

SUMMARY
Congenital contractural arachnodactyly (CCA) is caused by mutations within fibrillin-2 (FBN2), which is crucial for microfibril structure. Affected individuals may have contractures, chest wall deformities, scoliosis, abnormal ear folding and elongated limbs. We describe a novel FBN2 mutation in a woman with CCA who also has pulmonary nontuberculous mycobacterial infection. The population with pulmonary nontuberculous mycobacterial infections shares phenotypic features with CCA, such as elongated body habitus, scoliosis and pectus deformities. While it is unlikely that FBN2 defects account for susceptibility to nontuberculous mycobacterial infection in the majority of cases, the overlap between these two diseases suggests some shared pathophysiology.
Keywords: Mycobacterial, atypical; Mycobacterium avium Complex; fibrillin 2 (congenital contractural arachnodactyly) protein, human; fibrillin
CASE REPORT
A 59 year-old Caucasian woman with frequent pneumonias and prior right middle lobectomy developed weight loss, fever and cough. She was diagnosed with pulmonary Mycobacterium avium complex (MAC) infection by bronchial alveolar lavage culture and commenced treatment with ethambutol, rifabutin and clarithromycin. Her symptoms subsequently improved and antimicrobials were discontinued after 18 months. During this period, her sputum cultures were negative and have remained so off of antibiotics.
She was tall (170.2 cm) and thin (63.5 kg) (body mass index=21.9) with elongated ears, arachnodactyly with contractures and scoliosis. Sweat chloride and interferon-gamma receptor function were normal. Computerized tomography of the chest showed nodular disease and bronchiectasis of the right middle zone and lower lobe (Figure 1A). Transthoracic echocardiography showed an atrial septal aneurysm, mildly dilated aortic root, and borderline anterior mitral valve prolapse.
Figure 1.
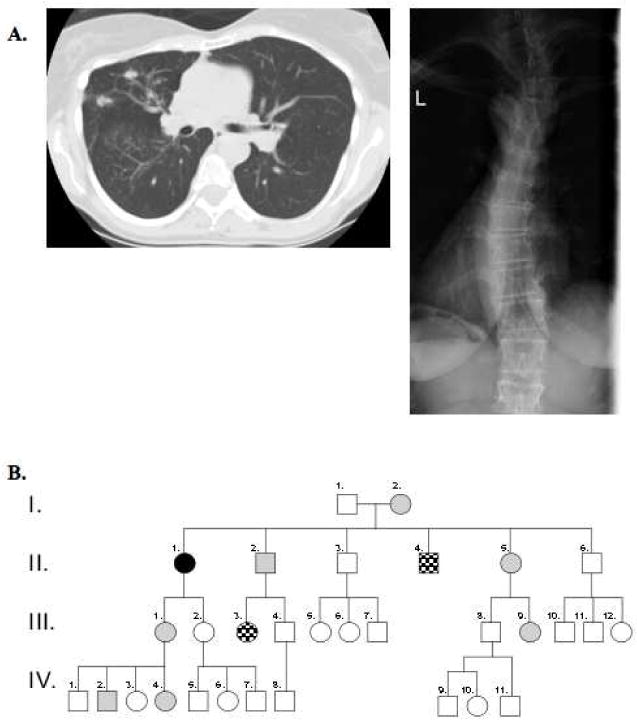
A. Computerized tomography of the chest depicting bronchiectasis and xray showing scoliosis in our patient. B. Pedigree of affected patient and family members. The proband described in this report is identified by a solid black circle (II.1). Gray shading denotes family members who have a CCA morphotype. Checkered shading denotes family members who have a CCA morphotype and are found to have the C1246G mutation in FBN2, identical to that of our proband (II.4 and III.3). Unshaded shapes represent family members who lack physical features suggestive of CCA.
She had 3 siblings (II.2, II.4, II.5), a mother (I.2), a daughter (III.1), and two grandchildren (IV.2, IV.4) with similar morphologic features (Figure 1B). Her father (I.1) did not share these features but had bronchiectasis, necessitating right pneumonectomy. He later contracted pulmonary tuberculosis. No other family members have been diagnosed with mycobacterial infections or bronchiectasis. Their family was previously described for the cardiac findings of congenital contractural arachnodactyly (CCA) prior to the knowledge that fibrillin-2 (FBN2) mutations cause CCA.1
Genomic DNA was submitted for FBN2 analysis (Connective Tissue Gene Tests). A heterozygous missense mutation at nucleotide 3736T>G was found within a calcium-binding epidermal growth factor-like domain of exon 29. This novel mutation is likely disease-causing because it lays within the region shared by most reported CCA mutations and changes a conserved cysteine to a glycine (C1246G), which is predicted to disrupt microfibril structure. Two additional family members (II.4 and III.3) with similar morphotypes were confirmed to have this mutation. No other family members underwent genetic testing.
DISCUSSION
CCA is an autosomal dominant disorder due to missense and splice site mutations in FBN2.2,3 Its phenotype overlaps with Marfan syndrome (MFS) caused by mutations in fibrillin-1 (FBN1). Clinical manifestations of CCA include contractures, arachnodactyly, dolichostenomelia, scoliosis, crumpled ears and pectus deformities.2
FBN1 and FBN2 encode glycoproteins that form extracellular microfibrils, which are necessary for organ tethering, local cellular stability, and binding to molecules that regulate cellular actions.2 Both contain regions with homology to latent transforming growth factor-beta 1 binding protein and calcium-binding domains with homology to epidermal growth factor (EGF).3 The predicted pathologic mutation of our patient lays within an EGF-like domain that contains cysteine residues, which facilitate calcium binding.
Recent studies suggest fibrillins can influence transforming growth factor-beta 1 (TGF-β1) signaling. Lungs from Fbn1 deficient mice lacked distal alveolar septation and had abnormally large distal airway caliber, and tissues had increased levels of TGF-β.4 Potential interactions of TGF-β1 and fibrillin-2 have yet to be systematically evaluated in humans with CCA or animal models with disrupted Fbn2.
FBN2 is expressed early in embryogenesis in elastic cartilage, aortic tunica media and bronchial epithelium.5 Fbn2 deficient mice have forelimbs contractures, bilateral syndactyly and disorganized microfibrils.6 Embryonic rat lungs treated with a Fbn2 antisense oligodeoxynucleotide had underdeveloped lung bud branches and collapsed conducting airways.7
CCA is not typically associated with pulmonary abnormalities. In contrast, MFS is associated with cystic disease and pneumothoraces, and older reports of bronchiectasis and pneumonias.8 It is possible that a subset of MFS patients described in the older literature actually had CCA, as they can be difficult to differentiate on phenotype alone.
Pulmonary MAC infections in individuals lacking a defined immunodeficiency have been described in persons (often women) who are otherwise healthy, nonsmoking and older.9 In one series of 63 patients with pulmonary MAC, 11% had pectus excavatum, 51% scoliosis and slender body habitus.9 This phenotype resembles the characteristics seen in CCA and MFS.
Genetic susceptibility to pulmonary nontuberculous mycobacteria (PNTM) infections remains elusive. There are associations with heterozygous mutations in the cystic fibrosis transmembrane conductance regulator but their mechanisms are unclear. Genes that regulate interferon gamma and interleukin-12 synthesis and response are strongly associated with disseminated, but not isolated pulmonary mycobacterial infections. However, the existence of a clear morphotype apparent in early adulthood associated with the development of PNTM infection in later adulthood suggests a dominant single gene effect with both somatic and immunologic components.9,10 The pathways around fibrillin and TGFβ metabolism may be highly relevant to this disease complex.
The patient’s father lacked both features of CCA and the typical phenotype seen in pulmonary MAC infections. He had bronchiectasis and developed pulmonary tuberculosis at a time when it was more prevalent in the general population.
We describe the first association of PNTM infection with a FBN2 mutation causing CCA. Although these mutations have not been classically associated with bronchiectasis or PNTM infections, the overlapping phenotype between the two syndromes is striking. Better understanding of fibrillin genes, their mutations and TGFβ metabolism should increase our knowledge of the pathogenesis of PNTM susceptibility.
Acknowledgments
FUNDING
Financial support:
This project has been funded in part with federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
This research was supported [in part] by the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Footnotes
COMPETING INTERESTS
None.
Contributor Information
Michelle L. Paulson, Email: paulsonm@niaid.nih.gov.
Kenneth N. Olivier, Email: olivierk@niaid.nih.gov.
Steven M. Holland, Email: sholland@niaid.nih.gov.
References
- 1.Anderson RA, Koch S, Camerini-Otero RD. Cardiovascular findings in congenital contractural arachnodactyly: report of an affected kindred. Am J Med Genet. 1984;18:265–271. doi: 10.1002/ajmg.1320180210. [DOI] [PubMed] [Google Scholar]
- 2.Callewaert BL, Loeys BL, Ficcadenti A, et al. Comprehensive clinical and molecular assessment of 32 probands with congenital contractural arachnodactyly: report of 14 novel mutations and review of the literature. Hum Mutat. 2009;30:334–341. doi: 10.1002/humu.20854. [DOI] [PubMed] [Google Scholar]
- 3.Frédéric MY, Monino C, Marschall C, et al. The FBN2 gene: new mutations, locus-specific database (Universal Mutation Database FBN2), and genotype-phenotype correlations. Hum Mutat. 2009;30:181–190. doi: 10.1002/humu.20794. [DOI] [PubMed] [Google Scholar]
- 4.Neptune ER, Frischmeyer PA, Arking DE, et al. Dysregulation of TGF-β activation contributes to pathogenesis in Marfan syndrome. Nat Genet. 2003;33:407–411. doi: 10.1038/ng1116. [DOI] [PubMed] [Google Scholar]
- 5.Zhang H, Hu W, Ramirez F. Developmental expression of fibrillin genes suggests heterogeneity of extracellular microfibrils. J Cell Biol. 1995;129:1165–1176. doi: 10.1083/jcb.129.4.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arteaga-Solis E, Gayraud B, Lee SY, Shum L, Sakai L, Ramirez F. Regulation of limb patterning by extracellular microfibrils. J Cell Biol. 2001;154:275–281. doi: 10.1083/jcb.200105046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang Q, Ota K, Tian Y, et al. Cloning of rat fibrillin-2 cDNA and its role in branching morphogenesis of embryonic lung. Dev Biol. 1999;212:229–242. doi: 10.1006/dbio.1999.9331. [DOI] [PubMed] [Google Scholar]
- 8.Wood JR, Bellamy D, Child AH, Citron KM. Pulmonary disease in patients with Marfan syndrome. Thorax. 1984;39:780–784. doi: 10.1136/thx.39.10.780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim RD, Greenberg DE, Ehrmantraut ME, et al. Pulmonary nontuberculous mycobacterial disease: prospective study of morphotype, laboratory and genetic features of a distinct syndrome. Am J Respir Crit Care Med. 2008;178:1066–1074. doi: 10.1164/rccm.200805-686OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Colombo RE, Hill SC, Claypool RJ, Holland SM, Olivier KN. Familial clustering of pulmonary nontuberculous mycobacterial disease. Chest. 2010;137(3):629–34. doi: 10.1378/chest.09-1173. [DOI] [PMC free article] [PubMed] [Google Scholar]