

Abstract
First-generation, E1-deleted adenovirus subtype 5 (Ad5)-based vectors, although promising platforms for use as cancer vaccines, are impeded in activity by naturally occurring or induced Ad-specific neutralizing antibodies. Ad5-based vectors with deletions of the E1 and the E2b regions (Ad5 [E1-, E2b-]), the latter encoding the DNA polymerase and the pre-terminal protein, by virtue of diminished late phase viral protein expression, were hypothesized to avoid immunological clearance and induce more potent immune responses against the encoded tumor antigen transgene in Ad-immune hosts. Indeed, multiple homologous immunizations with Ad5 [E1-, E2b-]-CEA(6D), encoding the tumor antigen carcinoembryonic antigen (CEA), induced CEA-specific cell-mediated immune (CMI) responses with antitumor activity in mice despite the presence of preexisting or induced Ad5-neutralizing antibody. In the present phase I/II study, cohorts of patients with advanced colorectal cancer were immunized with escalating doses of Ad5 [E1-, E2b-]-CEA(6D). CEA-specific CMI responses were observed despite the presence of preexisting Ad5 immunity in a majority (61.3 %) of patients. Importantly, there was minimal toxicity, and overall patient survival (48 % at 12 months) was similar regardless of preexisting Ad5 neutralizing antibody titers. The results demonstrate that, in cancer patients, the novel Ad5 [E1-, E2b-] gene delivery platform generates significant CMI responses to the tumor antigen CEA in the setting of both naturally acquired and immunization-induced Ad5-specific immunity.
Electronic supplementary material
The online version of this article (doi:10.1007/s00262-013-1400-3) contains supplementary material, which is available to authorized users.
Keywords: Immunotherapy, Ad5 vector, CEA, Cell-mediated immunity
Introduction
Cancer immunotherapy achieved by delivering tumor-associated antigens (TAA) has recently demonstrated survival benefits [1, 2]; however, limitations to these strategies exist and more immunologically potent vaccines are needed. To address the low immunogenicity of self-tumor antigens, a variety of advanced, multi-component vaccination strategies including co-administration of adjuvants and immune-stimulating cytokines have been employed [3, 4]. Alternatives include the use of recombinant viral vectors that inherently provide innate pro-inflammatory signals while simultaneously engineered to express the antigen of interest. Of particular interest are adenovirus serotype-5 (Ad5)-based immunotherapeutics that have been repeatedly used in humans to induce robust T-cell-mediated immune (CMI) responses all while maintaining an extensive safety profile [5–7]. In addition, Ad5 vectors can be reliably manufactured in large quantities and are stable for storage and delivery for outpatient administration [6–8]. Nonetheless, a major obstacle to the use of first-generation (E1-deleted) Ad5-based vectors is the high frequency of preexisting anti-adenovirus type 5 neutralizing antibodies. These antibodies can be present in a potential vacinee due to either prior wild-type adenovirus infection [8, 9] or induction of adenovirus neutralizing antibodies by repeated injections with Ad5-based vaccines, each resulting in inadequate immune stimulation against the target TAA [10].
Attempts to overcome anti-Ad immunity have included use of alternative Ad serotypes and/or alternations in the Ad5 viral capsid protein, each with limited success and the potential for significantly altering biodistribution of the resultant vaccines. Therefore, a completely novel approach was attempted by further reducing the expression of viral proteins from the E1-deleted Ad5 vectors, proteins known to be targets of preexisting Ad immunity. Specifically, a novel recombinant Ad5 platform has been described with deletions in the early 1 (E1) gene region and additional deletions in the early 2b (E2b) gene region (Ad5 [E1-, E2b-]) [11]. Deletion of the E2b region (that encodes DNA polymerase and the pre-terminal protein) results in decreased viral DNA replication and late phase viral protein expression. This vector platform has been previously reported to successfully induce CMI responses in animal models of cancer and infectious disease [10, 12–18], and more importantly, this recombinant Ad5 gene delivery platform overcomes the barrier of Ad5 immunity and can be used in the setting of preexisting and/or vector-induced Ad immunity [10, 12–19], thus enabling multiple homologous administrations of the vaccine. We have constructed and tested an Ad5 [E1-, E2b-] platform containing a gene insert for the tumor antigen carcinoembryonic antigen (CEA) with a modification that enhances T-cell responses (Ad5 [E1-, E2b-]-CEA(6D) [12, 16, 19, 20]. Multiple immunizations with this Ad5 platform induced CEA-specific CMI responses with antitumor activity despite the presence of existing Ad5 immunity in mice [12, 16]. We now present results of a first-in-man, phase I/II clinical trial to determine the safety and immunogenicity of dose escalation of the Ad5 [E1-, E2b-]-CEA(6D) vector in advanced stage colorectal cancer patients to determine whether CMI could be induced and whether there was an effect on clinical outcome relative to the existence of preexisting Ad5 immunity.
Methods
Construction and production of Ad5 [E1-, E2b-]-CEA(6D)
The cDNA sequence containing the modified CEA with the CAP1(6D) mutation was produced at Duke University [21]. Clinical grade Ad5 [E1-, E2b-]-CEA(6D) was constructed as previously described [12] and manufactured using the E.C7 cell line [12] under GMP at SAFC, Carlsbad, California, and provided by Etubics Corporation.
Protocol schema and patient treatment
The clinical study was performed under an FDA-approved Investigational New Drug Exemption (IND14325) and registered at ClinicalTrials.gov (NCT01147965). Participants were recruited from medical oncology clinics at Duke University Medical Center, Durham, NC, and Medical Oncology Associates, Spokane, WA. Patients provided informed consent approved by the respective Institutional Review Boards (IRB). Eligibility requirements included metastatic cancer expressing CEA and adequate hematologic, renal, and hepatic function. Trial participants were required to have received treatment with standard therapy known to have a possible overall survival benefit or refused such therapy. Exclusion criteria included chemotherapy or radiation within the prior 4 weeks, history of autoimmune disease, viral hepatitis, HIV, or use of immunosuppressives. Patients who had been receiving bevacizumab or cetuximab for at least 3 months prior to enrollment were permitted to continue receiving these antibodies. Prior CEA immunotherapy was permitted. The study employed a standard 3 + 3 dose escalation strategy with dose-limiting toxicities (DLT) defined as grade 3 or 4 major organ toxicity. The Ad5 [E1-, E2b-]-CEA(6D) doses were delivered to patients as follows: cohort 1: dose of 1X109 VP in 0.5 ml subcutaneously (SQ) in the same thigh every 3 weeks for 3 treatments; cohort 2: dose of 1X1010 VP in 0.5 ml SQ every 3 weeks for 3 treatments; cohort 3: dose of 1 × 1011 in 0.5 ml SQ every 3 weeks for 3 treatments. Following the establishment of the dose of 1 × 1011 VP as safe, an additional 12 patients received Ad5 [E1-, E2b-]-CEA(6D) at this dose and schedule (phase II cohort). After completing the phase II cohort, an additional cohort (cohort 5) of six patients received a dose of 5 × 1011 VP in 2.5 ml SQ every 3 weeks for 3 treatments to determine safety of the highest achievable dose. PBMCs were collected from patients just prior to the immunizations at weeks 0, 3, 6, and three weeks following the last treatment. The PBMCs were frozen in liquid nitrogen until ELISPOT assays were performed. In cohort 5, fresh PBMCs were analyzed in preliminary flow cytometry assays for polyfunctional CD8+ T lymphocytes.
Assessment of clinical activity
Clinical activity was assessed according to Response Evaluation Criteria in Solid Tumors (RECIST 1.0 criteria [22]) using computed tomography (CT) or magnetic resonance imaging (MRI) scans obtained at baseline and after treatments were completed. Toxicity was assessed according to the National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE) version 4.0 [23]. Peripheral blood CEA levels, hematology, serum chemistries, and anti-nuclear antibody titers were compared at baseline and 3 weeks following the final treatment. Survival was measured from the day of the first immunization until death from any cause.
Analysis of CMI responses by ELISPOT assay
An ELISPOT assay for IFN-γ-secreting lymphocytes was adapted from our previous animal studies and performed as described [12]. Briefly, isolated PBMCs (2 × 105 cells/well) from individual patient samples were incubated 36–40 h with a CEA peptide pool (15mers with 11aa overlap covering full-length CEA with the 6D modification; 0.1 μg/well) to stimulate IFN-γ-producing T cells. CMI responses to Ad5 were determined after exposure of patient PBMC to Ad5 null (empty vector). Cells stimulated with concanavalin A (Con A) at a concentration of 0.25 μg/well served as positive controls. Colored spot-forming cells (SFC) were counted using an Immunospot ELISPOT plate reader (Cellular Technology, Shaker Heights, OH), and responses were considered to be positive if 50 SFC were detected/106 cells after subtraction of the negative control and SFC were ≥twofold higher than those in the negative control wells.
Determination of Ad5 neutralizing antibody (NAb) titers
Endpoint Ad5 NAb titers were determined as previously described [12–14]. Briefly, dilutions of heat-inactivated test sera in 100 μL of DMEM containing 10 % fetal calf serum were mixed with 4 × 107 VP of Ad5 [E1-]-null and incubated for 60 min at room temperature. The samples were added to microwells containing HEK293 cells cultured in DMEM containing 10 % heat-inactivated calf serum at 2 × 103 cells/well for 24 h at 37 °C in 5 % CO2. The mixture was incubated for an additional 72 h at 37 °C in 5 % CO2. An MTS tetrazolium bioreduction assay (Promega Corp. Madison, WI) [24] was used to measure cell killing and endpoint Ad5 NAb titers. Endpoint titers with a value less than 1:25 were assigned a value of 0.
Statistics
Statistical analyses comparing immune responses were performed employing the Mann–Whitney test (PRISM, GraphPad). Survival comparisons were made employing Kaplan–Meier plots (PRISM, GraphPad). Ad5 NAb titer and CEA-specific CMI were analyzed as continuous variables. The association of Ad5 NAb titer with change in CEA-specific CMI was tested with the Spearman correlation coefficient. The association of Ad5 NAb titer with survival was tested with the Wald test of the proportional hazards model. All tests used a two-sided alpha of 0.05.
Results
Patient demographics and safety and tolerability
Thirty-two patients with metastatic colorectal cancer, median age 57.5 (range 38–77) who had failed a median of three prior chemotherapeutic regimens (range 2–5), had a performance status of 90 % (range 70–100 %), and had three sites of metastatic disease (range 1–4), were enrolled (Table 1). The majority were able to receive all three immunizations. All four patients who stopped immunizations early did so due to significant disease progression. There was no dose-limiting toxicity and no serious adverse events (SAE) that resulted in treatment discontinuation at any vaccine dose level. The most common toxicity (see Supplemental Table 1) was a self-limited, injection site reaction. Other reactions occurred with less than a 10 % incidence and included fever, flu-like symptoms, anorexia, chills, nausea, and headache. These symptoms were also self-limiting and did not require intervention other than symptomatic measures such as acetaminophen. Routine hematology and chemistry studies showed no significant biologic changes during the immunization period (Supplemental Table 2). In particular, the total lymphocyte count remained stable (pre and post). Overall, comparisons of ANA titers at baseline and 3 weeks after the last immunization revealed no significant difference in values across all patient groups (Supplemental Table 2).
Table 1.
Patient demographics
| Patient ID/cohort | Dose (VP) | Dx | Age | Sex | KPS | # prior CTx | Mets (# of sites) | # of doses | ++Disease Status after tx | Survival (Months) |
|---|---|---|---|---|---|---|---|---|---|---|
| 002/1 | 109 | C | 67 | M | 70 | >3 | 4 | 3 | PD | 3 (−) |
| 003/1 | 109 | R | 63 | M | 100 | 5 | 2 | 3 | PD | 9 (−) |
| 004/1 | 109 | C | 53 | F | 100 | 2 | 3 | 3 | PD | 11 (−) |
| 005/2^ | 1010 | C | 60 | M | 100 | 3 | 3 | 3 | SD | 12 (+) |
| 007/2 | 1010 | C | 52 | M | 80 | 2 | 5 | 1 | PD | 1 (−) |
| 008/2 | 1010 | C | 42 | F | 100 | 3 | 3 | 3 | PD | 12 (+) |
| 010/2 | 1010 | C | 58 | M | 90 | 3 | 3 | 3 | PD | 12 (−) |
| 011/3 | 1011 | R | 50 | M | 100 | 5 | 1 | 3 | PD | 12 (+) |
| 012/3 | 1011 | C | 48 | M | 100 | 1 | 2 | 3 | PD | 12 (+) |
| 013/3 | 1011 | R | 62 | M | 100 | 3 | 2 | 3 | PD | 4 (−) |
| 500/3 | 1011 | C | 55 | M | 80 | 4 | 3 | 3 | PD | 12 (+) |
| 015/3 | 1011 | C | 58 | F | 80 | 3 | 4 | 3 | PD | 10 (−) |
| 016/3@ | 1011 | C | 53 | F | 100 | 3 | 4 | 3 | PD | 6 (−) |
| 017/3* | 1011 | R | 52 | F | 90 | 3 | 2 | 3 | PD | 3 (−) |
| 501/II | 1011 | R | 54 | M | 90 | 1 | 1 | 3 | PD | 12 (+) |
| 502/II | 1011 | C | 66 | F | 80 | 1 | 2 | 2 | PD | 3 (−) |
| 019/II | 1011 | C | 69 | M | 90 | 1 | 3 | 3 | PD | 12 (+) |
| 020/II^ | 1011 | C | 59 | M | 100 | 5 | 4 | 3 | SD | 12 (+) |
| 021/II^ | 1011 | C | 51 | F | 100 | 4 | 3 | 3 | PD | 12 (+) |
| 506/II | 1011 | C | 77 | F | 80 | 2 | 2 | 3 | PD | 3 (−) |
| 023/II | 1011 | C | 51 | F | 100 | 3 | 4 | 3 | PD | 4 (−) |
| 504/II | 1011 | C | 57 | M | 90 | 3 | 3 | 3 | PD | 12 (+) |
| 507/II | 1011 | R | 58 | M | 90 | 2 | 2 | 3 | PD | 12 (+) |
| 024/II | 1011 | C | 67 | M | 90 | 2 | 3 | 3 | PD | 12 (+) |
| 025/II | 1011 | C | 62 | F | 100 | 2 | 4 | 3 | PD | 7 (−) |
| 026/II | 1011 | C | 53 | M | 100 | 3 | 2 | 2 | PD | 4 (−) |
| 030/5 | 5 × 1011 | C | 38 | M | 90 | 4 | 3 | 3 | PD | 10 (+) |
| 031/5 | 5 × 1011 | R | 72 | F | 90 | 4 | 2 | 3 | SD | 9 (+) |
| 032/5@ | 5 × 1011 | R | 53 | M | 90 | 4 | 3 | 3 | PD | 6 (−) |
| 033/5 | 5 × 1011 | R | 48 | F | 90 | >3 | 2 | 3 | PD | 5 (−) |
| 034/5 | 5 × 1011 | C | 62 | M | 100 | 5 | 4 | 3 | PD | 7 (+) |
| 035/5 | 5 × 1011 | C | 60 | F | 90 | 3 | 5 | 2 | PD | 2 (−) |
Dx diagnosis, C colon, R rectal cancer, KPS Karnofsky performance status, PD progressive disease, SD stable disease
* concurrent cetuximab; ^concurrent bevacizumab; @ concurrent panitumumab
++Represents disease status at 9 weeks post-initiation of immunizations
(+) Alive; (−) Dead at last follow-up
Determination of Induced CMI Responses to CEA
ELISPOT analysis was performed on cryopreserved PBMC samples drawn before each immunization and after the completion of the final immunization to assess CEA-specific CMI responses. We observed a dose–response effect with the highest magnitude CEA-specific CMI responses occurring in patients who received the highest dose of Ad5 [E1-, E2b-]-CEA(6D) (Fig. 1). Of the doses received, 0/3 (0 %) patients in cohort 1 exhibited positive CEA-directed CMI responses, 1/4 (25 %) patient in cohort 2 exhibited positive CEA-directed CMI responses, 10/19 (53 %) patients in cohort 3/phase II exhibited positive CEA-directed CMI responses, and 4/6 (67 %) patients in cohort 5 exhibited positive CEA-directed CMI responses. The time course of induction of CEA-specific CMI (Supplemental Fig. 1) demonstrated that there may be plateau in the magnitude of CEA CMI prior to the last dose although small numbers could affect this finding. In the largest group of patients who received the same dose (cohort 3 plus phase II), we observed a significant increase over baseline in the average CEA-directed CMI responses at the week 6 evaluation (P < 0.05, Mann–Whitney test), averaging 94 SFC/106 PBMC, which increased further by the week 9 evaluation (Supplementary Fig. 1). One patient (patient ID 13) had a highly elevated baseline CEA-specific immune response (1100 SFC) and had elevated CMI at week six (2305 SFC) but did not return for week 9 evaluation and therefore was not included in CEA CMI data analysis.
Fig. 1.

CEA-directed CMI responses in treated patients. CMI (IFN-γ secretion) was assessed at baseline (pre) and after administrations of Ad5 [E1-, E2b-]-CEA(6D) (post). The highest CMI responses (regardless of time point) observed in the patients after treatment revealed a dose response. The highest CMI levels occurred in patients that received the highest dose of 5 × 1011 VP (Cohort 5). The CMI responses for cohort 3/phase II and cohort 5 were significantly elevated (Mann–Whitney test) as compared to their baseline (pre) values. Specificity of the responses was demonstrated by the lack of reactivity with the irrelevant antigens β-galactosidase and HIV-gag (data not shown). For positive controls, PBMCs were exposed to concanavalin A (data not shown). Horizontal line and error bar indicate the mean ± SEM for each cohort
We also measured Ad5 NAb and CMI against Ad5 and correlated it with CEA-specific CMI. Each patient had their serum and PBMC sample tested at baseline (prior to treatment) and at 9 weeks after completion of 3 treatments. Nineteen of 31 patients (61.3 %) tested in this study had Ad5 neutralizing activity in serum samples prior to the onset of treatment with the CEA(6D)-expressing Ad vaccine. The mean pre-treatment Ad5 NAb titer value obtained among all patients was 1:189 ± 1:71 SEM (geometric mean 1:21), and the mean pre-treatment Ad5 NAb titer among seropositive patients was 1:308 ± 1:108 (geometric mean 1:146). Analysis of serum samples from patients who received 3 immunizations revealed Ad5 NAb titers that were significantly increased (P < 0.0001, Mann–Whitney test) by week 9 (mean 1:4767 ± 1:1225 SEM) (geometric mean 1:1541) when compared with their respective baseline values (Fig. 2a). Analysis of PBMC for CMI responses to Ad5 also revealed a significant increase (P < 0.01, Mann–Whitney test) in Ad5-directed CMI responses after immunizations with Ad5 [E1-, E2b-]-CEA(6D) (Fig. 2b). Only ELISPOT assays were performed for CMI, and we did not assess the relative contribution of CD4+ and CD8+ T cells; thus, it is unclear whether both cell types are responding or whether responses are associated preferentially from one group.
Fig. 2.

Ad5 immune responses. Ad5 NAb titers a and CMI responses b to Ad5 were determined in patients at baseline (week 0) and 3 weeks (week 9) after the third immunization. The number of IFN-γ-secreting PBMCs from patients that were specific for Ad5 was determined by ELISPOT. Both the Ad5 NAb titers and Ad5 CMI responses were significantly elevated at week 9 (Mann–Whitney test). Horizontal line and error bar indicate the mean ± SEM
Comparison of week 9 CEA-directed CMI responses from patients with low baseline preexisting Ad5 immunity (Ad5 NAb ≥200) versus those with high baseline Ad5 immunity (Ad5 NAb >200) revealed no significant difference in responses (P > 0.4, Mann–Whitney test). Further, when the highest CEA-specific CMI responses were compared with preexisting or vector-induced Ad5 NAb activity, there was no correlation between levels of CEA CMI and Ad5 NAb activity (Fig. 3). These data indicate that immunizations with Ad5 [E1-, E2b-]-CEA(6D) were able to induce CEA-specific immune responses in colorectal cancer patients despite the presence of existing and/or immunization-induced Ad5 immunity.
Fig. 3.

CEA-specific immunity in patients and comparisons with Ad5 immunity. Correlation between preexisting Ad5 NAb activity and highest levels of induced CEA CMI responses a. Correlation between vector-induced Ad5 NAb activity and CEA CMI responses b. The r 2 values revealed no correlation between preexisting or vector-induced Ad5 NAb activity and CEA CMI ELISPOT responses
Clinical outcomes
Carcinoembryonic antigen levels in serum at baseline and week 9 were assessed in patients. Among those with CEA levels available at baseline and follow-up, three had no increase in CEA levels at the end of the immunization period while the remaining patients showed increased CEA levels. There were three patients with stable disease who remained so during the 9-week study period. All other patients experienced some level of progressive disease (Table 1). Patients in cohorts 1, 2, 3, and phase II who received at least 2 treatments (n = 25) were followed for survival and Kaplan–Meier plots and survival probabilities performed. Patients in cohort 5 (n = 6) have not completed the 12-month follow-up period and, therefore, were not evaluated for survival by Kaplan–Meier plots. Six patients in cohorts 1 and 2 experienced a 12-month survival probability of 33.3 % (Fig. 4). Nineteen patients in the combined group of cohort 3 and phase II experienced a 12-month survival probability of 52.6 % (Fig. 4). With a median follow-up of 12 months, all 25 patients as a group (cohorts 1, 2, 3, and phase II) experienced a 12-month survival probability of 48 % (Fig. 4). There was no association between Ad5 NAb and survival using Ad5 NAb both as a continuous variable and as a variable dichotomized between <200 and ≥200 (P values 0.48 and 0.44, respectively). These data indicate that preexisting Ad5 NAb did not significantly impact survival outcomes following immunization with the Ad5 [E1-, E2b-]-CEA(6D) vaccine.
Fig. 4.
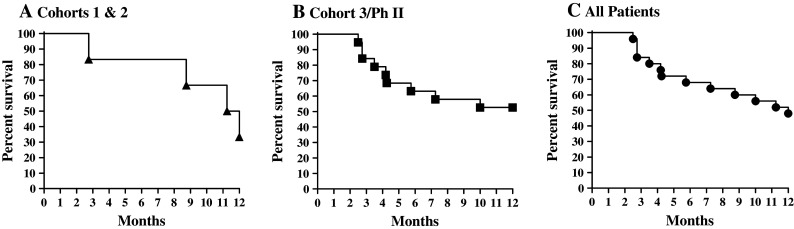
Kaplan–Meier survival plots of patients treated with Ad5 [E1-, E2b-]-CEA(6D). Patients treated at least two times with Ad5 [E1-, E2b-]-CEA(6D) were followed for survival. Panel a represents 6 patients in cohorts 1 and 2 that were followed for survival. There were 4 events in this group. Panel b represents 19 patients in cohort 3 and phase II that were followed for survival. There were 9 events in this group. Panel c represents all 25 patients (cohorts 1,2, 3, and phase II) that were followed for survival. There were 13 events in this group
Discussion
Adenoviral vectors have significant potential for use as cancer therapeutic vaccines because of their propensity to induce robust adaptive immune responses specifically against transgene products in general; however, recombinant first-generation Ad5 [E1-] vectors used in homologous prime/boost regimens have been greatly limited in their potential efficacy due to the presence of preexisting Ad5 immunity as well as vector-induced immunity [7–10]. Specifically, Ad5-directed immunity mitigates immune responses to TAA that have been incorporated into earlier generation Ad5 [E1-]-based platforms [10]. The Ad5 [E1-, E2b-] platform utilized in the present study was intended to accommodate a homologous prime–boost regimen, by avoiding presentation of antigens that are the targets of preexisting Ad5 immunity [2, 8, 25–28]. Since CEA has been identified as one of the priority cancer antigens by the National Cancer Institute [29], we investigated this TAA as a transgene to be incorporated into the new Ad5 [E1-, E2b-] vector platform for use as a cancer therapeutic vaccine. CEA expression in adults is normally limited to low levels in the gastrointestinal epithelium, whereas CEA is over-expressed in adenocarcinomas of the colon and rectum and in many breast, lung, and pancreas cancers [30, 31]. We chose the HLA A2-restricted CAP1(6D) modification of CEA because, compared with the wild-type CAP1 epitope, CAP1(6D) has been shown to enhance the sensitization of CTLs [19, 20] and has been included in our recent CEA-based vaccine constructs [32, 33]. Although we did not test for HLA type because we used full-length CEA that is not HLA-restricted, A*0201 is the allele observed most frequently in Caucasians (allele frequency 0.2717) and is common in other populations [34]. However, in expanded trials, we plan to test patients for HLA type and assess whether or not there may be a relationship between HLA type and clinical and/or CMI responses.
Previously, we tested multiple subcutaneous immunizations employing three administrations of a single dose level (1 × 1010 VP) of this class of Ad5 vaccine expressing the TAA CEA, (Ad5 [E1-, E2b-]-CEA(6D)) in a preclinical murine model of CEA-expressing cancer. In mice with preexisting Ad5 immunity, we demonstrated the induction of potent CEA-directed CMI responses that resulted in anti-tumor activity and noted that these CMI and anti-tumor responses were significantly greater than those responses induced by a current generation Ad5 [E1-]-based vector vaccine [12, 16]. We have also demonstrated in additional animal models (both cancer and infectious disease targeted) [10, 12–18] that multiple subcutaneous immunizations with vaccines based on the new Ad5 [E1-, E2b-] platform induce CMI responses that were superior to those of current generation Ad5 [E1-]-based vaccines, can overcome the barrier of Ad5 immunity, and can be utilized in multiple immunization regimens requiring a generation of robust CMI responses. In our present report, the greatest magnitude of CEA-directed CMI responses occurred in patients receiving the highest dose of the vector. We observed that a CEA-directed CMI response was induced in a dose–responsive manner despite the presence of preexisting and/or vector-induced Ad5 immunity. We did not assess CAP1(6D)-specific CMI responses in this phase I/II clinical study and plan to assess CAP1(6D) and other CEA epitope-directed CMI responses in our expanded clinical trials. No CEA-directed antibody responses were observed either pre- or post-vaccination employing an ELISA technique [21]. In a preliminary analysis (data not shown), we also observed a population of polyfunctional CD8+ T cells (those that secrete more than one cytokine when activated) after immunizations, a sign of greater functionality of T cells induced by the vaccine. These data support the use of the Ad5 [E1-, E2b-]-CEA(6D) vector in homologous prime–boost regimens designed to induce and increase CEA-directed CMI responses in patients with advanced colorectal adenocarcinoma, as well as any number of other vaccine amenable diseases or applications.
Although the precise mechanism(s) of how the Ad5 [E1-, E2b-] vector platform accomplishes tumor antigen-specific immune induction in the setting of existing or induced Ad5 immunity is not fully understood at present, we believe there are factors that contribute to the favorable activity of this new platform. As compared to earlier generation Ad5 [E1-] vectors containing deletion in the early 1 (E1) gene region, the Ad5 [E1-, E2b-] vector platform with additional deletions in the early 2b (E2b) gene region exhibits significantly reduced inflammatory responses directed at the vector [11, 35, 36]. This can result in longer transgene expression and a reduction in elimination of transgene expressing cells (e.g., antigen-presenting cells) that would otherwise occur due to induced inflammatory responses [35, 37]. Since Ad5 late gene antigen expression is significantly reduced as compared to earlier generation Ad5 platforms [8, 11], this could enable the Ad5 [E1-, E2b-] platform to evade Ad5 immune-mediated neutralizing activity for significantly longer periods of time resulting in greater longevity and amplification of TAA expression. In addition, the E2b gene product, polymerase, is a known target of human cellular memory immune responses to Ad5 infection and its elimination from the vaccine could be furthering its capability in the setting of preexisting Ad5 immunity [38]. The extended and/or greater expression of TAA by the vector in this milieu could result in a more effective immune response against the target antigen. However, it is also possible that this vector configuration produces better transgene expression, different biodistribution, or different innate/adaptive immune effects that impact the effectiveness of this vector, rather than escape from preexisting immunity.
Our patient demographics, albeit limited in size, compares favorably with previously published studies of patients with chemotherapy-refractory colorectal cancer [39–41]. Of interest is the observation that treated patients in our study exhibited favorable survival probability. Overall, all 25 patients treated at least two times with Ad5 [E1-, E2b-]-CEA(6D) exhibited a 12-month survival probability of 48 % and this was achieved despite the presence of significant levels of preexisting Ad5 neutralizing antibody titers. However, the true impact of this new immunotherapy on overall survival will only be determined in a statistically controlled and randomized trial with larger numbers of patients.
In other clinical trials, immunotherapeutic agents have been found to increase overall survival without having a direct impact on time to objective disease progression, a trend noted in our study as well [1, 42–44]. By engaging the patient’s immune system, active immunotherapeutics, such as Ad5 [E1-, E2b-]-CEA(6D), could induce continuous immunologic anti-tumor responses over a long period of time that could result in a “deceleration” or alteration in specific aspects of the rapid growth rate or spread of the tumor not measured by standard response assessments [39, 45]. Indeed, we have observed slower tumor progression in Ad5 immune mice harboring established CEA-expressing tumors following treatment with Ad5 [E1-, E2b-]-CEA(6D) [12]. Moreover, it has been noted that overall survival might be the only true parameter for the determination of clinical efficacy of any potential cancer (immune) therapy [46].
As with any new treatment modality, safety is an important factor. In this phase I/II trial, we demonstrate that the Ad5 [E1-, E2b-]-CEA(6D) could be manufactured to scale, as well be easily and repeatedly administered by conventional subcutaneous injection techniques. The most common adverse effects were site of injection reactions and flu-like symptoms consisting of fever, chills, headache, and nausea. There was no impact on blood hematology or serum chemistries, and overall, the treatments were well tolerated. Specifically, no SAE were noted, and no treatments were stopped due to adverse events, indicating that a dose limitation to use of Ad5 [E1-, E2b-]-CEA(6D) in this clinical application had not been met.
These data suggest that patients with advanced colorectal cancer which are treated with Ad5 [E1-, E2b-]-CEA(6D) do not have serious adverse effects and may experience extension of life even if they have preexisting immunity to Ad5; however, this study had a small number of patients in a trial that was not randomized against a control population. The results of this trial are encouraging enough to advance to a large, randomized, single-agent trial. The observation that some of the patients experienced an increase in CMI which is dose dependent could be an indication that this may play a role in their clinical outcome. We plan to initiate a large multicenter trial which should give us the opportunity to evaluate in greater detail the influence of Ad5 [E1-, E2b-]-CEA(6D) treatment on safety, overall survival, time to progression following treatment, the levels of induction of CMI, and the relationship of induced CMI responses with clinical outcome.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
This research was supported by SBIR Grants 1R43CA134063 and 2R44CA134063 from the NCI and by SBIR Research Contracts HHSN261200900059C and HHSN261201100097C from the NCI. The authors wish to thank Dr. Bercedis Peterson for statistical analysis and Ms. Carol Jones for her assistance with grants management. The authors would also like to thank the nursing staff at the Duke Comprehensive Cancer Center and Medical Center and Medical Oncology Associates for care of the patients in the phase I/II clinical trial.
Conflict of interest
The following authors declare financial conflict of interest: Elizabeth S. Gabitzsch, Younong Xu, Stephanie Balcaitis, Rajesh Dua, Susan Nguyen, Joseph P. Balint, Jr., Frank R. Jones are employees of Etubics Corporation. All other authors do not have any conflict of interest.
References
- 1.Kantoff P, Higano CS, Shore ND, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363:411–422. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- 2.Kantoff PW, Schuetz TJ, Blumenstein BA, et al. Overall survival analysis of a phase II randomized controlled trial of a Poxviral-based PSA-targeted immunotherapy in metastatic castration-resistant prostate cancer. J Clin Oncol. 2010;28:1099–1105. doi: 10.1200/JCO.2009.25.0597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vergati M, Intrivici C, Huen N-Y, Schlom J, Tsang KY. Strategies for cancer vaccine development. J Biomed Biotechnol. 2010;pii:596432. doi: 10.1155/2010/596432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Palena C, Schlom J. Vaccines against human carcinomas: strategies to improve antitumor immune responses. J Biomed Biotechnol. 2010;pii:380697. doi: 10.1155/2010/380697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bangari DS, Mittal SK. Development of nonhuman adenoviruses as vaccine vectors. Vaccine. 2006;24:849–862. doi: 10.1016/j.vaccine.2005.08.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Campos SK, Barry MA. Current advances and future challenges in adenoviral vector biology and targeting. Curr Gene Ther. 2007;7:189–204. doi: 10.2174/156652307780859062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tatsis N, Ertl HCJ. Adenoviruses as vaccine vectors. Mol Ther. 2004;10:616–629. doi: 10.1016/j.ymthe.2004.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seregin SS, Amalfitano A. Overcoming pre-existing Adenovirus immunity by genetic engineering of Adenovirus-based vectors. Expert Opin Biol Ther. 2009;9:1–11. doi: 10.1517/14712590903307388. [DOI] [PubMed] [Google Scholar]
- 9.Nwanegbo E, Vardas E, Gao W, et al. Prevalence of neutralizing antibodies to adenoviral serotypes 5 and 35 in the adult populations of The Gambia, South Africa, and the United States. Clin Diagn Lab Immunol. 2004;11:351–357. doi: 10.1128/CDLI.11.2.351-357.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gabitzsch ES, Jones FR. New recombinant Ad5 vector overcomes Ad5 immunity allowing for multiple safe, homologous immunizations. J Clin Cell Immunol. 2011 [Google Scholar]
- 11.Amalfitano A, Hauser MA, Hu H, Serra D, Begy CR, Chamberlain JS. Production and characterization of improved adenovirus vectors with the E1, E2b, and E3 genes deleted. J Virol. 1998;72:926–933. doi: 10.1128/jvi.72.2.926-933.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gabitzsch ES, Xu Y, Balint JP, Jr, Hartman ZC, Lyerly HK, Jones FR. Anti-tumor immunity despite immunity to adenovirus using a novel adenoviral vector Ad5 [E1-, E2b-]-CEA. Cancer Immunol Immunother. 2010;59:1131–1135. doi: 10.1007/s00262-010-0847-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gabitzsch ES, Xu Y, Balcaitis S, Balint JP, Jr, Jones FR. An Ad5 [E1-, E2b-]-HER2/neu vector induces immune responses and inhibits HER2/neu expressing tumor progression in Ad5 immune mice. Cancer Gene Ther. 2011;18:326–335. doi: 10.1038/cgt.2010.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gabitzsch ES, Xu Y, Balint JP, Jr, Balcaitis S, Sanders-Beer B, Jones FR. Induction and comparison of SIV immunity in Ad5 Naïve and Ad5 immune non-human primates using an Ad5 [E1-, E2b-] based vaccine. Vaccine. 2011;29:8101–8107. doi: 10.1016/j.vaccine.2011.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jones FR, Gabitzsch ES, Xu Y, et al. Prevention of influenza virus shedding and protection from lethal H1N1 challenge using a consensus 2009 H1N1 HA and NA adenovirus vector vaccine. Vaccine. 2011;29:7020–7026. doi: 10.1016/j.vaccine.2011.07.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Osada T, Yang XY, Hartman ZC, et al. Optimization of vaccine responses with an E1, E2b and E3-deleted Ad5 vector circumvents pre-existing anti-vector immunity. Cancer Gene Ther. 2009;16:673–682. doi: 10.1038/cgt.2009.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gabitzsch ES, Xu Y, Yoshida LH, et al. A preliminary and comparative evaluation of a novel Ad5 [E1-, E2b-] recombinant based vaccine used to induce cell mediated immune responses. Immunol Lett. 2009;122:44–51. doi: 10.1016/j.imlet.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gabitzsch ES, Yu X, Yoshida LH, Balint J, Amalfitano A, Jones FR. Novel adenovirus type 5 vaccine platform induces cellular immunity against HIV-Gag, Pol, Nef despite the presence of Ad5 immunity. Vaccine. 2009;27:6394–6398. doi: 10.1016/j.vaccine.2009.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zaremba S, Barzaga E, Zhu M, Soares N, Tsang KY, Schlom J. Identification of an enhancer agonist cytotoxic T lymphocyte peptide from human carcinoembryonic antigen. Cancer Res. 1997;57:4570–4577. [PubMed] [Google Scholar]
- 20.Tangri S, Ishioka GY, Huang X, et al. Structural features of peptide analogs of human histocompatibility leukocyte antigen class I epitopes that are more potent and immunogenic than wild-type peptide. J Exp Med. 2001;194:833–846. doi: 10.1084/jem.194.6.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morse MA, Hobeika AC, Osada T, et al. An alphavirus vector overcomes the presence of neutralizing antibodies and elevated numbers of Tregs to induce immune responses in humans with advanced cancer. J Clin Invest. 2010;120:3234–3241. doi: 10.1172/JCI42672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumors: revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45:228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 23.CTEP Cancer Therapy Evaluation Program. CTCAE and CTC Website (2010) http://ctep.cancer.gov/protocolDevelopment/electronic_applications/ctc.htm. Accessed 10 Feb 2012
- 24.Cory AH, Owen TC, Barltrop JA, Cory JG. Use of an aqueous soluble tetrazolium/formazan assay for cell growth assays in culture. Cancer Commun. 1991;3:207–212. doi: 10.3727/095535491820873191. [DOI] [PubMed] [Google Scholar]
- 25.Sanda MG, Smith DC, Charles LG, et al. Recombinant vaccinia-PSA (PROSTVAC) can induce a prostate specific immune response in androgen-modulated human prostate cancer. Urology. 1999;53:260–266. doi: 10.1016/S0090-4295(98)00539-1. [DOI] [PubMed] [Google Scholar]
- 26.Eder JP, Kantoff PW, Roper K, et al. A phase I trial of a recombinant vaccinia virus expressing prostate-specific antigen in advanced prostate cancer. Clin Cancer Res. 2000;6:1632–1638. [PubMed] [Google Scholar]
- 27.Gulley J, Chen AP, Dahut W, et al. Phase I study of a vaccine using recombinant vaccinia virus expressing PSA (rV-PSA) in patients with metastatic androgen-independent prostate cancer. Prostate. 2002;53:109–117. doi: 10.1002/pros.10130. [DOI] [PubMed] [Google Scholar]
- 28.Kaufman HL, Wang W, Manola J, et al. Phase II randomized study of vaccine treatment of advanced prostate cancer (E7897): a trial of the Eastern Cooperative Oncology Group. J Clin Oncol. 2004;22:2122–2132. doi: 10.1200/JCO.2004.08.083. [DOI] [PubMed] [Google Scholar]
- 29.Cheever MA, Allison JP, Ferris AS, et al. The prioritization of cancer antigens: a National Cancer Institute pilot project for the acceleration of translational research. Clin Cancer Res. 2009;15:5323–5337. doi: 10.1158/1078-0432.CCR-09-0737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Berinstein NL. Carcinoembryonic antigen as a target for therapeutic anticancer vaccines: a review. J Clin Oncol. 2002;20:2197–2207. doi: 10.1200/JCO.2002.08.017. [DOI] [PubMed] [Google Scholar]
- 31.Hammarstrom S. The carcinoembryonic antigen (CEA) family: structures, suggested functions and expression in normal and malignant tissues. Semin Cancer Biol. 1999;9:67–81. doi: 10.1006/scbi.1998.0119. [DOI] [PubMed] [Google Scholar]
- 32.Morse MA, Clay TM, Hobeika AC, et al. Phase I study of immunization with dendritic cells modified with recombinant fowlpox encoding carcinoembryonic antigen and the triad of costimulatory molecules CD54, CD58, and CD80 in patients with advanced malignancies. Clin Cancer Res. 2005;11:3017–3024. doi: 10.1158/1078-0432.CCR-04-2172. [DOI] [PubMed] [Google Scholar]
- 33.Morse MA, Hobeika AC, Osada T, et al. Depletion of human regulatory T cells specifically enhances antigen-specific immune responses to cancer vaccines. Blood. 2008;112:610–618. doi: 10.1182/blood-2008-01-135319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cao K, Hollenbach J, Shi X, Shi W, Chopek M, Fernández-Viña MA. Analysis of the frequencies of HLA-A, B, and C alleles and haplotypes in the five major ethnic groups of the United States reveals high levels of diversity in these loci and contrasting distribution patterns in these populations. Hum Immunol. 2001;62:1009–1030. doi: 10.1016/S0198-8859(01)00298-1. [DOI] [PubMed] [Google Scholar]
- 35.Amalfitano A, Parker RJ. Separating fact from fiction: assessing the potential of modified adenovirus vectors for use in human gene therapy. Curr Gene Ther. 2002;2:111–133. doi: 10.2174/1566523024605618. [DOI] [PubMed] [Google Scholar]
- 36.Everett RS, Hodges BL, Ding EY, Xu F, Serra D, Amalfitano A. Liver toxicities typically induced by first-generation adenoviral vectors can be reduced by use of E1, E2b-deleted adenoviral vectors. Hum Gene Ther. 2003;14:1715–1726. doi: 10.1089/104303403322611737. [DOI] [PubMed] [Google Scholar]
- 37.Hodges BL, Serra D, Hu H, Begy CA, Chamberlain JS, Amalfitano A. Multiply deleted [E1, polymerase-, and pTP-] adenovirus vector persists despite deletion of the preterminal protein. J Gene Med. 2000;2:250–259. doi: 10.1002/1521-2254(200007/08)2:4<250::AID-JGM113>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 38.Joshi A, Tang J, Kuzma M, et al. Adenovirus DNA polymerase is recognized by human CD8+ T cells. J Gen Virol. 2009;90:84–94. doi: 10.1099/vir.0.002493-0. [DOI] [PubMed] [Google Scholar]
- 39.Jonker DJ, O’Callaghan CJ, Karapetis CS, et al. Cetuximab for the treatment of colorectal cancer. N Engl J Med. 2007;357:2040–2048. doi: 10.1056/NEJMoa071834. [DOI] [PubMed] [Google Scholar]
- 40.Karapetis CS, Khambata-Ford S, Jonker DJ, et al. K-ras mutations and benefit from Cetuximab in advanced colorectal cancer. N Engl J Med. 2008;359:1757–1765. doi: 10.1056/NEJMoa0804385. [DOI] [PubMed] [Google Scholar]
- 41.Van Cutsem E, Peeters M, Siena S, et al. Open-label Phase III trial of panitumumab plus best supportive care compared with best supportive care alone in patients with chemotherapy-refractory metastatic colorectal cancer. J Clin Oncol. 2007;25:1658–1664. doi: 10.1200/JCO.2006.08.1620. [DOI] [PubMed] [Google Scholar]
- 42.Small EJ, Schellhammer PF, Higano CS, et al. Placebo-controlled phase III trial of immunologic therapy with sipuleucel-T (APC8015) in patients with metastatic, asymptomatic hormone refractory prostate cancer. J Clin Oncol. 2006;24:3089–3094. doi: 10.1200/JCO.2005.04.5252. [DOI] [PubMed] [Google Scholar]
- 43.Higano CS, Schellhammer PF, Small EJ, et al. Integrated data from 2 randomized, double-blind, placebo-controlled, phase 3 trials of active cellular immunotherapy with sipuleucel-T in advanced prostate cancer. Cancer. 2009;115:3670–3679. doi: 10.1002/cncr.24429. [DOI] [PubMed] [Google Scholar]
- 44.Gulley JL, Arlen PM, Madan RA, et al. Immunologic and prognostic factors associated with overall survival employing a poxviral-based PSA vaccine in metastatic castrate-resistant prostate cancer. Cancer Immunol Immunother. 2010;59:663–674. doi: 10.1007/s00262-009-0782-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gulley JL, Madan RA, Schlom J. Impact of tumor volume on the potential efficacy of therapeutic vaccines. Curr Oncol. 2011;8:150–157. doi: 10.3747/co.v18i3.783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Palucka K, Ueno H, Fay J, Banchereau J. Dendritic cells and immunity against cancer. J Intern Med. 2011;269:64–73. doi: 10.1111/j.1365-2796.2010.02317.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.