

Abstract
We describe seven Turkish children with DOCK8 deficiency who have not been previously reported. Three patients presented with typical features of recurrent or severe cutaneous viral infections, atopic dermatitis, and recurrent respiratory or gastrointestinal tract infections. However, four patients presented with other features. Patient 1-1 featured sclerosing cholangitis and colitis; patient 2-1, granulomatous soft tissue lesion and central nervous system involvement, with primary central nervous system lymphoma found on follow-up; patient 3-1, a fatal metastatic leiomyosarcoma; and patient 4-2 showed no other symptoms initially besides atopic dermatitis. Similar to other previously reported Turkish patients, but in contrast to patients of non-Turkish ethnicity, the patients’ lymphopenia was primarily restricted to CD4+ T cells. Patients had homozygous mutations in DOCK8 that altered splicing, introduced premature terminations, destabilized protein, or involved large deletions within the gene. Genotyping of remaining family members showed that DOCK8 deficiency is a fully penetrant, autosomal recessive disease. In our patients, bone marrow transplantation resulted in rapid improvement followed by disappearance of viral skin lesions, including lesions resembling epidermodysplasia verruciformis, atopic dermatitis, and recurrent infections. Particularly for patients who feature unusual clinical manifestations, immunological testing, in conjunction with genetic testing, can prove invaluable in diagnosing DOCK8 deficiency and providing potentially curative treatment.
Keywords: DOCK8, combined immunodeficiency, epidermodysplasia verruciformis, sclerosing cholangitis, CNS lymphoma, leiomyosarcoma
INTRODUCTION
DOCK8 deficiency is an autosomal recessive combined immunodeficiency that includes the autosomal recessive form of hyperimmunoglobulinemia E syndrome (HIES). [1–4] Loss-of-function mutations in the DOCK8 gene, which encodes for a poorly understood atypical guanine nucleotide exchange factor on human chromosome 9, underlie this disease. Like the autosomal dominant form of HIES caused by STAT3 mutations, which is also known as Job’s syndrome, DOCK8 deficiency exhibits eczema, recurrent pneumonias, elevated serum IgE, and sometimes boils and mucocutaneous candidiasis. However, unlike Job’s syndrome, DOCK8 deficiency features intractable cutaneous viral infections with associated cancers, widespread infections (sinusitis, otitis media, sepsis), and lacks dental, skeletal, or connective tissue abnormalities. Neurologic abnormalities such as CNS vasculitis have been reported [2, 5]. Immunological abnormalities include lymphopenia, defective antibody function, and variable serum immunoglobulin levels.
Despite distinctive clinical features, patients may present with unusual findings or may only be mildly affected, leading to diagnostic uncertainty. The cases reported here illustrate instances where additional immunological and genetic testing were useful.
METHODS
Clinical and Laboratory Evaluations
Seven patients with high clinical suspicion for DOCK8 deficiency but negative HIV testing were studied, along with their immediate family members. Subjects provided written informed consent and were investigated under Institutional Review Board approved research protocols in accordance with the Helsinki Declaration. Lymphocyte immunophenotyping by flow cytometry was performed on whole blood followed by RBC lysis. Lymphocyte proliferation to mitogens (PHA, ConA) was measured by 3H-thymidine incorporation. Serum immunoglobulin isotypes were quantitated by nephelometry, except for IgE which was measured by radioallergosorbent test. Antibody titers were measured by ELISA. All tests were performed in the Clinical Pathology Laboratory and the Pediatric Immunology Laboratory at Hacettepe University Hospital.
Mutational Analyses
DOCK8 sequencing and array comparative genomic hybridization (CGH) were performed on genomic DNA from peripheral blood mononuclear cells (PBMC) of probands and all living immediate family members, as described [1]. Where indicated, total RNA was isolated from PBMC or proliferating T cells and reverse transcribed into cDNA using SuperScript III© First Strand Synthesis SuperMix kit (Invitrogen). PCR amplification of cDNA spanning exons 19 to 24 was performed using primers 5′-TGTACACACCCAGGACAACC-3′ and 5′-ACCAGAAGCTCAAAGAAGAACC-3′. Amplified products were separated by gel electrophoresis, or used for cDNA sequencing [1]. Variants were named using GenBank Reference Sequences NC_000009.10 (DNA), NM_001193536.1 (mRNA; previously NM_203447.1), and NP_0011800465.1 (protein; previously NP_982272.1) based upon NCBI Build 36.3, which correspond to the isoform that is predominantly expressed in primary T cells (Genbank NM991479.1, HM991480.1). The deleterious effects of missense mutations were predicted using the PolyPhen-2 HumDiv, PolyPhen-2 HumVar, and SIFT software packages. Mutations were not found in 50 healthy Turkish normal controls.
Overexpression Studies
Full-length wildtype DOCK8, cloned in the pCMV6-Entry vector that has C-terminal fusion Myc and Flag epitope tags, was purchased from Origene (cat# CW100311). A C1447R mutant version of this construct was generated by site-directed mutagenesis, with confirmation by Sanger sequencing. In brief, PCR amplification of full length DOCK8 plasmid with primers containing the point mutation was performed using AccuPrime™ Pfx SuperMix (Invitrogen), and PCR products were treated with Dpn1 (New England Biolabs) followed by transformation into XL10-Gold Ultracompetent Cells (Stratagene). Plasmid DNA was purified by PureLink HiPure Filter Plasmid Kit (Invitrogen) and 1.5 μg transfected into 0.8 million human embryonic kidney 293T cells using Lipofectamine 2000 (Invitrogen). Cells were lysed in 2% SDS buffer, and 8 μg of protein lysate was separated per lane. Immunoblotting for DOCK8 or β-actin was otherwise performed as described [1]. DOCK8 expression was also confirmed by immunoblotting using the M2 monoclonal antibody specific to the Flag tag (Sigma-Aldrich).
Literature Review and Analyses
We analyzed data from DOCK8-deficient patients in this report and in previous publications[1, 2, 6–11]. We only included patients with DOCK8 mutations defined on both alleles (ARH001, 2, 3, 4, 5, 8, 9, 10.2, 10.4, 10.8, 10.9, 11.3, 12, 13.3, 13.4, 14, 15, 16.6, and 16.7) [2]. For estimation of disease penetrance, we analyzed data from all living family members of our additional patients, as well as data from a previous publication for the families of ARH008, 10, 11, and 12 [2]. The latter dataset included all living family members except for the parents of ARH010.2/10.3/10.4, ARH010.3, and ARH010.6. The younger sister of ARH011.3, ARH011.4, and ARH011.5 was genotyped as a heterozygous carrier for inclusion in the present analysis (H. Jing and H. C. Su, unpublished). For contingency table analyses of lymphopenia in Turkish versus non-Turkish populations, we analyzed data from DOCK8-deficient patients in this report and in previous publications where ethnicity was reported [1, 2, 7, 9–11]. We classified a patient as Turkish if this ethnicity was indicated, or as non-Turkish if a different ethnicity was indicated, and then used the two-tailed Fisher’s exact test as part of the Prism software package.
RESULTS
Patient Presentations
Our seven additional Turkish patients are from four consanguineous families, whose pedigrees are shown in Figure 1. Their demographics and clinical features are described in Table I. In general, patients had early onset of disease, which first manifested as atopic dermatitis between 3 months to 2 years of age. For very young infants, such as patient 4-2, atopic dermatitis was the only initial clinical manifestation. Atopic dermatitis was soon followed by recurrent otitis externa, otitis media, respiratory tract infections, gastrointestinal tract infections, and food allergies. Patients developed recurrent herpes simplex virus (HSV) infections including eczema herpeticum, disseminated molluscum contagiosum, or severe varicella-zoster virus infection. However, in four patients, additional skin lesions resembling epidermodysplasia verruciformis (EV) were diagnosed. Those patients presented with pinkish hypopigmented flat or atrophic papules (patient 1-1) (Figure 2), or tan to pink-colored macules and hyperkeratotic papules (patients 2-1, 3-1, and 3-2), over the trunk and extremities. Histopathology of representative lesions showed hyperkeratosis and acanthosis consistent with EV (Figure 2). This particular manifestation of human papillomavirus infections has not been previously described for DOCK8 deficiency.
Figure 1.

Pedigrees of the families studied. Genotypes are indicated below each individual, where “+” designates normal DOCK8 allele, and “m” or “Δ” designates mutated or deleted DOCK8 allele, respectively. Numbers about the pedigrees correspond to the family identifiers, whereas patient identifiers are indicated below the symbols. A closed symbol designates an affected individual, whereas an open symbol designates an unaffected individual. Squares designate males; circles, females; arrows, probands; and slashes, deceased individuals.
Table I.
Summary of the seven additional DOCK8-deficient patients studied
| Demographics | |||||||
|---|---|---|---|---|---|---|---|
| Patient | 1-1 | 2-1 | 2-2 | 3-1 | 3-2 | 4-1 | 4-2 |
| Age (yr)† | 15 | 12† | 8 | 8† | 5 | 9 | 2.5 |
| Sex | Male | Female | Male | Female | Female | Female | Male |
| Age of onset of symptoms (yr) | 2 | 0.5 | unknown | 0.5 | 1.75 | 0.25 | 0.58 |
| Clinical Features | |||||||
| Skin | |||||||
| Atopic dermatitis | (+) | (+) | (+) | (+) | (+) | (+) | (+) |
| Lesions resembling epidermodysplasia verruciformis | (+) | (+) | (+) | (+) | (−) | (−) | (−) |
| Recurrent Herpes Simplex Virus infections | (−) | (+) | (+) | (−) | (+) | (+) | (+) |
| Severe primary varicella | (−) | (−) | (−) | (−) | (+) | (−) | (−) |
| Molluscum contagiosum | (−) | (−) | (−) | (−) | (−) | (+) | (+) |
| Other | Seborrheic dermatitis | Eczema herpeticum | |||||
| Respiratory tract | |||||||
| Recurrent respiratory tract infections | (+) | (+) | (+) | (+) | (+) | (+) | (+) |
| Otitis media | (−) | (+) | (+) | (−) | (+) | (−) | (−) |
| Mastoiditis | (−) | (+) | (+) | (−) | (−) | (−) | (−) |
| Sinusitis | (−) | (+) | (+) | (−) | (−) | (+) | (−) |
| Pneumonia/ bronchitis | (+) | (+) | (−) | (+) | (+) | (+) | |
| Bronchiectasis | (−) | (+) | (−) | (−) | (−) | (−) | (−) |
| Otitis externa | (+) | (−) | unknown | (−) | (+) | (−) | (−) |
| Gastrointestinal tract | |||||||
| Poor growth | (+) | (−) | (−) | (−) | (−) | (+) | (−) |
| Chronic diarrhea | (+) | (−) | (−) | (+) | (−) | (+) | (−) |
| Giardiasis | (+) | (−) | unknown | (−) | (−) | (+) | (−) |
| Cryptosporidium infections | (−) | (−) | (−) | (−) | (−) | (+) | (−) |
| Other | Colitis, sclerosing cholangitis, portal vein dilation | Buccal granulomatous inflammation and ulceration | Oral candidiasis, Sclerosing cholangitis | ||||
| Neurological | (−) | Presented with nystagmus, spasticity, ataxia, due to CNS lymphoma | (−) | (−) | (−) | (−) | (−) |
| Allergies or specific IgE | (−) | (−) | Egg | ND | Egg, lentil, milk | Egg, hazelnut, milk, peanut | Egg, hazelnut, milk |
| Malignancy | Diffuse large B cell CNS lymphoma | Metastatic multifocal leiomyosarcoma | |||||
| Other | Recently underwent matched related donor HCT | Recently underwent matched related donor HCT at another center | Underwent matched related donor HCT | ||||
Age at death.
ND, not determined
Figure 2.

Skin lesions in patient 1-1. Human papillomavirus-associated pinkish, hypopigmented flat papules on dorsal hands (A) and buttocks (B). C) Histopathology shows hyperkeratosis and acanthosis, with vacuolated cells in the stratum malpighii (arrow), keratinocytes having abundant slightly basophilic cytoplasm (small arrow), and occasional large, round, empty-appearing nuclei (arrowhead). Patients 2-1, 2-2, 3-1, and 3-2 had similar lesions.
Some cases initially drew clinical attention away from the immune system. Patients 1-1 and 3-1 had prominent gastrointestinal symptoms. Patient 1-1 had poor growth, chronic diarrhea, and active colitis as revealed by endoscopy. Magnetic resonance (MR) cholangiopancreatography showed portal vein dilatation, sclerosing cholangitis, and thickened pancreatic parenchyma, which may be caused by persistent giardiasis. Patient 4-1 also developed sclerosing cholangitis in the setting of persistent cryptosporidium and giardia infections. Patient 3-1, who was previously reported without a molecular diagnosis, was 6 years old when admitted with chronic abdominal pain, bloody stools, weight loss, and hepatomegaly [12]. Imaging revealed multiple nodules within liver lobes, gallbladder wall, left suprarenal retroperitoneum, right lower quadrant intestines, and lungs, and she was diagnosed with metastatic multifocal leiomyosarcomatosis. She died 13 months later after limited surgical resection plus treatment with cisplatin, adriamycin, vincristine, and cyclophosphamide.
Another unusual case was illustrated by Patient 2-1 who had prominent neurological features. Patient 2-1 presented at age 9 with a 3x4 cm granulomatous lesion on her buccal mucosa. Histopathology of buccal mucosa showed chronic granulomatous inflammation and ulceration. Magnetic resonance imaging (MRI) demonstrated a T2-hyperintense lesion in the left buccal region and retromolar trigone. The lesion had an ill-defined margin and showed enhancement on post-Gadolinium (Gd) series (Figure 3A, B). The oral lesion improved after debridement and treatment with corticosteroids. However, during follow-up she developed dizziness and unsteadiness while walking. She displayed nystagmus, spasticity, increased deep tendon and pathological reflexes bilaterally, and a wide-based spastic-ataxic gait. Cranial MRI revealed bilateral pulvinar T2 hyperintensities on T2-weighted imaging and cerebellar and cerebral volume loss (Figure 3C). BK and JC viral studies were negative. Over the subsequent nine months she partially improved without treatment, but repeat cranial MRI disclosed a lesion that showed rim enhancement and restricted diffusion on the left temporo-occipital parenchyma (Figure 3D, 3E). Imaging studies were suggestive of an abscess and she was treated with wide-spectrum antibiotics and antifungal agents. Despite these treatments, her course was progressive. Histopathologic examination of excisional biopsy via craniotomy performed 1.8 years after the onset of neurological symptoms demonstrated an EBNA+ diffuse large B cell lymphoma. The patient received systemic chemotherapy with high-dose methotrexate, cyclophosphamide, vincristine, prednisolone, intrathecal chemotherapy, and cranial irradiation. She died four months after the diagnosis of CNS lymphoma due to progressive disease.
Figure 3.

Cranial MRI in patient 2-1. A) Transverse T2 weighted (W) fat-suppressed (left) and B) contrast enhanced (ce) fat-suppressed T1W (right) spin-echo (SE) images, at initial presentation. A T2-hyperintense and Gadolinium-enhancing lesion with irregular border in the left buccal space and retromolar trigone is seen (arrowheads in A, B). C). Transverse T2W turbo SE images, at five months, show bilateral pulvinar T2 hyperintensities (arrowheads) and cerebral volume loss. D) Axial Gadolinium-enhanced T1W SE shows a lesion with rim enhancement (long arrow). E) Axial diffusion-weighted imaging (DWI), at eighteen months, shows hyperintensity in the parenchyma and in the lesion, especially at the periphery.
Patients 1-1, 3-2, and 4-1 were treated with intravenous immunoglobulin (IVIG), which markedly decreased their frequency of infections and improved their skin lesions, including those associated with HSV. Additionally, patients 4-2 and 1-1, who had matched related donors available, underwent bone marrow transplantation 10 and 2 months ago, respectively. Patient 4-2 is now healthy with resolution of atopic dermatitis, viral and other recurrent infections. In patient 1-1, skin lesions started to improve during pre-conditioning chemotherapy and continued to clear after bone marrow transplantation. For both patient 4-2 and 1-1, details of their clinical progress after transplantation will be reported in a separate manuscript. Patient 2-2 underwent bone marrow transplantation at another medical center several weeks ago. Patients 3-2 and 4-1 lack matched donors and have not yet been transplanted.
Immunological Evaluations
Standard immunological evaluations were performed on all patients with the exception of patient 2-2, for whom we were unable to obtain peripheral blood. Values are shown in Figure 4 and summarized in Table II. Determinations for Table II were made based upon the majority of readings for any given patient, or if only two readings were available, their average. All tested patients had low numbers of CD4+ T cells and high serum IgE (peaks ranged from 412 to 20,000 IU/mL). Nearly all had decreased serum IgM, variably impaired specific antibodies, and mild to moderate eosinophilia. Several patients had T cell lymphopenia. However, lymphocyte proliferation to T cell mitogens was within normal limits, except for patient 3-1 who already had multifocal leiomyosarcomatosis when tested. Serum IgG and IgA were elevated in several patients. NK cell numbers were decreased in one patient, but neither CD8+ T cells nor B cells were consistently decreased in any patient, and in a few patients B cells were instead increased. Retrospective statistical analysis of all patients in the literature in which ethnicity was reported showed that lymphopenia affecting CD4+ T cells, CD8+ T cells, and B cells occurred less frequently in Turkish as compared to non-Turkish patients (Table III). Overall, immunological findings were similar to previous observations in other DOCK8-deficient patients, except that in the Turkish cohort lymphopenia occurred less frequently and was primarily restricted to CD4+ T cells, and T lymphocyte proliferation to mitogens was normal.
Figure 4.

Peripheral blood enumerations of lymphocyte subsets, quantitative serum immunoglobulin isotypes, and eosinophils. Patient 1-1, open circle; patient 2-1, open square; patient 3-1, cross; patient 3-2, X; patient 4-1, open inverted triangle; patient 4-2, filled-in circle. Shaded in grey are the ranges for normal age-matched controls. For lymphocytes and their subsets, published 10th to 90th percentiles are shown in grey. For immunoglobulin isotypes, the 95 % confidence interval is shown. For eosinophils, arbitrarily thresholds for mild, moderate, and severe eosinophilia are as indicated.
Table II.
Laboratory evaluation of additional DOCK8-deficient patients
| Patient | 1-1 | 2-1 | 2-2 | 3-1 | 3-2 | 4-1 | 4-2 |
|---|---|---|---|---|---|---|---|
| Lymphocyte numbers | Normal | Normal | ND | ↓ | Normal | ↓ | Normal |
| T cells | ↓ | Normal | ND | ↓ | ↓ | Normal | ↓ |
| CD4 T cells | ↓ | ↓ | ND | ↓ | ↓ | ↓ | ↓ |
| CD8 T cells | Normal | ↑ | ND | Normal | Normal | Normal | Normal |
| NK cells | Normal | ↑ | ND | ↓ | ↓ | Normal | Normal |
| B cells | Normal | Normal | ND | Normal | ↑ | Normal | ↑ |
| Lymphocyte proliferation to T cell mitogens (PHA, ConA) | Normal | Normal | ND | ↓ | Normal | Normal | Normal |
| Eosinophil numbers | ↑ | ↑ | ND | Normal | ↑ | ↑ | ↑ |
| IgG | ↑ | ↑ | ND | ↑ | ↑ | Normal | Normal |
| IgA | ↑ | ↑ | ND | ↓ | Normal | ↑ | Normal |
| IgM | ↓ | ↓ | ND | Normal | ↓ | ↓ | ↓ |
| IgE | ↑ | ↑ | ND | ↑ | ↑ | ↑ | ↑ |
| Hepatitis B virus surface Ab | (−) | ND | ND | (+) | ND | (−) | (+) |
| Rubella IgG | (+) | ND | ND | ND | (−) | ND | (−) |
| Cytomegalovirus IgG | (+) | ND | ND | ND | (+) | ND | ND |
| Epstein-Barr Virus viral capsid antigen IgG | (−) | ND | ND | (+) | ND | ND | (+) |
| Hepatitis A Virus IgG | (+) | ND | ND | ND | ND | ND | (+) |
| Herpes Simplex Virus-1 IgG | (+) | (+)* | ND | ND | ND | (+) | (+)* |
| Varicella-Zoster Virus IgG | (−) | (+) | ND | ND | ND | ND | (−) |
| Genetic mutation in DOCK8 | Homozygous c.2401G>C → p.A746_M800del | Homozygous c.4698C>G → p.Thr1566X plus c.2389A>G → p.Val797Met | Homozygous c.4698C>G → p.Thr1566X plus c.2389A>G → p.Val797Met | Homozygous c.4339T>C → p.Cys1447Arg | Homozygous c.4339T>C → p.Cys1447Arg | Chr9:g.(153,190_ 194,193)_(315,7 72_322,456) homozygous deletion spanning from exons 1 to 9 | Chr9:g.(153,190_ 194,193)_(315,7 72_322,456) homozygous deletion spanning from exons 1 to 9 |
Age at death.
ND, not determined.
IgG to HSV-2 also detected
Table III.
Lymphopenia in Turkish as compared to non-Turkish DOCK8-deficient patients
| Turkish | Non-Turkish | ||||||
|---|---|---|---|---|---|---|---|
| Lymphocyte subset | # Affected | # Unaffected | % Affected | # Affected | # Unaffected | % Affected | P-value* |
| CD4 T cells | 15 | 8 | 65 | 24 | 0 | 100 | 0.0016 |
| CD8 T cells | 5 | 17 | 23 | 16 | 8 | 67 | 0.0037 |
| NK cells | 8 | 14 | 36 | 12 | 10 | 55 | NS |
| B cells | 0 | 23 | 0 | 10 | 14 | 42 | 0.0006 |
Numbers of affected or unaffected DOCK8-deficient patients, who have lymphopenia of the indicated lymphocyte subsets.
P-value calculated by Fisher’s exact test.
NS, not statistically significant.
Genetic Evaluations
We found novel homozygous DOCK8 mutations and large deletions in the genomic DNA of our seven additional patients (Table II). These were predicted to alter splicing (family 1), introduce premature termination with probable nonsense-mediated decay (family 2), alter protein structure (family 3), or lead to loss of protein expression (family 4). For families 1 and 3, in which effects were not obvious, we performed additional experimental validation. We found that in family 1, the mutation in the consensus 5′ splice donor site of intron 20 yielded an aberrantly spliced, smaller product (Figure 5A). Sequencing of cDNA showed that this product corresponded to an in-frame deletion of 55 amino acids just after the conserved DHR1 domain (Table II). In family 3, the cysteine to arginine missense mutation in exon 35 between the conserved DHR1 and DHR2 domains was predicted to exert protein destabilizing effects. When equivalent amounts of plasmid encoding either mutant or wildtype DOCK8 were overexpressed in 293T cells (which do not endogenously express DOCK8), the level of mutant DOCK8 protein was markedly decreased (Figure 5B and data not shown). Thus, in all cases, the mutations resulted in loss of normal DOCK8 expression with predicted loss of function, as is characteristic of this disease.
Figure 5.
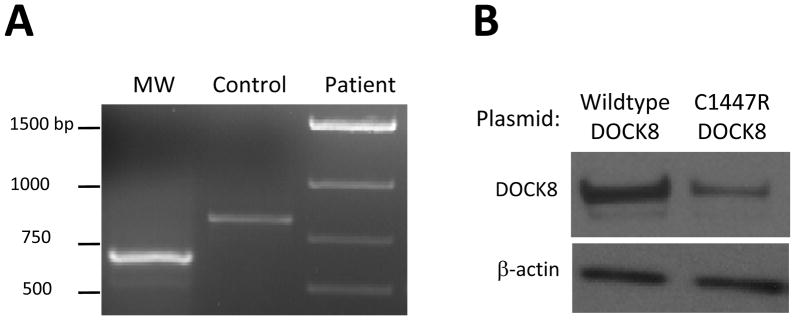
DOCK8 expression data for mutations in patients from families 1 and 3. A) PCR amplification of exons 19 to 24 of DOCK8 from cDNA shows a smaller product from patient 1-1, which corresponds to an aberrantly spliced mRNA. B) Immunoblotting shows destabilized DOCK8 protein in 293T cells that have been transiently transfected with a plasmid encoding the full-length mutant (C1447R), as compared to wildtype, DOCK8. Patients 3-1 and 3-2 only express this mutant form of DOCK8.
We next studied the immediate families of the seven patients (Figure 1). All living family members were healthy, with no history of serious viral infections, bacterial infections, skin infections, atopic dermatitis, asthma, allergies, or malignancy. The living family members were either homozygous for the normal DOCK8 gene or heterozygous carriers, and these genotyping results are shown in the pedigrees below each corresponding family member (Figure 1). A sibling of patient 1-1 died at 40 days of age with vomiting and abdominal distension. Because of a lack of additional clinical features to support a diagnosis of DOCK8 deficiency, we considered this death to be unrelated. By contrast, a sibling of patients 2-1 and 2-2 who had similar skin lesions died at age 10 years because of an intra-abdominal tumor, whom we judged as likely to have been affected with DOCK8 deficiency. No DNA samples were available from these two deceased individuals for genotyping.
It has been assumed that the autosomal recessive inheritance and lack of discernable symptoms in tested carriers are consistent with a fully penetrant model of disease. However, prior studies have been limited to four fully genotyped families, raising the possibility of ascertainment bias [2]. When data from those families are combined with data from our four additional families, we observed that all individuals either homozygous for the normal DOCK8 allele (n = 6) or heterozygous for the mutant DOCK8 allele (n = 26), who ranged from ages 4 to 49 years old, were healthy. Furthermore, of 15 individuals who were homozygous for the mutant DOCK8 allele, all 15 developed disease, supporting a disease penetrance of ~100%. Nevertheless, as illustrated by our patients, the range of disease manifestations varied, depending upon patient age.
DISCUSSION
DOCK8 mutations are responsible for a combined immunodeficiency that was formerly characterized as autosomal recessive HIES. A total of 52 patients reported in the literature, including our seven additional patients, had homozygous or compound heterozygous DOCK8 mutations [1, 2, 6–11]. Their most common findings are summarized in Table IV. Patients typically present with intractable cutaneous viral infections, atopic dermatitis often accompanied by otitis externa or Staphylococcus aureus skin infections, recurrent respiratory tract infections, mucocutaneous candidiasis, allergic disease, as well as high serum IgE, low serum IgM, eosinophilia, and lymphopenia [1, 2, 5]. Gastrointestinal infections, malignancies, and neurological abnormalities have been sometimes reported in DOCK8 deficiency. However, the EV-like skin lesions, liver and pancreatic abnormalities, leiomyosarcoma, and CNS lymphoma in our additional patients have not been previously described in this disease. Interestingly, the increasing constellation of features seemed to reflect increasing patient age, consistent with the progressive nature of this disease (Table I).
Table IV.
Frequency of findings in DOCK8-deficient patients reported in the literature
| Feature | Number of patients affected* | % of patients affected |
|---|---|---|
| Virus infections, 1 or more | 46 | 88 |
| HSV | 28 | 54 |
| MCV | 19 | 37 |
| HPV | 19 | 37 |
| VZV | 13 | 25 |
| Respiratory tract infections | 49 | 94 |
| Atopic dermatitis | 46 | 88 |
| Bacterial skin infections | 34 | 65 |
| Mucocutaneous candidiasis | 28 | 54 |
| Gastrointestinal tract infections | 6 | 12 |
| Cancers | 9 | 17 |
| Neurological abnormalities | 11 | 21 |
| Low serum IgM | 38§ | 75 |
| High serum IgE | 50§ | 98 |
| Eosinophilia | 46¶ | 92 |
| CD4 T cell lymphopenia | 42¶ | 84 |
| CD8 T cell lymphopenia | 22‡ | 45 |
| NK cell lymphopenia | 21† | 45 |
| B cell lymphopenia | 9¶ | 18 |
Total number of patients evaluated was 52 except where indicated.
of 51 patients evaluated.
of 50 patients evaluated.
of 49 patients evaluated.
of 47 patients evaluated.
The differential diagnosis for DOCK8 deficiency includes autosomal dominant HIES caused by STAT3 mutations, as well as other conditions that can be associated with high serum IgE, with or without lymphopenia, such as Comèl-Netherton syndrome, Wiskott-Aldrich syndrome (WAS), IPEX (Immunodysregulation, Polyendocrinopathy, Enteropathy, X-linked) syndrome, acquired immunodeficiency syndrome (AIDS), and severe combined immunodeficiency (SCID). Comèl-Netherton syndrome, due to autosomal recessive SPINK5 mutations, features a distinctive “bamboo” appearance of the hair shaft (trichorrhexis invaginata), as well as eczema, allergic disease, recurrent bacterial infections of the skin, respiratory and gastrointestinal tracts, particularly with Staphylococcus aureus, high IgE, and impaired functional antibodies [13]. WAS, due to mutations in the X-linked gene WAS, can present in boys with eczema, bloody diarrhea associated with thrombocytopenia, recurrent sinopulmonary infections, skin viral infections including HSV and molluscum contagiosum, autoimmunity, hematological malignancy, and immune abnormalities including high IgE, low IgM, variably impaired functional antibodies, and progressive T cell lymphopenia with decreased T cell proliferation [14]. IPEX, due to mutations in the X-linked gene FOXP3, also presents in boys with eczema, allergic disease, Staphylococcus aureus sepsis, high IgE, and eosinophilia [15]. AIDS can present with pruritic dermatitis, Staphylococcus aureus skin infections, mucocutaneous candidiasis, eosinophilia, high IgE, and CD4+ T cell and sometimes B cell lymphopenia [16]. “Leaky” SCID causing Omenn’s syndrome, usually due to autosomal recessive RAG1/RAG2 mutations, presents with erythroderma, recurrent infections, autoimmunity, eosinophilia, high IgE, low IgM, and variable lymphopenia [17]. Opportunistic infections such as CMV, Pneumocystis jirovecii, or EV-like skin lesions are also remarkable in WAS, AIDS, or SCID [18] and can occur in DOCK8 deficiency.
Other considerations in the differential diagnosis include EV and WHIM (Warts, Hypogammaglobulinemia, Infections, Myelokathexis) syndrome, which do not present with high serum IgE. WHIM syndrome, due to autosomal dominant CXCR4 mutations, can also present with lymphopenia involving B cells and sometimes T cells [19]. EV, an HPV genodermatosis, due to autosomal recessive EVER1/EVER2 mutations, generally does not feature cell-mediated immune defects [20]. It is possible that other case reports of EV with high IgE, low IgM, and lymphopenia, but lacking confirmatory mutational analyses, were instead cases of DOCK8 deficiency [21–23].
Given that the clinical phenotype of DOCK8 deficiency overlaps with many other disorders and also shows varied features among patients even within the same family, additional immunological testing can be a useful adjunct for diagnosis. However, our results, expanding upon previously published work, also demonstrates heterogeneity of the cellular phenotype among patients. For example, although CD4+ T cell lymphopenia appeared nearly universal in all patients, involvement of other lymphocyte subsets occurred less frequently in Turkish patients. Thus, in cases where immunological testing remains equivocal, genetic sequencing is essential to establish a diagnosis. Although most patients have large deletions, or smaller point mutations or indels that cause nonsense-mediated decay of mRNA or premature terminations, mutations that result in abnormal or destabilized protein with loss of normal DOCK8 expression also can occur. Identification of additional cases and research for disease-modifying genes or environmental factors will aid in future management of this clinically heterogeneous disease.
CONCLUSIONS
Our seven additional patients extend the clinical phenotype and help establish that DOCK8 deficiency is highly penetrant with variable multi-organ disease manifestations, and that disease complications, which include CNS involvement and development of malignancy, are associated with increasing patient age. Immunologically, our patients provide additional evidence of gene modifier or environmental factors that modulate the effects of lymphopenia on different cellular subsets. Furthermore, our additional patients establish for the first time that disease can be associated with DOCK8 mutations that alter protein structure or stability. Together, this knowledge will improve the diagnosis of patients and aid in their management. Once patients are diagnosed, aggressive antimicrobial treatment, treatment with IVIG, and cancer surveillance are indicated. Furthermore, allogeneic hematopoietic cell transplantation, which was reported as curative in four patients, also resulted in rapid healing and clearance of viral and other infections in our patients, and should be considered as early as possible given the progressive course of the disease [6–9].
Acknowledgments
We thank the physicians who referred their patients to us and the patients and their relatives who participated in this study. We also thank Baran Erman for isolating RNA and performing the RT-PCR reaction on PBMC from patient 1-1. This work was supported in part by the Intramural Research Program of the National Institutes of Health, the National Institute of Allergy and Infectious Diseases, through the Lymphocyte Molecular Genetics Unit (LMGU) program. Sequencing was performed by the Genomics Unit of the Rocky Mountain Laboratories Research Technologies Section of the National Institute of Allergy and Infectious Diseases.
References
- 1.Zhang Q, Davis JC, Lamborn IT, Freeman AF, Jing H, Favreau AJ, Matthews HF, Davis J, Turner ML, Uzel G, Holland SM, Su HC. Combined immunodeficiency associated with DOCK8 mutations. N Engl J Med. 2009;361:2046–2055. doi: 10.1056/NEJMoa0905506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Engelhardt KR, McGhee S, Winkler S, Sassi A, Woellner C, Lopez-Herrera G, Chen A, Kim HS, Lloret MG, Schulze I, Ehl S, Thiel J, Pfeifer D, Veelken H, Niehues T, Siepermann K, Weinspach S, Reisli I, Keles S, Genel F, Kutuculer N, Camcioglu Y, Somer A, Karakoc-Aydiner E, Barlan I, Gennery A, Metin A, Degerliyurt A, Pietrogrande MC, Yeganeh M, Baz Z, Al-Tamemi S, Klein C, Puck JM, Holland SM, McCabe ER, Grimbacher B, Chatila TA. Large deletions and point mutations involving the dedicator of cytokinesis 8 (DOCK8) in the autosomal-recessive form of hyper-IgE syndrome. J Allergy Clin Immunol. 2009;124:1289–1302.e1284. doi: 10.1016/j.jaci.2009.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang Q, Su HC. Hyperimmunoglobulin E syndromes in pediatrics. Curr Opin Pediatr. 2011;23:653–658. doi: 10.1097/MOP.0b013e32834c7f65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Su HC, Jing H, Zhang Q. DOCK8 deficiency. Ann N Y Acad Sci. 2011;1246:26–33. doi: 10.1111/j.1749-6632.2011.06295.x. [DOI] [PubMed] [Google Scholar]
- 5.Renner ED, Puck JM, Holland SM, Schmitt M, Weiss M, Frosch M, Bergmann M, Davis J, Belohradsky BH, Grimbacher B. Autosomal recessive hyperimmunoglobulin E syndrome: a distinct disease entity. J Pediatr. 2004;144:93–99. doi: 10.1016/S0022-3476(03)00449-9. [DOI] [PubMed] [Google Scholar]
- 6.Gatz SA, Benninghoff U, Schutz C, Schulz A, Honig M, Pannicke U, Holzmann KH, Schwarz K, Friedrich W. Curative treatment of autosomal-recessive hyper-IgE syndrome by hematopoietic cell transplantation. Bone Marrow Transplant. 2010 doi: 10.1038/bmt.2010.169. [DOI] [PubMed] [Google Scholar]
- 7.Bittner TC, Pannicke U, Renner ED, Notheis G, Hoffmann F, Belohradsky BH, Wintergerst U, Hauser M, Klein B, Schwarz K, Schmid I, Albert MH. Successful long-term correction of autosomal recessive hyper-IgE syndrome due to DOCK8 deficiency by hematopoietic stem cell transplantation. Klin Padiatr. 2010;222:351–355. doi: 10.1055/s-0030-1265135. [DOI] [PubMed] [Google Scholar]
- 8.McDonald DR, Massaad MJ, Johnston A, Keles S, Chatila T, Geha RS, Pai SY. Successful engraftment of donor marrow after allogeneic hematopoietic cell transplantation in autosomal-recessive hyper-IgE syndrome caused by dedicator of cytokinesis 8 deficiency. J Allergy Clin Immunol. 2010 doi: 10.1016/j.jaci.2010.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barlogis V, Galambrun C, Chambost H, Lamoureux-Toth S, Petit P, Stephan JL, Michel G, Fischer A, Picard C. Successful allogeneic hematopoietic stem cell transplantation for DOCK8 deficiency. J Allergy Clin Immunol. 2011 doi: 10.1016/j.jaci.2011.03.025. [DOI] [PubMed] [Google Scholar]
- 10.Chu EY, Freeman AF, Jing H, Cowen EW, Davis J, Su HC, Holland SM, Turner ML. Cutaneous Manifestations of DOCK8 Deficiency Syndrome. Arch Dermatol. 2011 doi: 10.1001/archdermatol.2011.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dasouki M, Okonkwo KC, Ray A, Folmsbeel CK, Gozales D, Keles S, Puck JM, Chatila T. Deficient T Cell Receptor Excision Circles (TRECs) in autosomal recessive hyper IgE syndrome caused by DOCK8 mutation: Implications for pathogenesis and potential detection by newborn screening. Clin Immunol. 2011 doi: 10.1016/j.clim.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boybeyi O, Akcoren Z, Oguz B, Akyuz C, Sanal O, Ergin S, Ersoy-Evans S, Tanyel FC. Multifocal leiomyosarcomatosis in a 6-year-old child with epidermodysplasia verruciformis and immune defect. J Pediatr Surg. 2009;44:e5–8. doi: 10.1016/j.jpedsurg.2009.04.011. [DOI] [PubMed] [Google Scholar]
- 13.Renner ED, Hartl D, Rylaarsdam S, Young ML, Monaco-Shawver L, Kleiner G, Markert ML, Stiehm ER, Belohradsky BH, Upton MP, Torgerson TR, Orange JS, Ochs HD. Comel-Netherton syndrome defined as primary immunodeficiency. J Allergy Clin Immunol. 2009;124:536–543. doi: 10.1016/j.jaci.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thrasher AJ. New insights into the biology of Wiskott-Aldrich syndrome (WAS) Hematology Am Soc Hematol Educ Program. 2009:132–138. doi: 10.1182/asheducation-2009.1.132. [DOI] [PubMed] [Google Scholar]
- 15.Halabi-Tawil M, Ruemmele FM, Fraitag S, Rieux-Laucat F, Neven B, Brousse N, De Prost Y, Fischer A, Goulet O, Bodemer C. Cutaneous manifestations of immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome. Br J Dermatol. 2009;160:645–651. doi: 10.1111/j.1365-2133.2008.08835.x. [DOI] [PubMed] [Google Scholar]
- 16.Paganelli R, Scala E, Mezzaroma I, Pinter E, D’Offizi G, Fanales-Belasio E, Rosso RM, Ansotegui IJ, Pandolfi F, Aiuti F. Immunologic aspects of hyperimmunoglobulinemia E-like syndrome in patients with AIDS. J Allergy Clin Immunol. 1995;95:995–1003. doi: 10.1016/s0091-6749(95)70100-1. [DOI] [PubMed] [Google Scholar]
- 17.Villa A, Notarangelo LD, Roifman CM. Omenn syndrome: inflammation in leaky severe combined immunodeficiency. J Allergy Clin Immunol. 2008;122:1082–1086. doi: 10.1016/j.jaci.2008.09.037. [DOI] [PubMed] [Google Scholar]
- 18.Rogers HD, Macgregor JL, Nord KM, Tyring S, Rady P, Engler DE, Grossman ME. Acquired epidermodysplasia verruciformis. J Am Acad Dermatol. 2009;60:315–320. doi: 10.1016/j.jaad.2008.08.035. [DOI] [PubMed] [Google Scholar]
- 19.Kawai T, Malech HL. WHIM syndrome: congenital immune deficiency disease. Curr Opin Hematol. 2009;16:20–26. doi: 10.1097/MOH.0b013e32831ac557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ramoz N, Rueda LA, Bouadjar B, Montoya LS, Orth G, Favre M. Mutations in two adjacent novel genes are associated with epidermodysplasia verruciformis. Nat Genet. 2002;32:579–581. doi: 10.1038/ng1044. [DOI] [PubMed] [Google Scholar]
- 21.Guilhou JJ, Malbos S, Barneon S, Habib A, Baldet P, Meynadier J. Epidermodysplasia verruciformis (2 cases). Immunological study (author’s transl) Ann Dermatol Venereol. 1980;107:611–619. [PubMed] [Google Scholar]
- 22.Azzimonti B, Mondini M, De Andrea M, Gioia D, Dianzani U, Mesturini R, Leigheb G, Tiberio R, Landolfo S, Gariglio M. CD8+ T-cell lymphocytopenia and lack of EVER mutations in a patient with clinically and virologically typical epidermodysplasia verruciformis. Arch Dermatol. 2005;141:1323–1325. doi: 10.1001/archderm.141.10.1323. [DOI] [PubMed] [Google Scholar]
- 23.Gul U, Soylu S, Yavuzer R. Epidermodysplasia verruciformis associated with isolated IgM deficiency. Indian J Dermatol Venereol Leprol. 2007;73:420–422. doi: 10.4103/0378-6323.37064. [DOI] [PubMed] [Google Scholar]