAGTCAC-3′, complementary to positions 6337 to 6313 of M13mp18) labeled with FAM at the 5′ end. To make the 84/25mer and 25/25mer substrates, the labeled primers (500 nM) were mixed with the 84mer or 25mer (5′-GTGACTGGGAAAACCCTGGCGTTAC-3′) oligonucleotide (1.0 µM) in 20 µl of buffer (7 mM Tris–HCl pH 8.0, 50 mM NaCl and 7 mM MgCl2). The tube was boiled for 5 min and then cooled gradually to room temperature. The standard polymerization reaction mixture (10 µl) contained 20 mM Tris–HCl (pH 8.8, 25°C), 8 mM MgSO4 (or MnSO4), 10 mM KCl, 10 mM (NH4)2SO4, 0.1% Triton-X 100, 0.25 mM dNTP, 50 or 200 nM labeled substrate as specified and 23 nM enzymes. The reaction was performed at 60°C for 2 min or as specified. After the reaction, 10 µl of stop buffer containing 95% formamide, 10 mM EDTA and 1.0 mg/ml of bromophenol blue was added to the mixture and the tube was heated in boiling water for 5 min. The samples were loaded onto a 15% polyacrylamide gel containing 7 M urea and 1× TBE buffer (89 mM Tris–HCl, 89 mM boric acid and 2 mM EDTA pH 8.0) and electrophoresed for 1.5 h. Reaction products were visualized using a FluorImager 585 (Amersham Biosciences).
Assay of 3′–5′ exonuclease activity
The 25mer ssDNA labeled at the 5′ end with FAM, 25/25mer dsDNA and the 84/25mer substrate were used for the 3′–5′ exonuclease assay. The reaction mixture and procedures used to assay the 3′–5′ exonuclease activity of the wild type and the mutants were the same as for the primer extension assay, except that dNTP was not added, and the enzyme amount was 92 nM. The reaction was performed at 60°C for 10 min or as specified. After electrophoresis, the products were visualized and quantified using a FluorImager 585 (Amersham Biosciences).
Assay of the endonuclease activity
Purified enzyme of PolDPho (58 nM) was mixed with 0.5 µg of circular M13mp18 ssDNA or 1.0 µg of circular pUC18 dsDNA in 20 mM Tris–HCl pH 8.8, 8 mM MnSO4 (or MgSO4), 10 mM KCl, 10 mM (NH4)2SO4 and 0.1% Triton-X 100. The mixture (20 µl) was incubated at 60°C for 60 min or as specified. The reaction was stopped by adding a 1/10 volume of 10× stop buffer containing 10 mM EDTA and 3% SDS. After purification using the QIAquick mini column (Qiagen), the reaction products were separated on a 0.7% agarose gel containing ethidium bromide and visualized under UV light.
RESULTS
Identification of residues critical for the 3′–5′ exonuclease activity
In order to map the catalytic residues for the enzymatic activities, we previously constructed, co-expressed, and analyzed various alanine mutants of PolD complexes with mutations in DP1Pho (E291A/ D292A, D506A and D548A) and DP2Pho [E43A/D49A, E55A, E69A/E77A, E103A, E115A/E123A, D166A, E189A/E190A, E209A, E224A/E228A/D235A, E254A, E333A, E403A, D445A, E461A, D500A, E660A, E1201A/E1204A/D1205A, D1221A/D1223A/D1224A, D1239A/E1241A/E1242A, E1349A/D1351A/D1353A, E1357A and D1373A, as well as those reported (28)]. We were able to obtain purified proteins after heat treatment and using a nickel column. We identified Asp1122 and Asp1124 to be the catalytic residues for DNA polymerization (28). However, none of the mutant proteins led to complete loss of the 3′–5′ activity (data not shown). The results indicated that the residues in DP1Pho and DP2Pho are not directly involved in the catalysis of the 3′–5′ nuclease activity.
According to computer analysis and structural prediction, the C-terminal of DP1 contains five Mre11-like nuclease motifs presumably important for catalysis (33,37). Within the motifs, seven histidine, aspartic acid and asparagine residues are putatively involved in metal binding, and the remaining two in catalytic assistance (33,37). The partial amino sequence alignment of DP1s from four Euryarchaetota species is shown in Figure 1A, and the structure of Mre11 from P.furiosus at the catalytic site is illustrated in Figure 1B. In DP1Pho, the seven possible metal binding residues (shown in red) are D363, H365, D407, N453, H500, H563 and H565, and two putative assistance residues (shown in green) are D410 and H454 respectively (Fig. 1A and B).
Figure 1.


Site-directed mutation sites at PD1Pho in the PolD complex. (A) Partial sequence alignment of the DP1s from Euryarchaeota species. The DP1 sequences are from four Euryarchaeota species, P.horikoshii (P.hori, accession number, AP000001–127), Methanococcus jannaschii (M.jann, U67516–16), Archaeoglobus fuljidus (A.fulj, AE000979–13) and Halobacterium sp. NRC-1 (Hal.sp, AE005122–10). The alignment was carried out using ClustalW software. The motifs (I–V) conserved among the phosphoesterase super-family are boxed. The corresponding motifs from Mre11 of P.furiosus (Mre11Pfu) are shown above the boxes. The residues colored in red are putatively involved in metal binding and those in green are putative catalytic assistance residues according to the structure of Mre11 from P.furiosus. The arrows indicate residues chosen for the site-directed mutagenesis in this study. (B) Stereoview showing the catalytic site of Mre11Pfu. Structural data (1II7, ref. 37) were retrieved from the Protein Data Bank and the figure was made with Molscript (51) and Raster3D (52). The residues involved in metal binding are shown in red and those putatively involved in catalytic assistance in green. The bound dAMP is shown in yellow. The residues of Mre11Pfu are labeled together with their corresponding residues of DP1Pho (in brackets). Two manganese ions and a water molecule are shown in blue and cyan respectively.
To confirm if these motifs are functional in PolD or not, these residues together with five other residues (D421, N479, H492, E553 and D557) conserved in DP1s from known Euryarchaeota species (28,32) were also chosen for the site-directed mutagenesis (Fig. 1A, Table 1). The mutant DP1 and wild-type DP2 were co-expressed using the co-expression vectors as for the wild-type PolD and other mutants (Fig. 1, Table 1) (28,32). All the mutant protein complexes were expressed and purified as the wild type with an anion exchange column, a nickel column and a gel filtration column after heat-treatment at 85°C for 30 min (see Materials and Methods). The SDS–PAGE analysis of representative mutant proteins is shown in Figure 2. For all the mutants, bands corresponding to the large subunit (DP2) and small subunit (DP1) were present, indicating that the protein complexes were formed after the purification steps, although there was variation in relative band intensity between DP2 and DP1. While the band intensity of D410A, N453A, H454A, N479A, H492A, D557A and H565A was similar to the wild type, there seemed to be more DP1 in D363A, H365A and D407A, but less full DP1 in H500A and H563A than the wild type, and putative degraded bands were observed in H500A and H563 (Fig. 2A). Presumably, this difference in band patterns was due to variation in the degradation of the DP1 subunit during purification.
Figure 2.

SDS–PAGE analysis of the representative mutant proteins of the PolDPho complex (A), the small subunit DP1Pho expressed alone (B) and the large subunit DP2Pho (C). The gels (10–15%) were stained with Coomassie Brilliant Blue R-250 after electrophoresis. The protein samples were purified using Hitrap Q, nickel affinity and Superdex-200 columns as described in the Materials and Methods. The band positions of the full-length of the small subunit (DP1Pho) and the large subunit (DP2Pho) are indicated with arrows. Due to the instability of DP2Pho expressed alone, the upper minor band in panel C corresponds to the full-length polypeptide.
The mutants were firstly subjected to 3′–5′ exonuclease and DNA polymerization analysis using 84/25mer as the substrate and magnesium as the cofactor. All the fourteen mutants showed strong DNA polymerization activity as did the wild-type protein (Fig. 3A and data not shown). However, we did not detect 3′–5′ exonuclease activity for the mutants D363A, H365A, N453A, H454A, H500A and H563A (Fig. 3B). The mutants D407A and H565A showed activity reduced to ∼12 and 25% of that of the wild type (Fig. 3B and C). D410A (Fig. 3B and C) and the five remaining mutants, D421A, N479A, H492A, E553A and D557A showed nearly the same activity as the wild-type PolDPho (data not shown). It is interesting that all the mutants (except D410A) at residues predicted to participate in the exonuclease catalysis (Fig. 1B, Table 1) showed a loss (D363A, H365A, N453A, H454A, H500A and H563A) or reduced activity (D407A and H565A), but none of the mutants at residues conserved only in DP1s (D421A, N479A, H492A, E553A and D557A) were affected in the exonuclease activity. This result strongly indicates that the C-terminal Mre11-like domain of DP1Pho is probably directly involved in the 3′–5′ exonuclease activity of PolDPho.
Figure 3.

Comparison of DNA polymerization (A) and the 3′–5′ exonuclease (B) activities of the wild type and mutants assayed using magnesium as the cofactor. The substrate used was a 84/25mer recess-ended dsDNA. The reaction time for the DNA polymerization and exonuclease activity was 2 and 30 min, respectively. (C) Quantification of the exonuclease activities of the wild-type PolDPho and the mutants using the 84/25mer substrate as shown in panel B. Each value was calculated based on the results of at least three independent reactions.
D363, H365 and H454 are the essential residues revealed by comparison of the 3′–5′ exonuclease activities of the mutants using manganese as the cofactor
To determine the roles of the critical residues, the 3′–5′ exonuclease activities of the mutants using manganese were compared. The use of manganese is due to the fact that Mre11 nuclease utilizes manganese as cofactor (37–43). As shown in Figure 4, the exonuclease activities of the wild type (Fig. 4A and B) as well as the mutants D407A, D410A and H565A (Fig. 4A) were enhanced, compared with those when magnesium was used (Fig. 3B and C). Interestingly, activity was detected in the mutants N453A, H500A and H563A (Fig. 4A). However, the mutants D363A, H365A and H454A still showed no degradation of the DNA (Fig. 4A). A similar result was obtained when a 25mer ssDNA was used (data not shown). It seems that the variation of the exonuclease activity of the mutants, particularly in the presence of manganese (Fig. 4), is not related to the variation of the degradation of the DP1 subunit (Fig. 2A). For example, H500A and H563A had more severe degradation than the others, but they had strong activity in the presence of manganese (Fig. 4). D363A and D407A had very similar band patterns to each other, but D363A was inactive in the exonuclease activity while D407A was active (Figs 3 and 4). It is therefore concluded that residues D363, H365 and H454 are essential, while D407, H453, H500, H563 and H565 are important but not essential, and D410 is not a critical residue for exonuclease activity.
Figure 4.

Comparison of the 3′–5′ exonuclease activities of the mutants assayed using manganese as the cofactor. The substrate used was a 8/25mer recess-ended dsDNA. (A) Gel profile of the exonuclease products using the protein samples. The reaction time was 30 min. (B) Quantification of the exonuclease activities of the wild-type PolD and the mutants. Each value was calculated based on the results of at least three independent reactions.
Requirement of PolD complex formation for efficient catalysis of the 3′–5′ endonuclease activity
In order to understand the roles of the subunits for the nuclease catalytic activity of PolD, we used separately expressed and purified DP1Pho and DP2Pho proteins for the analysis (Fig. 2B and C). When magnesium was used, there was no detectable exonuclease activity (Fig. 5, lane 3) for DP1Pho; however, when manganese was used the exonuclease activity was detectable (Fig. 5, lane 2), although it was much weaker than that of the PolDPho complex in the presence of manganese (Fig. 5, lane 8 and Fig. 4A) or magnesium (Fig. 3). There was no degradation of DNA by DP2Pho (Fig. 5, lane 5), indicating that DP2Pho is not directly involved in the exonuclease catalysis. Furthermore, we measured the DNA polymerization activity of each subunit. In the presence of dNTP, DP2Pho was able to synthesize DNA (Fig. 5, lanes 6 and 7), although the activity was much weaker than that of the PolDPho complex (lanes 9 and 10), while DP1Pho had no DNA polymerization activity (Fig. 5, lane 4). These results indicate that DP1Pho and DP2Pho are the nuclease and polymerization subunits respectively and that full activities, either the polymerization or the exonuclease, requires the formation of PolDPho complex.
Figure 5.
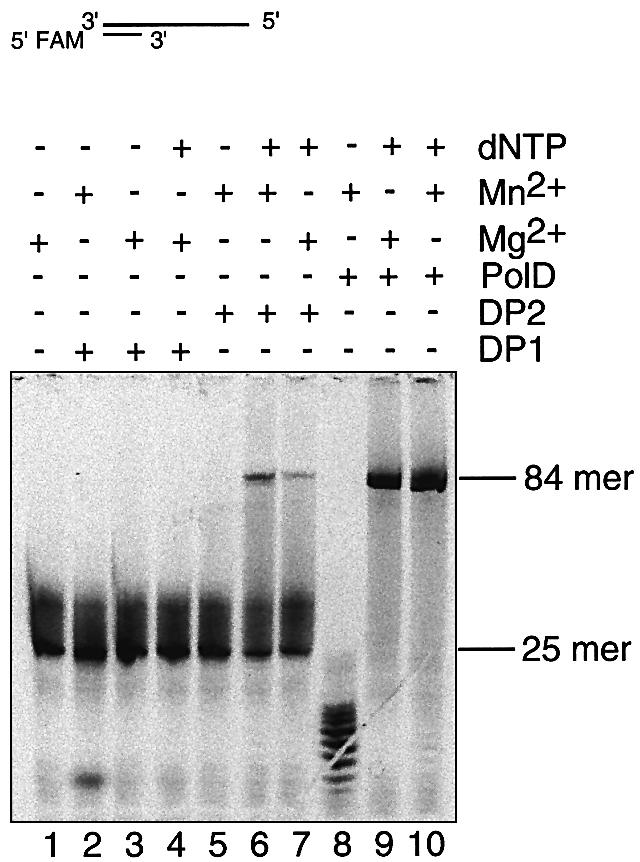
DP1Pho and DP2Pho as the nuclease and polymerization subunits respectively. The exonuclease and DNA polymerization activities were compared with different combinations of enzymes and metal ions using the 84/25mer substrate. MnSO4 or MgSO4 (10 mM) was used in the assay. The reaction time for lanes 1–7 was 30 min, for lane 8 was 10 min and for lanes 9–10 was 2 min. The band positions of the products (84mer) and substrate (25mer) are indicated.
The PolDPho complex has a manganese-dependent ssDNA endonuclease activity whose activity center might be the same as that for the 3′–5′ exonuclease
Because the Mre11 nuclease has a manganese-dependent ssDNA endonuclease activity, in addition to its 3′–5′ exonuclease activity (40,43), we then tested if PolDPho has this endonuclease activity as well. As shown in Figure 6A, the wild-type PolDPho was able to degrade circular M13 ssDNA in the presence of manganese. The complex of PolDPho was required for the digestion, since neither DP1Pho nor DP2Pho separately expressed and purified (Fig. 2B and C) was able to cut M13 ssDNA (data not shown). The wild-type PolDPho was not able to digest pUC18 circular dsDNA (data not shown). It is concluded that the PolDPho complex has a manganese-dependent ssDNA endonuclease activity like the Mre11 nuclease subfamily.
Figure 6.

Manganese-dependent ssDNA endonuclease activity of the wild-type PolDPho and the alanine mutants. (A) Degradation of circular ssDNA by the wild-type PolDPho in the presence of manganese. (B) Comparison of the ssDNA endonuclease activities of the alanine mutants. The arrows indicate the bands of the substrates M13 ssDNA.
We also checked the ssDNA endonuclease activity of the mutant proteins. As shown in Figure 6B, the mutants D363A, H365A, N453A, H454A and H500A had no detectable activity to digest the M13 ssDNA, D407A and H563A had weak but visible activity, while the mutants D410A and H565A had strong activity. The variation in ssDNA endonuclease activity among the mutants is similar to that of their corresponding 3′–5′ activities (Fig. 3). It seems likely that PolDPho uses the same catalytic center to perform both ssDNA endonuclease and 3′–5′ exonuclease activities.
A 21-aa peptide from DP2Pho stimulates the 3′–5′ exonuclease activity of DP1Pho
As DP2Pho is apparently involved in the exonuclease activity, it would be interesting if the domain of DP2Pho involved in the 3′–5′ exonuclease activity was identified. Previously we found that 1255–1332 of DP2 is the putative region involved in subunit interaction by analysis of truncated mutants (32). Interestingly, this sequence is the homologus region between PolD and the DNA polymerase epsilon of budding yeast (32). Within this region, a 21-aa fragment (1290-VKCNTKF RRPPLDGKCPICGG-1310, pI = 9.82) covering most of the putative zinc finger motif (1289-CVKCNTKFRRPPLDG KCPIC-1308, cysteine cluster II) has further been found to specifically interact with the whole and the N-terminal domain (1–200) of DP1Pho by yeast two-hybrid analysis and surface plasmon resonance analysis (X.-F.Tang et al., in preparation). Aparently, the 21-aa peptide is the candidate region which might be involved in the 3′–5′ exonuclease activity.
To test this hypothesis, we added the peptide in excess (100, 400 and 800× the protein in molar ratio) to the reaction mixtures and measured the 3′–5′ exonuclease activities (Fig. 7). A 21-aa control random peptide with a predicted pI of 9.74 (MKEGIPSVARFCLGKTPDRFG) was also used for the analysis. Although there was a slight stimulation of the exonuclease activity of DP1Pho on the addition of the random peptide, there was no apparent response to peptide concentrations (Fig. 7, lanes 1, 4–6). In contrast, the stimulation of activity by adding the 21-aa DP2Pho-derived peptide to DP1Pho was dramatic and responsive to the peptide amount (Fig. 7, lanes 1–3). These results show that the region of the cysteine cluster II is directly involved in the exonuclease catalysis, although the mechanism needs to be further investigated.
Figure 7.

Stimulation of the exonuclease activity of DP1Pho by the 21-aa peptide derived from the C-terminal of DP2Pho. The substrate used was the 84/25mer dsDNA and the ion was manganese (10 mM). All reactions were performed for 60 min. The numbers 100, 400 and 800 are the molar ratios between the peptide added and the enzyme.
The 3′–5′ exonuclease activity of the internal deletion mutants SL(1299–1308) and SL(1289–1298) is rescued by using manganese ion and enhanced by adding the peptides derived from the cysteine cluster II of DP2Pho
In our previous report, we demonstrated that deletion mutants lacking the 21-aa region, SL(1299–1308) and SL(1289–1298), lost the 3′–5′ exonuclease activity when magnesium was used (32). We tested the effect of manganese ion on the exonuclease activity. As shown in Figure 8, lanes 2 and 7, the exonuclease activity of SL(1299–1308) and SL(1289–1298) was dramatically increased by using manganese ion. Furthermore, the DP2Pho-derived peptide was able to stimulate the activity of the deletion mutant SL(1299–1308) in the presence of magnesium (Fig. 8, lanes 1 and 3) or manganese (Fig. 8, lanes 2 and 4), and SL(1289–1298) in the presence of manganese (Fig. 8, lanes 7 and 8). However, there was no stimulative effect when the random peptide was added to SL(1299–1308) (Fig. 8, lanes 2 and 6) and SL(1289–1298) (lanes 7 and 9). These results indicate that manganese is able to rescue the activity of the internal deletion mutants, and that the 21-aa DP2Pho-derived peptide is closely related to the 3′–5′ exonuclease activity and plays a stimulating role in the nuclease catalysis.
Figure 8.

Rescuing of the exonuclease activity of the internal deletion mutants SL(DEL1289–1298) and SL(DEL1299–1308) by adding the 21-aa peptide derived from the C-terminal of DP2Pho. The exonuclease activities with different combinations of proteins, metal ions and peptides were measured. The substrate used was the 84/25mer dsDNA and the ion concentration was 10 mM. All reactions were performed for 60 min. The number 400 is the molar ratio between the peptide added and the enzyme.
For a better comparison, a pair of 20-aa peptides were synthesized and used for analysis. One peptide (1289-CVKCNTKFRRPPLDGKCPIC-1308) is derived from the cysteine cluster II, and another (KRCFDLPCPRVTC KPKCNIG) has the same amino acid composition as the cysteine cluster II-derived 20-aa peptide but has an amino sequence order in random. The cysteine cluster II-derived 20-aa peptide had stimulative effect on the 3′–5′ exonuclease activity as the cysteine cluster II-derived 21-aa peptide, but the control 20-aa peptide did not have this effect, the same as the 21-aa random peptide (data not shown). In addition, we found that zinc ion enhanced the activating effect of the cysteine II-derived peptides, although only slightly (data not shown). These results indicate that the region within the cysteine cluster II is involved in the exonuclease catalysis and that the cysteine pairs are probably bound to zinc ion to form a correct conformation for the exonuclease catalysis.
DISCUSSION
The nuclease properties of PolD from P.horikoshii were studied. We have obtained biochemical evidence strongly supporting that the phosphoesterase domain located in the C-terminal of the small subunit of PolD is the catalytic domain responsible for the 3′–5′ exonuclease activity previously reported (26–29) and the ssDNA endonuclease activity identified in the present study.
There has been no report about site-directed mutagenesis of the residues in Mre11Pfu (37), however, there have been reports on the site-directed mutagenesis of several other enzymes of the phosphoesterase family (38,46–48), and our results are also in good agreement with these results. In a report on Mre11 of Saccharomyces cerevisiae, an alanine mutant at residue D16 (corresponding to D363 in DP1Pho) was made (38), and the in vitro exonuclease activity and in vivo phenotypes of the mutant during mitosis were analyzed. The ssDNA endonuclease activity of the D16A mutant was not detectable in the presence of manganese or other divalent cations (Mg2+, Ni2+, Ca2+, Sr2+, Co2+ and Zn2+), and the mutant showed a deficiency in the repair of DNA damage by methyl methanesulfonate and the maintenance of telomere length (38). In another report on Mre11 of S.cerevisiae (46), mutant Mre11–58, in which H213 (corresponding to H500 of DP1Pho) was changed to tyrosine, lost the ssDNA endonuclease and dsDNA exonuclease activities. Mre11–58 also showed full sensitivity to methyl methanesulfonate (46). In still another report on Mre11 of S.cerevisiae (47), the in vivo effects of the mutant alleles at the conserved motifs I, II, III and V (Fig. 1) were analyzed. The mutants were found to have increased spontaneous mitotic interhomologue recombination, to be sensitive to ionizing radiation, and to be defective in double-stranded DNA break repair. In a report on the mutational analysis of the bacteriophage lambda phosphatase (48), the asparagine mutants of residues D20, H22, D49, D52 and H76 (corresponding to D363, H365, D407A, D410 and H454 in DP1Pho, respectively) were analyzed. The kcat of D20N, H22N, D49N and H76N was reduced to 1/4000 or less of that of the wild-type protein, while the kcat of D52N was reduced to only 1/36. Interestingly, the km of the substrate among the mutants was not changed, but the km (Mn2+) of D20N, H22N, D49N and H76N increased to at least forty times that of the wild-type enzyme, while km (Mn2+) of D52N was not changed. The results suggested that all residues, D363, H365, D407 and H454 in DP1Pho (corresponding to D20, H22, D49 and H76 respectively in bacteriophage lambda phosphatase), except D410 in DP1Pho (corresponding to D52 in bacteriophage lambda phosphatase) might be involved in metal binding, not in substrate binding.
Although the nuclease of PolDPho is Mre11-like, the complex formation and manganese ion are needed for the full activities of ssDNA endonuclease and 3′–5′ exonuclease of PolDPho. In contrast, for the Mre11–Rad50 complex, Mre11 nuclease domain alone is sufficient to fulfill the 3′–5′ exonuclease and ssDNA endonuclease activity (37).
We observed stronger 3′–5′ exonuclease activity using manganese than magnesium of the wild-type PolDPho (Fig. 4). Striking difference was also observed between the use of two metal ions in the measurement of nuclease activity of the alanine mutants at the conserved motifs and the two internal deletion mutants (Figs 3, 4 and 8). Manganese was able to enhance the activity of D407A, D410A and H565A (Figs 3 and 4), and rescue the activities of N453A, H500A, H563A, SL(DEL1299–1308) and SL(1289–1308) (Figs 3, 4 and 8). In addition, the ssDNA endonuclease activity was only detected when manganese was used (Fig. 6). The results strongly suggest that manganese is probably the true metal ion cofactor for the nuclease activity of PolDPho. Then, we compared the DNA polymerization activity of DP2Pho (Fig. 5) and PolD complex (data not shown) using manganese and magnesium, no dramatic difference was observed. It remains unclear whether manganese or magnesium is used for the DNA polymerization in PolDPho. Manganese was not found to be favored in PCR using PolDPho, since the enzyme became unstable when manganese was used as cofactor (data not shown). It might be possible that in PolDPho DNA polymerase uses magnesium while the nuclease utilizes manganese as cofactor for catalysis. If so, it needs to be investigated how the metals are coupled in one molecule.
Perhaps the most interesting result in this study is the finding that the 21-aa subunit-interacting peptide directly stimulates the exonuclease catalysis. It is able to enhance the activity of DP1 as a single subunit (Fig. 7), and the internal deletion mutants SL(DEL1299–1308) and SL(1289–1308) lacking the sequence (Fig. 8). This sequence contains two cysteine pairs which presumably forms a zinc finger motif. It has homology to the C-terminal of yeast DNA polymerase epsilon, which was suggested to be important in cell cycle checkpoint control (49). In PolD, it was found to interact with DP1 as a whole and its N-terminal 1–200 fragment by yeast two-hybrid system and surface plasmon resonance analysis (X.-F.Tang et al., in preparation). Probably in polymerase epsilon of yeast, the homologous fragment has a similar role to regulate the exonuclease activity, for DNA repair and checkpoint control (49). It would be interesting if the molecular structure of the 21-mer peptide is solved by NMR.
The finding that PolD has a Mre11-like exonuclease domain suggests that DNA polymerization of PolD is perhaps coupled with DNA recombination through the Mre11–Rad50 complex. On the other hand, DP1 was found to interact with Rad51B in P.furiosus (50), therefore, DNA polymerization of PolD may be also linked to Rad51B-mediated recombination.
In conclusion, based on the results in this study, a model concerning the nuclease catalysis of PolDPho is proposed. The catalytic center is formed by the C-terminal Mre11-like domain of DP1Pho. The N-terminal 1–200 of DP1 inhibits the nuclease activity through regulation of DNA binding of the nuclease domain (32). The cysteine cluster II is deeply involved in the nuclease activity and plays a stimulatory role in the nuclease catalysis. It is probably that the peptide reduces the inhibitory effect of the N-terminal DP1Pho(1–200) on the catalysis of the C-terminal Mre11 nuclease domain (32), through direct binding to the N-terminal DP1(1–200). Manganese is required as a cofactor to coordinate the nuclease catalysis efficiently.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Emiko Yamamoto for technical help during this study and Yuji Urushibata for helpful discussions.
REFERENCES
- 1.Kunkel T.A. (1998) Exonucleolytic proofreading. Cell, 53, 837–840. [DOI] [PubMed] [Google Scholar]
- 2.Ito J. and Braithwaite,D.K. (1991) Compilation and alignment of DNA polymerase sequences. Nucleic Acids Res., 19, 4045–4057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kunkel T.A. and Bebenek,K. (2000) DNA replication fidelity. Annu. Rev. Biochem., 69, 497–529. [DOI] [PubMed] [Google Scholar]
- 4.Hubscher U., Maga,G. and Spadari,S. (2002) Eukaryotic DNA polymerases. Annu. Rev. Biochem., 71, 133–163. [DOI] [PubMed] [Google Scholar]
- 5.Brutlag D. and Kornberg,A. (1972) Enzymatic synthesis of deoxyribonucleic acid. XXXVI. A proofreading function for the 3′–5′ exonuclease activity in deoxyribonucleic acid polymerases. J. Biol. Chem., 247, 241–248. [PubMed] [Google Scholar]
- 6.Foury F. and Vanderstraeten,S. (1992) Yeast mitochondrial DNA mutators with deficient proofreading exonucleolytic activity. EMBO J., 11, 2717–2726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morrison A., Bell,J.B., Kunkel,T.A. and Sugino,A. (1991) Eukaryotic DNA polymerase: amino acid sequence required for 3′–5′ exonuclease activity. Proc. Natl Acad. Sci. USA, 88, 9473–9477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kuhn F.J.P. and Knopf,C.W. (1996) Herpes simple virus type I DNA polymerase: mutational analysis of the 3′–5′ exonuclease domain. J. Biol. Chem., 271, 29245–29254. [DOI] [PubMed] [Google Scholar]
- 9.Hwang Y.T., Liu,B.Y., Coen,D.M. and Hwang,C.B. (1997) Effects of mutations in the Exo III motif of the herpes simplex virus DNA polymerase gene on enzyme activities, viral replication and replication fidelity. J. Virol., 71, 7791–7798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu H., Naismith,J.H. and Hay,R.T. (2000) Identification of conserved residues contributing to the activities of adenovirus DNA polymerase. J. Virol., 74, 11681–11689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lancy E.D., Lifsics,M.R., Kehres,D.G. and Maurer,R. (1989) Isolation and characterization of mutants with deletions in dnaQ, the gene for the editing subunit of DNA polymerase III in Salmonella typhimurium. J. Bacteriol., 171, 5572–5580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bernad A., Blanco,L., Lazaro,J.M, Martin,G. and Salas,M. (1989) A conserved 3′–5′ exonuclease active site in prokaryotic and eukaryotic DNA polymerases. Cell, 59, 219–298. [DOI] [PubMed] [Google Scholar]
- 13.Blanco L., Bernad,A. and Salas,M. (1992) Evidence favoring the hypothesis of a conserved 3′–5′ exonuclease active site in DNA-dependent DNA polymerase. Gene, 112, 139–144. [DOI] [PubMed] [Google Scholar]
- 14.Kim Y., Eom,S.H., Wang,J., Lee,D.-S., Suh,S.W. and Steitz,T.A. (1995) Crystal structure of Thermus aquaticus DNA polymerase. Nature, 376, 612–616. [DOI] [PubMed] [Google Scholar]
- 15.Kiefer J.R., Mao,C., Hansen,C.J., Basehore,S.L., Hogrefe,H.H., Braman,J.C. and Beese,L.S. (1997) Crystal structure of a thermostable Bacillus DNA polymerase I large fragment at 2.1 Å resolution. Structure, 5, 95–108. [DOI] [PubMed] [Google Scholar]
- 16.Wang J., Sattar,A.K.M.A., Wang,C.C., Karam,J.D., Konigsberg,W.H. and Steitz,T.A. (1997) Crystal structure of a pol alpha family replication DNA polymerase from bacteriophage RB69. Cell, 89, 1087–1099. [DOI] [PubMed] [Google Scholar]
- 17.Zhao Y., Jeruzalmi,D., Moarefi,I., Leighton,L., Lasken,R. and Kuriyan,J. (1999) Crystal structure of an archaebacterial DNA polymerase. Struct. Fold Des., 7, 1189–1199. [DOI] [PubMed] [Google Scholar]
- 18.Hamdan S., Brown,S.E., Thompson,P.R., Yang,J.Y., Carr,P.D., Ollis,D.L., Otting,G. and Dixon,N.E. (2000) Preliminary X-ray crystallographic and NMR studies on the exonuclease domain of the theta subunit of Escherichia coli DNA polymerase III. J. Struct. Biol., 131, 164–169. [DOI] [PubMed] [Google Scholar]
- 19.Rodriguez A.C., Park,H.-W., Mao,C. and Beese,L.S. (2000) Crystal structure of a pol alpha family DNA polymerase from the hyperthermophilic archaeon Thermococcus sp. 9 degrees N-7. J. Mol. Biol., 299, 447–462. [DOI] [PubMed] [Google Scholar]
- 20.Hashimoto H., Nishoka,M., Fujiwara,S., Takagi,M., Imanaka,T., Inoue,T. and Kai,Y. (2001) Crystal structure of DNA polymerase from hyperthermophilic archaeon Pyrococcus kodakaraensis KOD1. J. Mol. Biol., 306, 467–477. [DOI] [PubMed] [Google Scholar]
- 21.Hamdan S., Carr,P.D, Brown,S.E., Ollis,D.L and Dixon,N.E. (2002) Structural basis for proofreading during replication of the Escherichia coli chromosome. Structure, 10, 535–546. [DOI] [PubMed] [Google Scholar]
- 22.Beese L.S. and Steitz,T.A. (1991) Structure basis for the 3′–5′ exonuclease activity of Escherichia coli DNA polymerase I: a two metal ion mechanism. EMBO J., 10, 25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brautigam C.A. and Steitz,T.A. (1998) Structure principles for the inhibition of the 3′–5′ exonuclease activity of Escherichia coli DNA polymerase I by phosphorothioates. J. Mol. Biol., 277, 363–377. [DOI] [PubMed] [Google Scholar]
- 24.Elisseeva E., Mandal,S.S. and Reha-Krantz,L.J. (1999) Mutational and pH studies of the 3′–5′ exonuclease activity of bacteriophage T4 DNA polymerase. J. Biol. Chem., 274, 25151–25158. [DOI] [PubMed] [Google Scholar]
- 25.Cann I.K., Komori,K., Toh,H., Kanai,S. and Ishino,Y. (1998) A heterodimeric DNA polymerase: evidence that members of Euryarchaeota possess a distinct DNA polymerase. Proc. Natl Acad. Sci. USA, 95, 14250–14255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Uemori T., Sato,Y., Kato,I., Doi,H. and Ishino,Y. (1997) A novel DNA polymerase in the hyperthermophilic archaeon, Pyrococcus furiosus: gene cloning, expression, and characterization. Genes Cells, 2, 499–512. [DOI] [PubMed] [Google Scholar]
- 27.Ishino Y., Komori,K., Cann,I.K. and Koga,Y. (1998) A novel DNA polymerase family found in Archaea. J. Bacteriol., 180, 2232–2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shen Y., Musti,K., Hiramoto,M., Kikuchi,H., Kawabayashi,Y. and Matsui,I. (2001) Invariant Asp1122 and Asp1124 are essential residues for polymerization catalysis of family D DNA polymerase from Pyrococcus horikoshii. J. Biol. Chem., 276, 27376–27383. [DOI] [PubMed] [Google Scholar]
- 29.Gueguen Y., Rolland,J., Lecompte,O., Azam,P., Romancer,G.L., Flament,D., Raffin,J. and Dietrich,J. (2001) Characterization of two DNA polymerases from the hyperthermophilic archaeon Pyrococcus abyssi. Eur. J. Biochem., 268, 5961–5969. [DOI] [PubMed] [Google Scholar]
- 30.Cann I.K. and Ishino,Y. (1999) Archaeal DNA replication: identifying pieces to solve a puzzle. Genetics, 152, 1249–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Myllykallio H., Lopez,P., Lopez-Garcia,P., Heilig,R., Saurin,W., Zivanovic,Y., Philippe,H. and Forterre,P. (2000) Bacterial mode of replication with eukaryotic-like machinery in a hyperthermophilic archaeon. Science, 288, 2212–2215. [DOI] [PubMed] [Google Scholar]
- 32.Shen Y., Tang,X. and Matsui,I. (2003) Subunit interaction and regulation of activity through terminal domains of the family D DNA polymerase from Pyrococcus horikoshii.J. Biol. Chem., 278, 21247–21257. [DOI] [PubMed] [Google Scholar]
- 33.Aravind L. and Koonin,E.V. (1998) Phosphoesterase domain associated with DNA polymerase of diverse origins. Nucleic Acids Res., 26, 3746–3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Strater N., Klabunde,T., Tucker,P., Witzel,H. and Krebs,B. (1995) Crystal structure of a purple acid phosphatase containing a dinuclear Fe(III)–Zn(II) active sites. Science, 268, 1489–1492. [DOI] [PubMed] [Google Scholar]
- 35.Griffith J.P., Kim,J.L., Kim,E.E, Sintchak,M.D., Thomson,J.A., Fitzgibbon,M.J., Fleming,M.A., Caron,P.R., Shiao,K. and Navia,M.A. (1995) X-ray structure of calcineurin inhibited by immunophilin–immunosuppressant FKBP12–FK506 complex. Cell, 82, 507–522. [DOI] [PubMed] [Google Scholar]
- 36.Kissinger C.R., Parge,H.E., Knighton,D.R., Lewis,C.T., Pelletier,L.A., Tempczyk,A., Kalish,V.J., Tucker,K.D., Showalter,R.E., Moomaw,E.W., Gastinel,L.N., Habuka,N., Chen,X., Maldonado,F., Barker,J.E., Bacquet,R. and Villafranca,J.E. (1995) Crystal structure of human calcineurin and the human FKBP12–FK506–calcineurin complex. Nature, 378, 641–644. [DOI] [PubMed] [Google Scholar]
- 37.Hopfner K.-P., Karcher,A., Craig,L., Woo,T.T., Carney,J.P. and Tainer,J.A. (2001) Structural biochemistry and interaction architecture of the DNA double-stranded break repair Mre11 nuclease and Rad50–ATPase. Cell, 105, 473–485. [DOI] [PubMed] [Google Scholar]
- 38.Furuse M., Nagase,Y., Tsubouchi,H., Murakami-Murofushi,K., Shibata,T. and Ohta,K. (1998) Distinct roles of two separable in vitro activities of yeast Mre11 in mitotic and meiotic recombination. EMBO J., 17, 6412–6425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Trujillo K.M., Yuan,S.Y., Lee,E.Y.P. and Sung,P. (1998) Nuclease activities in a complex of human recombination and DNA repair factors Rad50, Mre11 and p95. J. Biol. Chem., 273, 21447–21450. [DOI] [PubMed] [Google Scholar]
- 40.Usui T., Ohta,T., Oshiumi,H., Tomizawa,J., Ogawa,H. and Ogawa,T. (1998) Complex formation and functional versatility of Mre11 of budding yeast in recombination. Cell, 95, 705–716. [DOI] [PubMed] [Google Scholar]
- 41.Connelly J.C., Kirkham,L. and Leach,D.R. (1998) The SbcCD nuclease of Escherichia coli is a structural maintenance of chromosomes (SMC) family protein that cleaves hairpin DNA. Proc. Natl Acad. Sci. USA, 95, 7969–7974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Connelly J.C. and Leach,D.R.F. (2002) Tethering on the brink: the evolutionarily conserved Mre11–Rad50 complex. Trends Biochem. Sci., 27, 410–419. [DOI] [PubMed] [Google Scholar]
- 43.Hopfner K.-P., Karcher,A., Shin,D., Fairley,C., Tainer,J.A. and Carney,J.P (2000) Mre11 and Rad50 from Pyrococcus furiosus: cloning and biochemical characterization reveal an evolutionarily conserved multiprotein machine. J. Bacteriol., 182, 6036–6041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Constantinesco F., Forterre,P. and Elie,C. (2002) NurA, a novel 5′–3′ nuclease gene linked to rad50 and Mre11 homologs of thermophilic Archaea. EMBO Rep., 3, 537–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kadowaki H., Kadowaki,T., Wondisford,F.E. and Taylor,S.I. (1989) Use of polymerase chain reaction catalyzed by Taq DNA polymerase for site-specific mutagenesis. Gene, 76, 161–166. [DOI] [PubMed] [Google Scholar]
- 46.Tsubouchi H. and Ogawa,H. (1998) A novel mre11 mutation impairs processing of double-strand breaks of DNA during both mitosis and meiosis. Mol. Cell. Biol., 18, 260–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bressan D.A, Olivares,H.A., Nelms,B.E. and Petrini,H.J. (1998) Alteration of N-terminal phosphoesterase signature motifs inactivates Saccharomyces cerevisiae Mre11. Genetics, 150, 591–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhuo S., Clemens,J.C., Stone,R.L. and Dixon,J.E. (1994) Mutational analysis of a Ser/Thr phosphatase. Identification of residues important in phosphoesterase substrate binding and catalysis. J. Biol. Chem., 269, 26234–26238. [PubMed] [Google Scholar]
- 49.Dua R., Levy,D. and Campbell,J. (1998) Role of the putative zinc finger domain of Saccharomyces cerevisiae DNA polymerase in DNA replication and the S/M checkpoint pathway. J. Biol. Chem. 273, 30046–30055. [DOI] [PubMed] [Google Scholar]
- 50.Hayashi I., Morikawa,K. and Ishino,Y. (1999) Specific interaction between DNA polymerase II (PolD) and RadB, a Rad51/Dmc1 homolog, in Pyrococcus furiosus. Nucleic Acids Res., 27, 4695–4702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kraulis P.J. (1991) Molscript: a program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr., 24, 946–950. [Google Scholar]
- 52.Merrit E.A. and Bacon,D.J. (1997) Raster3D: photorealistic molecular graphics. Methods Enzymol., 277, 505–524. [DOI] [PubMed] [Google Scholar]