

Abstract
Advanced hepatocellular carcinoma (HCC) is an important cause of cancer mortality. Epithelial-mesenchymal transition (EMT) has been shown to be an important biological process in cancer progression and metastasis. We have focused on elucidating factors that induce EMT to promote carcinogenesis and subsequent metastasis in HCC using the BNL CL.2 (BNL) and BNL 1ME A. 7R.1 (1MEA) cell lines. BNL cells are normal hepatocytes whereas the 1MEA cells are HCC cells derived from chemical transformation of the BNL cells. Their morphological characteristics were examined. Expression levels of hepatocyte growth factor (HGF), markers of EMT and mediators of HGF signaling were determined and functional characteristics were compared. BNL cells were treated with HGF and effects on EMT-marker and mediators of HGF signaling were analyzed. BNL cells display characteristic epithelial morphology whereas 1MEA cells display mesenchymal characteristics. 1MEA cells express and secrete more HGF than BNL cells. There was significantly decreased expression of E-cadherin, albumin, AAT and increased expression of fibronectin, collagen-1, vimentin, snail and slug in 1MEA cells. There was also increased expression of cyclooxygenase-2 (COX-2), Akt and phosphorylated Akt (pAkt) in 1MEA cells. Moreover, 1MEA cells had increased migratory capacity inhibited by inhibition of COX-2 and Akt but not extracellular signal regulated kinase (ERK). Molecular mesenchymal characteristics of 1MEA cells were reversed by inhibition of COX-2, Akt and ERK. Treatment of BNL cells with HGF led to decreased expression of E-cadherin and increased expression of fibronectin, vimentin, snail, slug, COX-2, Akt, pAkt and increased migration, invasiveness and clonogenicity. We conclude that development of HCC is associated with upregulation of HGF which promotes EMT and carcinogenesis via upregulation of COX-2 and Akt. Consequently, HGF signaling may be targeted for therapy in advanced and metastatic HCC.
Keywords: Chemoprevention, Cyclooxygenase-2, Epithelial-mesenchymal transition, Hepatocellular carcinoma, Hepatocyte growth factor
Introduction
Hepatocellular carcinoma (HCC) is currently the third commonest cause of cancer mortality worldwide [1]. Advanced and metastatic HCC is a highly devastating disease with limited and largely ineffective treatment options. Consequently, there is still a great need for a better understanding of the underlying biology of advanced HCC and the efforts towards finding effective molecular targeted therapy should be intensified.
An important biological process that has been shown to underlie cancer progression, metastasis and recurrence is epithelial-mesenchymal transition (EMT) [2]. Indeed, metastasis of tumors has been associated with markers of EMT [3]. For example, loss of E-cadherin [4], gain of vimentin [5], gain of collagen I [6], gain of fibronectin [7] as well as expression of transcription factors of the snail family [8] have all been reported in cancers. The phenomenon of EMT results in epithelial cells losing their cell–cell adhesions, acquiring mesenchymal phenotype, becoming migratory and invasive and in the case of cancer cells the ultimate consequence is metastatic spread [4]. This process has also been implicated in the development of cancer chemoresistance [5].
Research has revealed that a number of factors promote EMT in various cells. These include growth factors such as hepatocyte growth factor (HGF), transforming growth factor beta (TGFβ), epidermal growth factor (EGF), fibroblast growth factor (FGF), insulin-like growth factor (IGF) and platelet derived growth factor (PDGF) [6-10]. The role of these growth factors in EMT was initially characterized in the context of embryogenesis, but it has since become widely recognized that EMT also occurs in the context of carcinogenesis.
In HCC, dysregulation of growth factor pathways is a well-known factor [11]. Further, there is now accumulating evidence that hepatocellular EMT takes place in the contexts of liver fibrosis and HCC [12-17]. So far, the established growth factors reported in literature to promote EMT in HCC are TGFβ and PDGF [16-18]. The c-met protooncogene and HGF have been reported to be associated with HCC [19-21] and a recent study reported whilst we were still conducting our own study has suggested that HGF may have a direct role in EMT induction in HCC [22]. Further clarification is still required and the underlying signaling mechanisms are still unknown.
HGF is a multifunctional cytokine that binds to the c-met proto-oncogene to promote cell proliferation, cell survival, cell migration, branching morphogenesis and migration in a variety of systems [23]. It has also been linked to invasion in many types of cancers [24]. Research has shown that HGF signals via a number of intracellular signaling mechanisms including phosphatidylinositol-3 kinase (PI3K)/protein kinase B (Akt) [25], extracellular signal regulated kinase (ERK) [25, 26] and cyclooxygenase-2 (COX-2) [27]. Although HGF activity in the liver has been linked to PI3K/Akt, ERK and COX-2 [23, 28], it is unknown whether they play a role in EMT in HCC.
Consequently, we have examined the direct role of HGF in promoting EMT and carcinogenesis in HCC and investigated the role of Akt, ERK or COX-2 signaling in this process.
Materials and methods
Cell culture and reagents
The BNL CL.2 (BNL) and BNL 1ME A. 7R.1 (1MEA) cell lines were obtained from the American Type Culture Collection (ATCC), Manassas, VA and used in this study. They are both hepatocytes derived from the liver of BALB/c mice. BNL is non-tumorigenic whereas 1MEA is tumorigenic. 1MEA was derived by chemical transformation of the BNL cell line using 3-methylcholanthrene epoxide [29]. Both cell lines were kept in culture in DMEM supplemented with 10% fetal bovine serum (FBS), 2 mM l-glutamine, nonessential amino acids, 100 mg/l penicillin and 100 mg/l streptomycin.
In proof of concept studies, BNL cells in culture were treated for 5 days with mouse HGF (5 ng/ml). Untreated cells were always used as controls. Also, selective inhibitors of COX-2 (1 μM celecoxib), ERK (25 μM PD 98059) and Akt (10 μM LY 294002) were used in mechanistic studies. All inhibitors were purchased from EMD Chemicals, NJ and dissolved in dimethyl sulfoxide (DMSO) and control cells were always treated with equivalent volumes of DMSO. Inhibitor concentrations used were those previously established to have specific activity [30].
Haematoxylin and eosin staining
Cells were plated onto microscope slides and cultured in complete medium overnight. After fixing in 1% formaldehyde for 1 min cells were washed in distilled water for 5 min, immersed in hematoxylin for 2 min, washed again twice in distilled water for 15 s and then 1 min, immersed in clarifier, washed again in distilled water and then immersed in bluing reagent, then distilled water and then 80% ethanol all for 30 s. The cells were then immersed in eosin for 1 min, dipped twice in 95% ethanol and then dehydrated in 2 changes of 100% ethanol and then 3 changes of xylene by immersion for 1 min each. Stained cells were visualized with a microscope equipped with imaging software. Images were acquired at ×20 magnification.
Immunoblotting
Cells were cultured in a monolayer in 6-well plates until 60–70% confluent. Immunoblotting was performed as previously described [31]. The specific primary antibodies used are listed in Table 1. Immunoreactive proteins were visualized by incubating in HRP-conjugated secondary antibodies. Chemiluminescence was detected by incubating in an equal-parts mixture of the SuperSignal West Pico stable peroxide solution and luminol/enhancer solution (Pierce, IL) and subsequently using an image processing machine.
Table 1.
List of antibodies used for immunoblotting
| Antibody | Catalog # and source | Concentration |
|---|---|---|
| Anti-HGF | #H00003082-001P, Abnova | 1:100 |
| Anti-E-cadherin | H-108: sc-7870, Santa Cruz | 1:200 |
| Anti-albumin | #126584, Calbiochem | 1:1000 |
| Anti-AAT | V1080, biømeda | 1:2000 |
| Anti-vimentin | H-84: sc-5565, Santa Cruz | 1:200 |
| Anti-fibronectin | ab2413-500, abcam | 1:1000 |
| Anti-collagen I | ab292-100, abcam | 1:500 |
| Anti-β-actin | A5441, Sigma | 1:2000 |
| Anti-ERK | #4695, Cell Signaling Technology | 1:2000 |
| Anti-pERK | #4370, Cell Signaling Technology | 1:2000 |
| Anti Akt | #9272, Cell Signaling Technology | 1:2000 |
| Anti p-Akt | #4070S, Cell Signaling Technology | 1:1000 |
| Anti-COX-2 | H-62: sc-7951, Santa Cruz | 1: 200 |
Quantitative real-time reverse transcriptase polymerase chain reaction (qPCR)
qPCR was performed as previously described [32]. The primers used for amplification are listed in Table 2. They were designed using the OligoPerfect™ Designer software (freely available at www.invitrogen.com). qPCR was performed using SYBR Green. Reactions were conducted in a 96-well spectrofluorometric thermal cycler (Applied Bio-systems, CA).
Table 2.
List of primers used for quantitative real-time PCR
| Gene | Primer sequences |
|---|---|
| GAPDH | f: 5′-TTTGTCAAGCTCATTTCCTG-3′ |
| r: 5′-TGGTCCAGGGTTTCTTACTC-3′ | |
| Vimentin | f: ′-ATTGTGGATTGTGAAGGTGA-3′ |
| r: 5′-TTCAAAAATGGTTGTGCAAT-3′ | |
| E-cadherin | f: 5′-CAGAAGGTTTTGTTTGAGCA-3′ |
| r: 5′-TGCATAGGGGATGATTTGTA-3′ | |
| Albumin | f: 5′-TCTCCAGAAATGCTCATACG-3′ |
| r: 5′-CCATAGTTTTCACGGAGGTT-3′ | |
| AAT | f: 5′-CCACCCTTTCCTTTCATAA-3′ |
| r: 5′-GGATGACATCTAGGGTGGTC-3′ | |
| Fibronectin | f: 5′-ATGACGATGGGAAGACCTAC-3′ |
| r: 5′-GGCTGGAAAGATTACTCTCG-3′ | |
| Collagen I | f: 5′-GTGCAATGCAATGAAGAACT-3′ |
| r: 5′-GAAGCAAAGTTTCCTCCAAG-3′ | |
| HGF | f: 5′-ATGATGTGGGGGACCAAA-3′ |
| r: 5′-CAACTTGTATGTCAAAATTACTTTGTG-3′ | |
| COX-2 | f: 5′-CTCTCAATGAGTACCGCAAA-3′ |
| r: 5′-CAAAGATAGCATCTGGACGA-3′ | |
| ERK | f: 5′-ACAGAGTACGTAGCCACACG-3′ |
| r: 5′-CATGGCACCTTATTTTTGTG-3′ | |
| Akt | f: 5′-TCACCTCTGAGACTGACACC-3′ |
| r: 5′-GCCCACAGTAAAAACATCCT-3′ |
HGF enzyme-linked immunosorbent assay (ELISA)
Cells were seeded at 1 × 106 cells/well in 6-well plates and cultured in complete medium for 72 h. To test for HGF release in medium, cells were serum-starved for 24 h before assessment of HGF secretion into serum-free medium using a specific mouse HGF ELISA (R&D Systems) and following manufacturer’s instructions.
Migration, invasion and clonogenic assays
1 × 104 cells were seeded into 6-well plates. At 60–70% confluency, the cell monolayer was wounded with a 200 μl-pipette, washed with PBS and medium replaced. Images were taken 24 h later. Images from three experiments were analyzed for percentage cell-covered area using the Wimasis Image Analysis software (Wimasis GmbH, Munich, Germany). For invasion experiments, 1 × 105 cells (untreated BNL cells vs. HGF-treated BNL cells vs. untreated 1MEA cells) in serum free medium were seeded into an insert with 6.5 mm Transwell membrane with 8 μm pores that had been pre-laid with 15 μl 10 μg/ml collagen. The bottom chamber contained serum-containing medium to serve as chemoattractant. After 24 h, medium in the insert was removed, cells were fixed with 10% formalin and stained with toluidine blue (Sigma, MO). Cells left on the upper surface of the insert were removed with moist soft paper or cotton wool swab. Cells on the under surface of the insert were visualized with microscopy and counted. For clonogenic assays, cells were seeded at a density of 2 × 104 in triplicate into 12-well plates containing agar and DMEM supplemented with 5% FCS.
After 12 days, colonies larger than 50 μm in diameter were counted using light microscopy.
Tumorigenicity studies
1 × 106 cells were transplanted into the flank of immunocompetent Balb/c mice following University of Florida IACUC approved protocols. Briefly, cells were harvested in serum-containing medium. After spinning down, the harvested cells were washed with PBS and resuspended in PBS at a concentration of 1 × 106 cells/100 μl and 100 μl was injected. Animals were humanely sacrificed after 3 weeks and tumor weight measured.
Statistical analysis
qPCR (normalized to GAPDH), immunoblotting, HGF ELISA, soft agar assay, invasion, migration and tumori-genicity experiments were performed at least three times. Results are expressed as mean ± SEM. Effects were compared with controls. Paired t tests were used to analyze the effect of experimental samples as compared to controls. P < 0.05 was considered significant.
Results
BNL and 1MEA cells are morphologically different
The morphological characteristics of BNL and 1MEA cells were examined initially by examination under a light microscope and then after using hematoxylin and eosin staining. There is obvious morphological difference between the two cell lines. BNL has more characteristic epithelial morphology whereas 1MEA has mesenchymal morphology (Fig. 1).
Fig. 1.
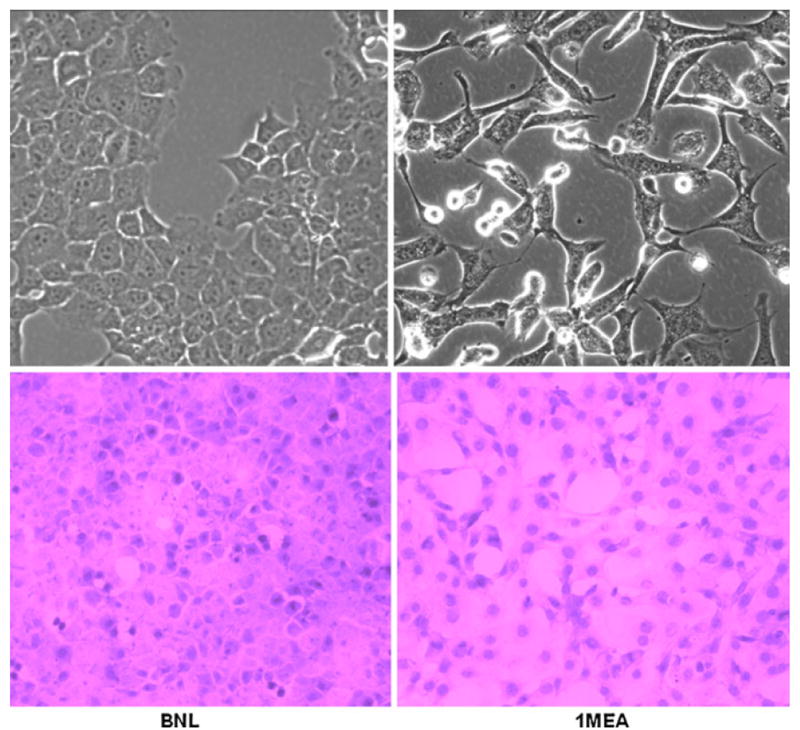
BNL and 1MEA cells are morphologically different. BNL cells display typical epithelial morphology whereas 1MEA cells show mesenchymal characteristics. Image magnification was ×40 objective for unstained cells and ×20 objective for H&E stained cells. Left BNL cells, right 1MEA cells
HGF is upregulated in 1MEA cells in comparison to BNL cells
Next, we examined the level of HGF expression in both BNL and 1MEA cells using both qPCR and immunoblotting. qPCR revealed that 1MEA cells express 200 times more HGF than BNL cells. Increased expression of HGF by 1MEA cells was confirmed by immunoblotting. Further, analysis of HGF secreted into medium using a specific mouse HGF ELISA revealed that HGF secretion by 1MEA cells is 2731.4 ± 87.2 pg/ml in comparison to 154.4 ± 20.6 pg/ml HGF secretion by BNL cells. HGF secretion by 1MEA cells is, therefore, about 17.7 fold higher than by BNL cells. c-Met receptor gene expression is, however, not significantly different (Fig. 2).
Fig. 2.
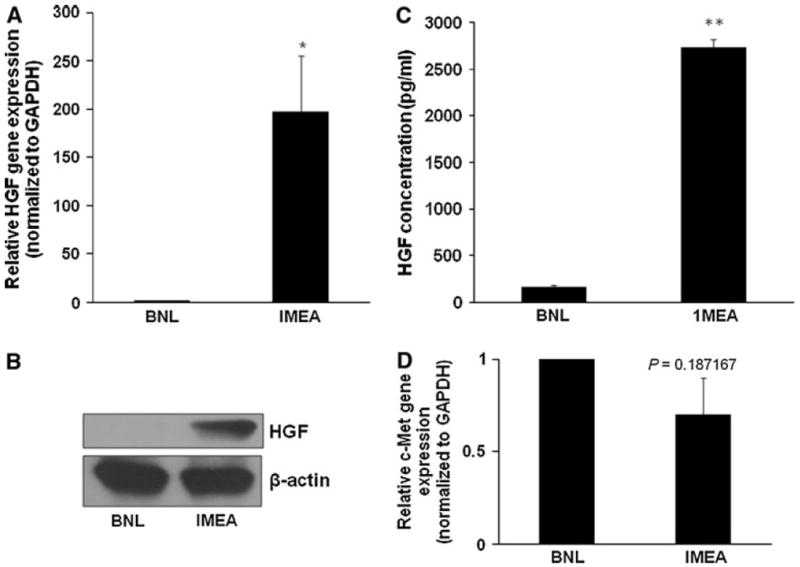
HGF is upregulated in 1MEA cells in comparison to BNL cells. a 1MEA cells express about 200 times more HGF than BNL cells as determined by qPCR (normalized to GAPDH). b 1MEA secretes 17.7-fold more HGF than BNL cells. c Increased HGF expression in 1MEA cells is confirmed by immunoblotting. d There is no significant difference between BNL and 1MEA c-Met receptor gene expression *P < 0.05; ** P <0.01, N = 3
1MEA cells display molecular mesenchymal characteristics in comparison to BNL cells
Then, we wanted to confirm whether the increased expression of HGF observed in the 1MEA cell line is associated with molecular mesenchymal characteristics. We examined expression levels of E-cadherin, albumin, fibronectin (gene and protein) as well as collagen-I, vimentin, AAT, snail and slug (gene). Our data show that 1MEA cells have significantly decreased E-cadherin, albumin and AAT expression, but significantly increased fibronectin, collagen-I, vimentin, snail and slug expression in comparison with BNL cells (Fig. 3; Supplementary Fig. 1). We also found that inhibition of COX-2 (1 μM celecoxib), ERK (25 μM PD 98059) and Akt (10 μM LY 294002) significantly reversed the mesenchymal characteristics of 1MEA cells (Supplementary Fig. 2). Mesenchymal cells typically have decreased E-cadherin expression [33], increased vimentin [34], fibronectin [35] and collagen-I expression [14]. Albumin expression is characteristic of hepatocytes and it is a known epithelial cell marker in the liver [36]. And preliminary data from our laboratory (unpublished) show that decreased AAT expression is characteristic of mesenchymal transformation in hepatocytes.
Fig. 3.
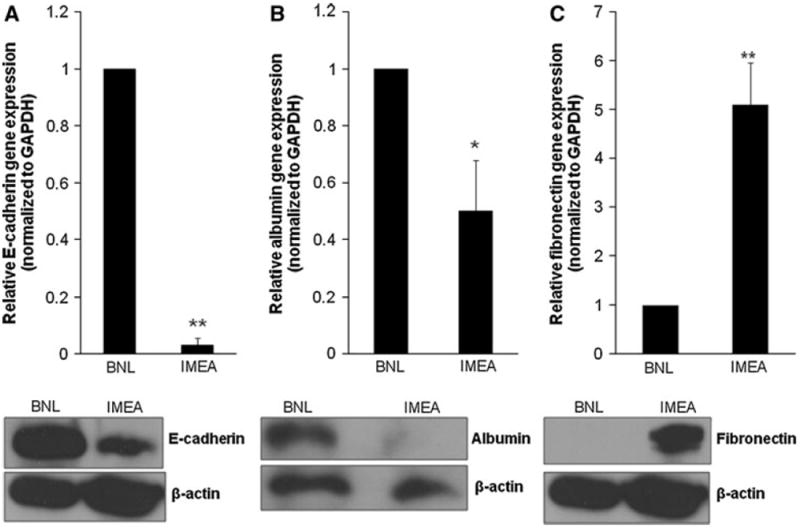
1MEA cells display molecular mesenchymal characteristics in comparison to BNL cells. a Decreased E-cadherin expression in 1MEA cells. b Decreased albumin expression in 1MEA cells. c Increased fibronectin expression in 1MEA cells. *P < 0.05; **P < 0.01, N = 3
Assessment of COX-2, ERK and Akt expression
To determine the underlying signaling mechanisms responsible for mediating HGF associated carcinogenesis and mesenchymal transformation, we examined the expression levels of COX-2 gene and protein, ERK gene and protein, phosphorylated ERK (p-ERK) protein, Akt gene and protein and p-Akt protein in both BNL and 1MEA cells. We found that 1MEA cells expressed significantly more COX-2, Akt, p-Akt, ERK and p-ERK than BNL cells (Fig. 4).
Fig. 4.
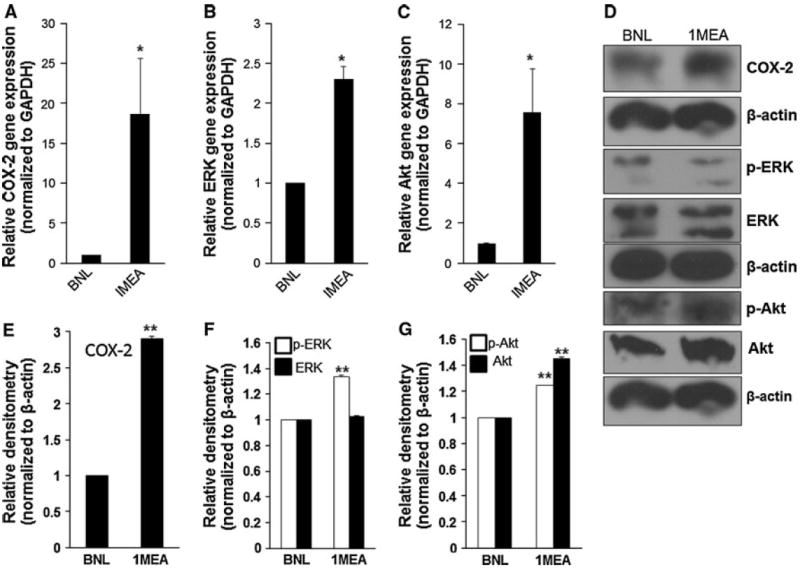
Assessment of COX-2, ERK and Akt expression. COX-2 (a, d, e), ERK and p-ERK (b, d, f) and Akt and p-Akt (c, d, g) expression are increased in 1MEA cells in comparison to BNL cells. *P < 0.05; **P < 0.01, N = 3
HGF promotes mesenchymal characteristics
To prove that the mesenchymal properties characteristic of 1MEA cells is being driven by HGF, we examined the effect of treating BNL cells with HGF on expression of mesenchymal characteristics. We found that HGF treatment led to significantly increased gene and protein expression of fibronectin and vimentin and significantly decreased gene and protein expression of E-cadherin by BNL cells. We further found that HGF promotes increased gene expression of snail and slug in BNL cells (Fig. 5). Also, light microscopic examination of morphology reveals that HGF treatment of BNL cells leads to mesenchymal morphology (Fig. 5).
Fig. 5.
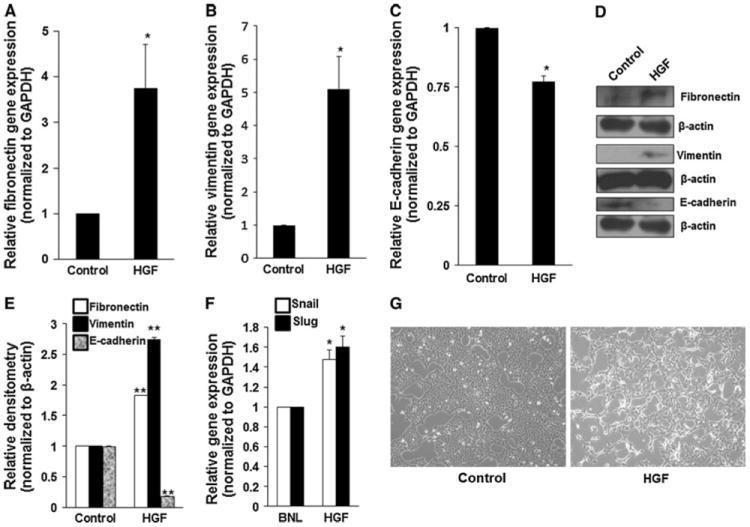
HGF promotes mesenchymal characteristics. BNL cells were treated with 5 ng/ml mouse hepatocyte growth factor for 5 days. HGF treatment led to increased expression of fibronectin (a, d, e), vimentin (b, d, e), snail and slug (f) and decreased expression of E-cadherin (c, d, e). *P < 0.05; **P < 0.01, N = 3. g HGF promotes mesenchymal morphology in BNL cells
HGF stimulates increased gene and protein expression of COX-2, ERK and Akt
Since our data showed that HGF overexpression in 1MEA cells is associated with increased expression of COX-2, ERK and Akt, we tested the hypothesis that HGF actually directly stimulates increased expression of these signaling intermediates. To do this, we examined the effect of HGF treatment on gene and protein expression of COX-2, ERK and Akt in BNL cells. We found that HGF treatment led to significantly increased expression of COX-2, ERK, p-ERK, Akt and p-Akt in BNL cells (Fig. 6). Further, selective inhibition of COX-2, Akt but not ERK reversed the HGF-induced expression of vimentin in BNL cells (Fig. 6).
Fig. 6.
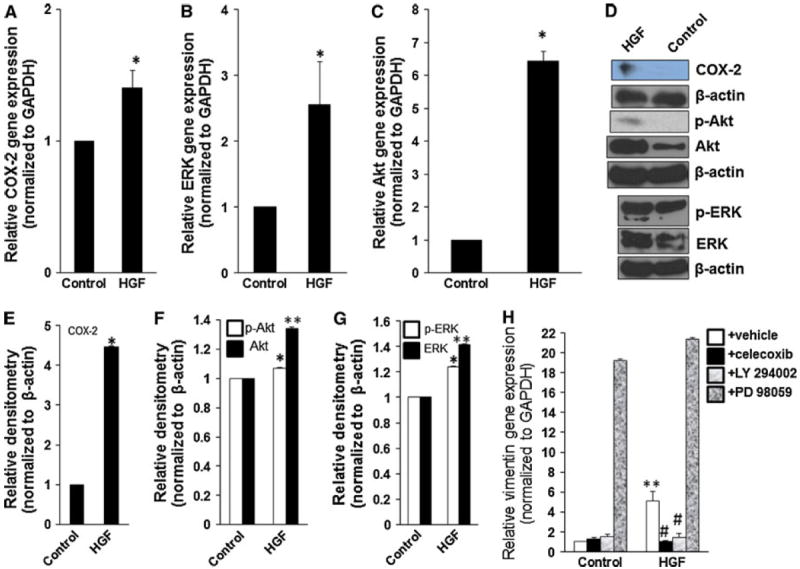
HGF stimulates increased expression of COX-2, ERK and Akt. BNL cells were treated with 5 ng/ml mouse hepatocyte growth factor for 5 days. HGF treatment led to increased expression of COX-2 (a, d, e), p-ERK and ERK (b, d, g) and p-Akt and Akt (c, d, f). h Selective inhibitors of COX-2 (1 μM celecoxib) and Akt (10 μM LY 294002) but not ERK (25 μM PD 98059) reversed HGF-induced gene expression of vimentin.*P < 0.05; **P < 0.01, N = 3
HGF promotes increased migration, invasion and clonogenicity
To determine if the increased expression of HGF by 1MEA cells in comparison to BNL cells affects the migratory capacity, we compared the migratory capacity of both cell lines using the wound healing migration assay and found that 1MEA cells have increased migratory capacity. We also found that treatment of 1MEA cells with selective inhibitors of COX-2 (1 μM celecoxib), and Akt (10 μM LY 294002) but not ERK (25 μM PD 98059) significantly inhibited the migratory capacity of 1MEA cells (Fig. 7a, b). Further, the direct functional effect of HGF on migratory capacity was examined by treating BNL cells with HGF and examining their migratory capacity using wound healing migration assays. These assays showed that HGF stimulated BNL cells to acquire significantly increased migratory capacity (Fig. 7c, d, e). Also, 1MEA cells are more invasive and clonogenic than BNL cells. HGF stimulated invasiveness and clonogenicity in BNL cells. Significantly, 1MEA cells form tumors in mice but BNL cells do not (Fig. 7; Supplementary Fig. 3).
Fig. 7.
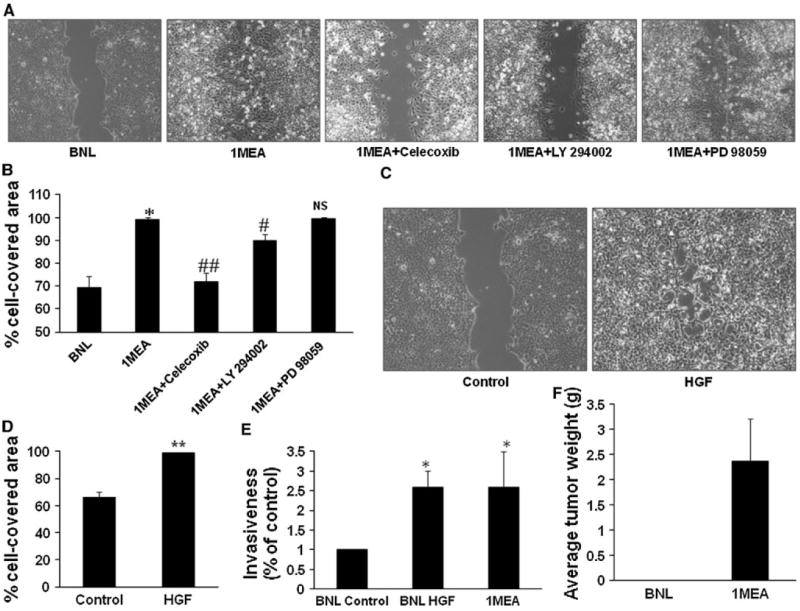
HGF promotes increased migration and invasiveness. a Migratory capacity of BNL and 1MEA cells were compared with wound healing assays. Selective inhibitors of COX-2 (1 μM celecoxib), ERK (25 μM PD 98059) and Akt (10 μM LY 294002) were used to determine the underlying mechanisms of 1MEA migration. Images (×10 objective) were taken after 24 h of culture. Images from three experiments were analyzed for percentage cell-covered area using the Wimasis Image Analysis software (Wimasis GmbH, Munich, Germany). Data is presented graphically in b. c Effect of HGF on migration was examined with wound healing assays in BNL cells. Left panel shows control cells and right panel shows cells treated with 5 ng/ml mouse hepatocyte growth factor. Image (×10 objective) was taken after 24 h. Images from three experiments were analyzed for percentage cell-covered area using the Wimasis Image Analysis software (Wimasis GmbH, Munich, Germany). Data is presented graphically in d. e Effect of HGF on BNL cell invasiveness was further examined using the in vitro Transwell double chamber invasion method. Cells that had invaded through collagen to the under surface of the insert were counted and data was analyzed and presented graphically in e. f 1MEA cells develop tumors but BNL cells do not. *P < 0.05; **P < 0.01, N = 3
Discussion
From the foregoing, it is evident that HGF upregulation in the 1MEA cancer cell line in comparison to its parent normal hepatocyte cell line, BNL, is directly related to the acquisition of mesenchymal and carcinogenic characteristics. This study proves, for the first time, that the underlying signaling mechanisms for this effect involve the Akt and COX-2 pathways.
BNL is a normal hepatocyte cell line derived from liver of BALB/c mice that is not tumorigenic when injected into mice. The 1MEA cell line is a HCC cell line that was directly derived from the BNL cell line by chemical transformation and is tumorigenic when injected into mice [37, 38]. Comparison of these two cell lines is, therefore, an excellent model for determining factors that may promote carcinogenesis in HCC. Moreover, our initial observations of morphology using both simple light microscopy and hematoxylin and eosin staining of both cell lines suggested that there is also a morphological difference between these cell lines. We observed that the BNL cells have typical epithelial morphology but 1MEA cells have mesenchymal morphology. This conclusion was further strengthened when our molecular characterization revealed that 1MEA cells have very clear mesenchymal characteristics (loss of E-cadherin, loss of albumin and gain of collagen-I, fibronectin and vimentin) in comparison to its parent cell line, BNL. Further, 1MEA cells are more migratory, invasive and clonogenic than BNL cells. Importantly, 1MEA cells are tumorigenic but BNL cells are not. These observations make the BNL vs. 1MEA model an excellent one for studying the factors that may promote EMT in HCC.
The difference in HGF expression between the two cell lines is striking. 1MEA cells express 200-fold more HGF than BNL cells. Further, 1MEA cells secrete over 17-fold more HGF than BNL cells. Taken together, these data suggest that HGF is a significant factor promoting the mesenchymal and tumorigenic properties of 1MEA cells mentioned above. This conclusion is supported by the findings of another recent study that studied the role of EMT in HCC [22]. Using a single clone of CD133 positive and CD45 negative cells isolated from the liver of Ptenloxp/loxp/Alb-Cre+ mice (P0), they sequentially transplanted them into mice and obtained two further passages (P1 and P2). Analysis of all three passages of cells (P0, P1 and P2) revealed that some P2 cells had acquired mesenchymal phenotype and secreted high levels of HGF. Further, P2 cells formed tumors that were more aggressive, invasive and metastatic [22]. Combining these data with ours, therefore, leads to the conclusion that HGF is a major driver of EMT and cancer progression in HCC. To prove that HGF actually plays a direct role in the acquisition of mesenchymal characteristics as the BNL cell line progressed to 1MEA we performed some proof of concept studies where we treated BNL cells with HGF. The result was that the BNL cells acquired mesenchymal properties (loss of E-cadherin expression and gain of fibronectin and vimentin expression and increased migratory, invasive and clonogenic capacity) thus proving that HGF plays a causal role in the mesenchymal and tumorigenic properties of 1MEA.
The signaling mechanisms underlying this HGF-induced EMT and carcinogenesis in HCC were previously unknown. We chose to examine whether Akt or ERK or COX-2 are possible mediating mechanisms because of their established association with progression of many cancers including HCC [23, 28, 31]. Our data show that the increased expression of HGF, mesenchymal and carcinogenic characteristics by 1MEA cells in comparison to the parent BNL cells is associated with significantly increased expression of Akt gene and protein, p-Akt protein and COX-2 gene and protein. Moreover, selective inhibitors of Akt, and COX-2 reversed the mesenchymal characteristics and migratory capacity of 1MEA cells. Although p-ERK protein expression was increased in 1MEA and the selective inhibitor of ERK was able to reverse the molecular mesenchymal characteristics of 1MEA cells, it was unable to inhibit its increased migratory capacity. The current data further show that HGF plays a direct causal role because HGF treatment also led to significantly increased protein expression of p-Akt, COX-2 and p-ERK by BNL cells. HGF-induced expression of vimentin in BNL cells was inhibited by inhibitors of COX-2 and Akt but not ERK. Taken together, these data strongly suggest that HGF-induced mesenchymal and carcinogenic properties in HCC is mediated by signaling via Akt and COX-2 pathways. The importance of ERK pathway as a mechanism in this model is unclear because of the inability of ERK inhibition to inhibit 1MEA migration and to reverse HGF-induced expression of vimentin. Further studies are required to establish whether ERK plays a role in HCC.
The determination that Akt and COX-2 pathways are involved in HGF-induced EMT and carcinogenesis in HCC is particularly important because it suggests that these intermediates are potential molecular targets for HCC therapy. The current outcome of advanced HCC treatment is largely dismal. The currently used multi-kinase inhibitor, sorafenib, is largely ineffective. It may be that activation of Akt and COX-2 pathways by HGF are important mechanisms in many patients with advanced HCC that account for the failure of sorafenib in these patients. Previous studies suggest that growth factor-induced intracellular signaling is sometimes organized into a cascade. For example, leptin signaling in esophageal adenocarcinoma involves sequential activation of a number of signaling intermediates wherein ERK and Akt activation are upstream of COX-2 [31]. A similar pathway was noted in sphingosine-1-phosphate activity in vascular smooth muscle cells [39]. Our data also shows that inhibition of ERK but not Akt activation also led to a significant inhibition of COX-2 expression. Consequently, it appears that COX-2 is downstream of ERK activation in this model of EMT in HCC. Therefore, inhibition of COX-2 may be a potentially effective treatment option in HCC, either as a chemopreventative or adjuvant therapeutic strategy. This is an especially attractive possibility that should be tested out clinically since many selective COX-2 inhibitors (including celecoxib that we used in our experiments) are currently in clinical use and have a relatively good safety profile. Moreover, routine use of selective COX-2 inhibitors may have chemopreventative benefits in HCC. Our data suggest that this possibility deserves further investigation.
In conclusion, our current data show that HGF directly promotes EMT and carcinogenic properties in HCC via separate activation of Akt and COX-2 pathways. ERK activation may be upstream of COX-2 in the process of EMT in HCC. Specific molecular targeting of these pathways may have clinical therapeutic and chemopreventative benefits in HCC.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health grant R01CA133086 to Chen Liu. Olorunseun O. Ogunwobi is a Postdoctoral Fellow on a NIH T32 grant.
Abbreviations
- COX-2
Cyclooxygenase-2
- ERK
Extracellular signal regulated kinase
- HGF
Hepatocyte growth factor
- DMSO
Dimethyl sulfoxide
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s10585-011-9404-x) contains supplementary material, which is available to authorized users.
Conflicts of interest None.
References
- 1.Bosch FX, Ribes J, Diaz M, et al. Primary liver cancer: worldwide incidence and trends. Gastroenterology. 2004;127(5):S5–S16. doi: 10.1053/j.gastro.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 2.Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer. 2009;9(4):265–273. doi: 10.1038/nrc2620. [DOI] [PubMed] [Google Scholar]
- 3.Yang J, Mani SA, Donaher JL, et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117(7):927–939. doi: 10.1016/j.cell.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 4.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119(6):1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Voulgari A, Pintzas A. Epithelial-mesenchymal transition in cancer metastasis: mechanisms, markers and strategies to overcome drug resistance in the clinic. Biochim Biophys Acta. 2009;1796(2):75–90. doi: 10.1016/j.bbcan.2009.03.002. [DOI] [PubMed] [Google Scholar]
- 6.Kaartinen V, Voncken JW, Shuler C, et al. Abnormal lung development and cleft palate in mice lacking TGF-beta-3 indicates defects of epithelial-mesenchymal interaction. Nat Genet. 1995;11(4):415–421. doi: 10.1038/ng1295-415. [DOI] [PubMed] [Google Scholar]
- 7.Khoury H, Dankort DL, Sadekova S, et al. Distinct tyrosine autophosphorylation sites mediate induction of epithelial mesenchymal like transition by an activated ErbB-2/Neu receptor. Oncogene. 2001;20(7):788–799. doi: 10.1038/sj.onc.1204166. [DOI] [PubMed] [Google Scholar]
- 8.Valles AM, Boyer B, Badet J, et al. Acidic fibroblast growth factor is a modulator of epithelial plasticity in a rat bladder carcinoma cell line. Proc Natl Acad Sci USA. 1990;87(3):1124–1128. doi: 10.1073/pnas.87.3.1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Naldini L, Weidner KM, Vigna E, et al. Scatter factor and hepatocyte growth factor are indistinguishable ligands for the MET receptor. EMBO J. 1991;10(10):2867–2878. doi: 10.1002/j.1460-2075.1991.tb07836.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weidner KM, Hartmann G, Naldini L, et al. Molecular characteristics of HGF-SF and its role in cell motility and invasion. EXS. 1993;65:311–328. [PubMed] [Google Scholar]
- 11.Breuhahn K, Longerich T, Schirmacher P. Dysregulation of growth factor signaling in human hepatocellular carcinoma. Oncogene. 2006;25:3787–3800. doi: 10.1038/sj.onc.1209556. [DOI] [PubMed] [Google Scholar]
- 12.Zeisberg M, Yang CQ, Martino M, et al. Fibroblasts derive from hepatocytes in liver fibrosis via epithelial to mesenchymal transition. J Biol Chem. 2007;282(32):23337–23347. doi: 10.1074/jbc.M700194200. [DOI] [PubMed] [Google Scholar]
- 13.Dooley S, Hamzavi J, Ciuclan L, et al. Hepatocyte-specific Smad7 expression attenuates TGF-beta-mediated fibrogenesis and protects against liver damage. Gastroenterology. 2008;135(2):642–659. doi: 10.1053/j.gastro.2008.04.038. [DOI] [PubMed] [Google Scholar]
- 14.Kaimori A, Potter J, Kaimori JY, et al. Transforming growth factor-beta 1 induces an epithelial-to-mesenchymal transition state in mouse hepatocytes in vitro. J Biol Chem. 2007;282(30):22089–22101. doi: 10.1074/jbc.M700998200. [DOI] [PubMed] [Google Scholar]
- 15.Lazarevich N, Cheremnova OA, Varga EV, et al. Progression of HCC in mice is associated with a downregulation in the expression of hepatocyte nuclear factors. Hepatology. 2004;39(4):1038–1047. doi: 10.1002/hep.20155. [DOI] [PubMed] [Google Scholar]
- 16.Gotzmann J, Fischer ANM, Zojer M, et al. A crucial function of PDGF in TGF-beta-mediated cancer progression of hepatocytes. Oncogene. 2006;25(22):3170–3185. doi: 10.1038/sj.onc.1209083. [DOI] [PubMed] [Google Scholar]
- 17.van Zijl F, Zulehner G, Petz M, et al. Epithelial-mesenchymal transition in hepatocellular carcinoma. Future Oncol. 2009;5(8):1169–1179. doi: 10.2217/fon.09.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fischer ANM, Herrera B, Mikula M, et al. Integration of Ras subeffector signaling in TGF-beta mediated late stage hepatocarcinogenesis. Carcinogenesis. 2005;26(5):931–942. doi: 10.1093/carcin/bgi043. [DOI] [PubMed] [Google Scholar]
- 19.Balaban YH, Us D, Hascelik G, et al. Hepatocellular carcinoma and cholangiocarcinoma are associated with high serum levels of hepatocyte growth factor. Indian J Gastroenterol. 2006;25(4):223–224. [PubMed] [Google Scholar]
- 20.Xie Q, Liu KD, Hu MY, et al. SF/HGF-c-Met autocrine and paracrine promote metastasis of hepatocellular carcinoma. World J Gastroenterol. 2001;7(6):816–820. doi: 10.3748/wjg.v7.i6.816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Son G, Hirano T, Seki E, et al. Blockage of HGF/c-Met system by gene therapy (adenovirus-mediated NK4 gene) suppresses hepatocellular carcinoma in mice. J Hepatol. 2006;45(5):688–695. doi: 10.1016/j.jhep.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 22.Ding W, You HN, Dang H, et al. Epithelial-to-mesenchymal transition of murine liver tumor cells promotes invasion. Hepatology. 2010;52(3):945–953. doi: 10.1002/hep.23748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Whittaker S, Marais R, Zhu AX. The role of signaling pathways in the development and treatment of hepatocellular carcinoma. Oncogene. 2010;29(36):4989–5005. doi: 10.1038/onc.2010.236. [DOI] [PubMed] [Google Scholar]
- 24.Matsumoto K, Nakamura T. Emerging multipotent aspects of hepatocyte growth factor. J Biochem. 1996;119(4):591–600. doi: 10.1093/oxfordjournals.jbchem.a021283. [DOI] [PubMed] [Google Scholar]
- 25.Li XY, Zhan XR, Lu C, et al. Mechanisms of hepatocyte growth factor-mediated signaling in differentiation of pancreatic ductal epithelial cells into insulin-producing cells. Biochem Biophys Res Commun. 2010;398(3):389–394. doi: 10.1016/j.bbrc.2010.06.078. [DOI] [PubMed] [Google Scholar]
- 26.Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103(2):211–225. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- 27.Moore AE, Greenhough A, Roberts HR, et al. HGF/Met signalling promotes PGE(2) biogenesis via regulation of COX-2 and 15-PGDH expression in colorectal cancer cells. Carcinogenesis. 2009;30(10):1796–1804. doi: 10.1093/carcin/bgp183. [DOI] [PubMed] [Google Scholar]
- 28.Abiru S, Nakao K, Ichikawa T, et al. Aspirin and NS-398 inhibit hepatocyte growth factor-induced invasiveness of human hepatoma cells. Hepatology. 2002;35(5):1117–1124. doi: 10.1053/jhep.2002.32676. [DOI] [PubMed] [Google Scholar]
- 29.Gonzales AJ, Goldsworthy TL, Fox TR. Chemical transformation of mouse liver cells results in altered cyclin D CDK protein complexes. Carcinogenesis. 1998;19(6):1093–1102. doi: 10.1093/carcin/19.6.1093. [DOI] [PubMed] [Google Scholar]
- 30.Ogunwobi OA, Beales ILP. Glycine-extended gastrin stimulates proliferation and inhibits apoptosis in colon cancer cells via cyclo-oxygenase-independent pathways. Regul Pept. 2006;134(1):1–8. doi: 10.1016/j.regpep.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 31.Ogunwobi O, Mutungi G, Beales ILP. Leptin stimulates proliferation and inhibits apoptosis in Barrett’s esophageal adenocarcinoma cells by cyclooxygenase-2-dependent, prostaglandin-E2-mediated transactivation of the epidermal growth factor receptor and c-Jun NH2-terminal kinase activation. Endocrinology. 2006;147(9):4505–4516. doi: 10.1210/en.2006-0224. [DOI] [PubMed] [Google Scholar]
- 32.Zhu HZ, Dong HJ, Eksioglu E, et al. Hepatitis C virus triggers apoptosis of a newly developed hepatoma cell line through antiviral defense system. Gastroenterology. 2007;133:1649–1659. doi: 10.1053/j.gastro.2007.09.017. [DOI] [PubMed] [Google Scholar]
- 33.Behrens J, Mareel MM, Vanroy FM, et al. Dissecting tumor cell invasion—epithelial cellsacquireinvasivepropertiesafterthe lossofuvomorulin-mediatedcell-cell adhesion. J Cell Biol. 1989;108(6):2435–2447. doi: 10.1083/jcb.108.6.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thompson EW, Torri J, Sabol M, et al. Oncogene-induced basement membrane invasiveness in human mammary epithelial cells. Clin Exp Metastasis. 1994;12(3):181–194. doi: 10.1007/BF01753886. [DOI] [PubMed] [Google Scholar]
- 35.Guan F, Handa K, Hakomori SI. Specific glycosphingolipids mediate epithelial-to-mesenchymal transition of human and mouse epithelial cell lines. Proc Natl Acad Sci USA. 2009;106(18):7461–7466. doi: 10.1073/pnas.0902368106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Campard D, Lysy PA, Najimi M, et al. Native umbilical cord matrix stem cells express hepatic markers and differentiate into hepatocyte-like cells. Gastroenterology. 2008;134(3):833–848. doi: 10.1053/j.gastro.2007.12.024. [DOI] [PubMed] [Google Scholar]
- 37.Har CH, Keong CK. Effects of tocotrienols on cell viability and apoptosis in normal murine liver cells (BNL CL.2) and liver cancer cells (BNL 1ME A.7R.1), in vitro. Asia Pac J Clin Nutr. 2005;14(4):374–380. [PubMed] [Google Scholar]
- 38.Shangguan DH, Meng L, Cao ZHC, et al. Identification of liver cancer-specific aptamers using whole live cells. Anal Chem. 2008;80(3):721–728. doi: 10.1021/ac701962v. [DOI] [PubMed] [Google Scholar]
- 39.Hsieh HL, Wu CB, Sun CC, et al. Sphingosine-1-phosphate induces COX-2 expression via PI3K/Akt and p42/p44 MAPK pathways in rat vascular smooth muscle cells. J Cell Physiol. 2006;207(3):757–766. doi: 10.1002/jcp.20621. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.