


Abstract
The phosphorylation of the RNA polymerase II (Pol II) C-terminal domain (CTD) has been shown to affect the initiation, and transition to elongation of the Pol II complex. The differential phosphorylation of serines within this domain coincides with the recruitment of factors important for pre-mRNA processing and transcriptional elongation. A role for tyrosine and threonine phosphorylation has yet to be described. The discovery of kinases that express a preference for specific residues within this sequence suggests a mechanism for the controlled recruitment and displacement of CTD-interacting partners during the transcription cycle. The last CTD repeat (CTD52) contains unique interaction sites for the only known CTD tyrosine kinases, Abl1/c-Abl and Abl2/Arg, and the serine/threonine kinase casein kinase II (CKII). Here, we show that removal or severe disruption of the last CTD repeat, but not point mutation of its CKII sites, results in its proteolytic degradation to the Pol IIb form in vivo, but does not appear to affect the specific transcription of genes. These results suggest a possible mechanism of transcription control through the proteolytic removal of the Pol II CTD.
INTRODUCTION
The large subunit of eukaryotic RNA polymerase II (Pol II LS/RBP1) differs strikingly from other eukaryotic polymerases through its possession of a unique C-terminal domain (CTD) consisting of repeats of the consensus heptapeptide sequence YSPTSPS (1). The consensus sequence is highly conserved across organisms, but the number of repeats appears to have increased through evolution (2). The repeat sequence of the CTD increasingly deviates from the consensus sequence with proximity to the C-terminus. The evolution of longer CTDs and non-consensus repeats has been speculated to attribute specific functions to different regions of the CTD (3). Non-consensus repeats may confer specificity for both the kinases that target them and the factors that subsequently bind or are displaced from them. A comparison of different organisms reveals the development of divergent sequences following the most distal, or ‘last’, CTD repeat (4). The mammalian last CTD repeat (CTD52) contains a total of 17 amino acids, forming what constitutes two potential casein kinase II (CKII) sites (5), and is essential for mediating the binding and phosphorylation of the CTD by the Abl1/c-Abl and Abl2/Arg tyrosine kinases (6,7).
Deletion of more than half of the repeats in the yeast or mouse CTD interferes with cell viability. Mice homozygous for a deletion of 13 repeats are smaller than wild-type littermates and have a high rate of neonatal lethality, suggesting that the CTD is important in growth regulation during mammalian development (8). Mammalian tissue culture experiments have demonstrated that a mutant containing 31 repeats, although capable of transcribing the hsp70A and c-fos genes (9), is growth limiting and cannot replace the endogenous Pol II LS with respect to long-term survival. A mutant containing a truncation of the CTD to five repeats is unable to transcribe a chromatin template in vivo, suggesting a role for the CTD in the correct initiation of transcription (10). Additionally, the correct processing of pre-mRNA is inhibited by truncation of the CTD (11).
The phosphorylation status of the CTD is essential for the regulation of transcription [reviewed in Dahmus (12), Palancade and Bensaude (13)]. Only the non-phosphorylated (IIA) form of Pol II can participate in the formation of a pre-initiation complex (PIC), while CTD phosphorylation is essential for transcriptional elongation (the IIO form) (14–16). The effectors of this regulation include several cyclin-dependent and stress-activated kinases, whose activities during certain stages of the transcription cycle may serve to regulate initiation, elongation and the binding of pre-mRNA processing factors to Pol II [for reviews see Bregman et al. (17), Kobor and Greenblatt (18), and Oelgeschlager (19)]. The phosphorylation of non-engaged Pol II by kinases such as ERK or CDK8/cyclin C may function to downregulate transcription by preventing the formation of new PICs (20–22). Phosphorylation of Pol II during the transition from initiation to elongation by CDK7/cyclin H of the general transcription factor TFIIH, and CDK9/cyclins T and K of the elongation factor P-TEFb, may relieve the inhibitory effects of the DSIF, NELF and mediator complexes [reviewed in Oelgeschlager (19)].
Serine is the predominant target of phosphorylation within the CTD, compared with low levels of threonine and tyrosine phosphorylation (14,23,24). The phosphorylation of Ser2 and Ser5 (YS2PTS5PS) differs dependent upon the position of the Pol II complex: a bias toward Ser5 phosphorylation is observed in promoter regions, while more Ser2 phosphorylation is observed during the transcription of coding regions (25). Such phosphorylation events may create specific phospho-amino acid motifs, thereby allowing the selective recruitment of accessory proteins to the CTD in a kinase-dependent manner. Each CTD repeat contains two potential sites (Ser/Thr–Pro) for proline-directed kinases. Phospho-Ser/Thr–Pro moieties may bind proteins containing WW domains, a phosphoprotein-binding module characterized by two invariant tryptophans, found in a wide range of signalling proteins (26,27). Similarly, following phosphorylation, the highly conserved tyrosine in every CTD repeat creates a potential site for the binding of SH2 domain-containing proteins. Indeed, the processive phosphorylation of the CTD by the Abl1 tyrosine kinase is facilitated by its own binding to the phospho-tyrosine motifs that it created, via its SH2 domain (28). However, although a requirement for phosphorylation of serines within the CTD has been shown to influence the binding of mRNA capping enzymes (29,30), splicing factors (31–33) and 3′ mRNA processing factors (34,35), no role has yet been attributed to phospho-tyrosine [reviewed in Proudfoot et al. (36) and Howe (37)].
It has long been postulated that Abl1 may affect transcription initiation as well as elongation: ectopic expression of Abl1 has been shown to activate the human immunodeficiency virus (HIV) promoter in the absence of Tat (6). Abl1 binds and phosphorylates the Pol II CTD following activation by ATM in response to ionizing radiation (IR), but the purpose of this interaction in the regulation of gene expression following IR is not known (23,38). The Abl1-related gene product, Abl2 (39), has also been shown to interact with and phosphorylate the Pol II CTD (7).
Phosphorylation of the CTD by CKII has been described previously (40), and is directed to only one of two possible sites within the last repeat (15). Nuclear CKII activity is required for cell cycle progression in the G1 phase of the cell cycle, and is implicated in the responses to genotoxic and other stresses [reviewed in Allende and Allende (41)]. The α-subunit of CKII co-localizes with productively transcribing Pol II, possibly via its interaction with the RAP74 subunit of TFIIF (42), the Pol II CTD or CDKII (43).
To investigate the purpose of these interactions, we have performed an in depth mutational analysis of the last CTD repeat in vivo. We show that removal or severe disruption of this domain has no effect on the specific transcription of genes, but results in the cleavage of the CTD from Pol II, and the appearance of the Pol IIb form.
MATERIALS AND METHODS
Cell lines and cell culture
Suspension cells were grown in RPMI 1640 medium supplemented with 10% fetal calf serum (FCS), 100 U/ml penicillin, 100 µg/ml streptomycin and 2 mM l-glutamine (Invitrogen), which in the following is referred to as growth medium. Stable cell lines were grown in the presence of 1 mg/ml G418 and 0.1 µg/ml tetracycline. Raji (44), Elijah and BL29 are human, Epstein–Barr virus (EBV)-positive Burkitt’s lymphoma (BL) cell lines. Rosi is a human, EBV-positive lymphoblastic B-cell line (LCL). Cells were transfected with Pol II LS* expression vectors by electroporation (10 µg of plasmid DNA/1 × 107 cells; 960 µF, 250 V) and polyclonal cell lines were established after selection with G418 and tetracycline. For the expression of the various recombinant large subunits of Pol II, 2 × 107 cells were washed three times with 40 ml of phosphate-buffered saline (PBS) supplemented with 1% FCS and subsequently re-suspended in 20 ml of growth medium. After 24 h, α-amanitin (2 µg/ml final concentration; Roche) was added to the medium to inhibit the endogenous Pol II.
Monolayers of HeLa cells were propagated on tissue culture dishes in Dulbecco’s modified Eagle’s medium supplemented with 10% FCS (DMEM, Gibco). When indicated, prior to harvesting, the cells were treated for 1 h with 100 µM dichloro-β-d-ribofuranosylbenzimidazole (DRB, Sigma) or with 500 ng/ml of actinomycin D (Sigma), or heat-shocked for 1 h at 45°C.
Nuclear run-on analysis
Isolation of nuclei and nuclear run-on reactions [in the presence of 2 µg/ml α-amanitin and 2.4% (w/v) sarkosyl] were carried out as previously described (10). After isolation of nuclear transcripts by Sephadex G-50 column filtration, labelled RNA was hybridized to an ATLAS human 1.2 array (Clontech) at 68°C for 48 h in 5 ml of Church buffer [0.5 M sodium phosphate pH 7.1, 7% (v/v) SDS, 0.1 mM EDTA/NaOH pH 8.0, Millipore quality H2O]. Arrays were extensively washed in succession with 1% SDS, 2× SSC; 0.5% SDS, 0.1× SSC; and 1× SSC, 1 mM EDTA at 55°C. Subsequently filters were treated for 15 min with 1× SSC, 1 mM EDTA, 2 µg/ml RNase A at room temperature and finally washed with 0.5% SDS, 0.1× SSC at 55°C. Thereafter, membranes were exposed to Kodak Biomax-MS film at –80°C with intensifying screens.
Preparation and analysis of proteins
For western blot analysis, whole-cell extracts were prepared using either Laemmli or RIPA buffer [50 mM Tris–HCl pH 7.0, 300 mM NaCl, 1% Triton X-100, 1% Na-deoxycholate, 1 mM EGTA, 1 mM phenylmethylsulfonyl fluoride (PMSF)], both of which were supplemented with protease inhibitors + EDTA (Protease Inhibitor Cocktail Tablets, Roche), as directed by the manufacturer, and phosphatase inhibitors (1 mM Na-orthovanadate, 1 mM Na-molybdate, 10 mM NaF). Cytosolic extracts or nuclear extracts were prepared as previously described (45). When indicated, the extracts were treated with 0.3, 1 or 3 U/µl of calf intestinal alkaline phosphatase (Amersham) as previously described (46). Separation was performed using 6% SDS–polyacrylamide gels. The membrane (Immobilon-P, Millipore) was stained with Ponceau S (Sigma) to verify the transfer of equal amounts of proteins.
Antibodies
The following antibodies were used during this study: high-affinity rat anti-haemagglutinin (HA) monoclonal antibody (mAb), 3F10 (Roche); mouse anti-CTD mAb, 8WG16, recognizes CTD regardless of its phosphorylation status (47); mouse anti-Pol II LS mAb, POL 3/3, recognizes the Pol II LS subunit at an evolutionarily conserved epitope located outside the CTD (48); horseradish peroxidase (HRP)-linked goat anti-rat IgG conjugate (Sigma); HRP-linked goat anti-rabbit conjugate (Promega); and HRP-linked goat anti-mouse conjugate (Promega). DEEN and DEEP polyclonal antisera were obtained from Eurogentec (Belgium) by immunizing rabbits with the Rpb1 C-terminal peptide TSPAISPDDSDEEN or a serine-phosphorylated version thereof (see Fig. 5). The specificity of the DEEN/DEEP antibodies was assayed as follows: a recombinant murine Rpb1 CTD (49) fused to GST was expressed in Escherichia coli cells and purified on glutathione–Sepharose 4B beads, according to the manufacturer’s instructions (Amersham). Purified GST–CTD (1 ng) was incubated for 30 min at 30°C with 1 mM ATP in the presence or absence of 20 µU of recombinant CKII (reconstituted holoenzyme αβ, Roche), in a total volume of 15 µl of GP buffer [20 mM sodium glycerophosphate, 5 mM MgCl2, 1 mM EGTA, 1 mM NaVO3, 10% glycerol, 1 mM dithiothreitol (DTT), 0.1% bovine serum albumin (BSA)]. CKII-phosphorylated and non-phosphorylated GST–CTD proteins were boiled in Laemmli sample buffer before SDS–PAGE/western blot analysis.
Figure 5.

CTD52 is phosphorylated by CKII in vivo. (A) Polyclonal antibodies were raised against peptides encoding CTD52 (DEEN) and a phosphorylated version thereof (DEEP). The monoclonal antibody 8WG16 recognizes the CTD consensus sequence, YSPTSPS. (B) DEEN and DEEP antibodies discriminate between non-phosphorylated and CKII-phosphorylated CTD: CKII-treated and non-treated GST–CTD were examined following western blot for their reactivity against DEEN and DEEP antibodies. The reactivity with 8WG16 is included as a loading control. (C) The immunoreactivity of Pol II from HeLa cells following DRB, actinomycin D (ActD) or heat shock (HS) treatment (∅ = non-treated control). The monoclonal antibody POL 3/3 recognizes all forms of the Pol II LS since it recognizes an epitope outside of the CTD. (D) Treatment with alkaline phosphatase regenerates DEEN activity: cytosolic (Cytosol-X) and nuclear (Nuclear-X) HeLa cell extracts were treated with increasing amounts of alkaline phosphatase, and their reactivity, along with that of whole extract (Whole-X), against DEEN and POL 3/3 antibodies was compared following western blotting.
Plasmids
A multiple cloning site (MCS) with the restriction sites AgeI, PmeI and NotI was introduced into the vector HAwt, supplied by W. Schaffner (50). Using this vector as template, overlap-PCR was performed using a primer that binds a sequence upstream of the last exon (A: 5′-GTCCCCAAACTCA CCCTGAA-3′), a primer that binds downstream of the last exon (B: 5′-CTCCTGCTGACGCACCTGTTCT-3′), and two primers that bind sequences flanking the termination codon, and containing the MCS (5′-GACCGGTTTAAACGCGG CCGCTGAGCGAACAGGGCGAAGAGCTGG-3′ and 5′- CGCCCTGTTCGCTCAGCGGCCGCGTTTAAACCGGTC GTTCTCCTC-3′). The 1.6 kb overlap-PCR product was inserted between the NgoMIV and ClaI sites of HAwt to create vector MM128-MCS. This modified sequence was subcloned into the vector LS*mock, as previously described (10), to produce LS*wtMCS. A fusion of LS*wtMCS with enhanced green fluorescent protein (EGFP) (LS*wtEGFP) was produced by insertion of a PCR product between the AgeI and NotI sites of LS*wtMCS. EGFP was amplified from the vector pEGFP-C1 (CLONTECH) using the primer pair 5′-AACGACC GGTTTGTGAGCAAGGGCGAGGAGCTGTTCACC-3′ and 5′-AAAAGGAAAGCGGCCGCGTCACTTGTACAGCTC GTCCATGCCGAG-3′.
For the production of CTD mutants, PCR was performed using LS*wtMCS as template, and the primers A and B (described above). The resulting 1.6 kb fragment, containing exon 29 of Pol II LS, was blunt-cloned into the HincII site of pUC19 (pUC19-CTD). Mutants of the distal CTD region were produced by insertion of phosphorylated linkers incorporating the modified repeat sequences between the StyI site present in the coding sequence for repeat 49, and the AgeI site present in the MCS. The exon 29 mutants (49+50, 49+52, 49+52 S9A, 49+52 S13A, 49+52 S9/13A, 49+NS52 and 49+50ATM) were removed from their pUC19-CTD subclone as NgoMIV and ClaI fragments, and cloned between the NgoMIV and ClaI sites of MM128-MCS, before cloning into LS*mock, as described above.
RESULTS
The construction and conditional expression of Pol II LS mutants
A tetracycline (Tc)-regulatable, EBV-based expression vector, LS*mock (Fig. 1A), containing the gene for the α-amanitin-resistant, Pol II LS (LS*wt) (10), was modified to enable easier mutation of the coding sequence for the CTD. An MCS was inserted into the last exon of the LS*wt sequence, adjacent to the stop codon, resulting in the addition of an extra seven amino acids to the CTD sequence (Fig. 1B). To examine the expression and in vivo localization of Pol II, a mutant fused to the EGFP was produced. Additionally, using the unique restriction enzyme sites present in the MCS and in the coding sequence for CTD repeat 49, it was possible to create mutants lacking repeats 50–52, or replace them with variations thereof. To examine the importance of the unique 52nd repeat (CTD52), we produced mutants containing a total of 50 CTD repeats, whereby the CTD52 was present (LS*49+52) or absent (LS*49+50).
Figure 1.

Establishment of cell lines conditionally expressing RNA Pol II LS* mutants. (A) A schematic drawing of the constructs used in this study. A construct containing the HA-tagged, α-amanitin-resistant large subunit of Pol II (LS*wt) was modified to include a multiple cloning site directly before the stop codon of the wild-type sequence (LS*wtMCS), allowing the insertion of the cDNA for EGFP in-frame with that of the wild-type Pol II sequence (LS*wtEGFP). Deletion mutants were produced by truncation of the CTD to 49 repeats before the addition of either the 50th (LS* 49+50) or 52nd repeat (LS*49+52). (B) Comparison of the amino acid sequence of the different mutants: Addition of an MCS results in the addition of a further seven amino acids to the wild-type CTD; mutants containing the same number of total repeats, but ending in repeat 50 or repeat 52; addition of EGFP creates a large C-terminal extension to the wild-type sequence. (C) A chemical ‘knock-in, knock-out’ system for the analysis of mutant Pol II LS*: expression of the α-amanitin-resistant polymerase is induced by removal of Tc from the cell culture medium; the endogenous Pol II is inhibited through the addition of α-amanatin 24 h following the induction of expression; localization was assessed a further 48 h later. (D) The expression and localization of an EGFP-tagged polymerase (LS*wtEGFP) in Raji cells examined using a fluorescence imaging system.
To test the properties of our mutated polymerases, their expression was induced by the removal of Tc from culture medium, followed by the addition of α-amanitin 24 h later, to deactivate the non-resistant, endogenous polymerase (Fig. 1C). In Raji cells, α-amanitin fully inhibits the endogenous Pol II within 24 h. Examination of Raji cells expressing LS*wtEGFP, using fluorescence microscopy, shows its complete nuclear localization after 48 h α-amanitin treatment (Fig. 1D).
CTD52 is essential for maintaining the viability of Raji cells
We know from previous studies that LS*wt, but not a mutant with just five repeats (LS*Δ5), can fully replace its endogenous cellular counterpart (LSwt) to maintain growth and viability in the presence of α-amanitin (10). To test whether mutation or extension of the distal CTD region influences viability, Pol II LS* mutants containing (LS*49+52, LS*wtMCS and LS*wtEGFP) or lacking CTD52 (LS*49+50) were compared (Fig. 2). The fusion of LS*wt with EGFP produces a large C-terminal extension of 238 amino acids following CTD52. As a control, a cell line ‘Mock’, transfected with empty vector (LS*mock), was also included. As observed in previous studies (10), the viability of all the cell lines dropped during the first 14 days, after which surviving cell lines (LS*49+52, LS*wtMCS and LS*wtEGFP) gradually recovered to full viability. Re-addition of Tc to a portion of the recovering cells at day 34 (LS*wtEGFP + Tc) confirmed that viability is conferred by the recombinant Pol II LS*. Despite possessing no resistant polymerase, the Mock control cell line still required 6 days before viability reached zero. This may be explained in part by the observation that the inhibition of Pol II transcription by α-amanitin leads to mRNA stabilization (10), a mechanism that may allow cells to survive a short-term block in transcription. More interestingly, however, a mutant lacking CTD52 also could not sustain the viability of Raji cells in the presence of α-amanitin: viability declines more rapidly than that of other mutants, reaching zero viability at day 12.
Figure 2.

A time course of cell viability in Raji cells expressing LS*mock, LS*49+50, LS*49+52, LS*wtMCS and LS*wtEGFP. α-Amanitin was added 24 h following the removal of Tc. The number of living (Nl) and dead cells (Nd) was determined by trypan blue staining. The percentage of viable cells (V) was calculated using the formula V = 100 × Nl/(Nl + Nd). The re-addition of Tc to a recovering cell line at day 34 (LS*wtEGFP + Tc) controls that the resistance to α-amanitin is dependent on the expression of the recombinant Pol II LS*. A representative example of several experiments is shown.
Removal of CTD52 does not affect the specific transcription of genes
Since CTD52 has been shown to be required for the interaction of Pol II with the tyrosine kinases Abl1 and Abl2 (6,7) and since Abl1 has been shown to influence transcriptional elongation at the HIV-1 promoter (6), the removal of this interaction may affect the specific transcription of genes. Given the problem of mRNA stabilization following α- amanitin treatment, nuclear run-on analysis was performed to assess the Pol II density on genes at a given time, after a given stimulus. Run-ons were performed using nuclei from a mutant containing (LS*49+52) or lacking (LS*49+50) CTD52, and the resultant RNA was hybridized to DNA arrays (Fig. 3). Abl1 has been shown to phosphorylate the Pol II CTD following activation by the ATM kinase, in response to IR (38). For this reason, this experiment was also performed using nuclei harvested 1 h post-IR, a time point at which the Pol II CTD is heavily tyrosine phosphorylated [data not shown; Baskaran et al. (38)]. Two (of six) fields, D and E from the same array are shown for each cell line and condition. No reproducible differences were found between the pattern of genes engaged by the different mutant polymerases. However, despite analysing the same number of nuclei from each cell line, a global decrease in signal for Pol II transcription can be seen for mutant LS*49+50 compared with LS*49+52. This decrease was concomitant with an ∼30% reduction in the label incorporated by LS*49+50 nuclei compared with LS*49+52. This suggests that although the pattern of gene transcription is the same, the density or activity of polymerases on these genes is reduced when CTD52 is absent.
Figure 3.

Run-on analysis of transcriptionally engaged Pol II in Raji cells expressing Pol II mutants LS*49+50 or LS*49+52. Radioactively labelled RNA from nuclear run-on reactions was hybridized to ATLAS human 1.2 arrays (Clontech). Of the six fields (A–F), two representative fields (D and E) are shown for each mutant. Cells were grown in the presence of α-amanitin 24 h prior to harvesting. The nuclei from cells additionally exposed to 12 Gy of γ-radiation (+IR) are compared with untreated cells (–IR). Nuclei were harvested 1 h post-irradiation. Experiments were performed with equal amounts of nuclei. A representative example of several experiments is shown.
Removal of CTD52 induces the appearance of the Pol IIb form
Could these results be explained by a lack of enough recombinant-Pol II LS*? All mutants used in this study contain an N-terminal HA tag to allow their expression to be distinguished from the endogenous proteins. Figure 4A demonstrates the Tc-regulatable expression of the mutants LS*49+50, LS*49+52 and LS*wtEGFP in Raji cells. For each mutant, it is possible to see the non-phosphorylated (IIa) and heavily phosphorylated (IIo) forms of the Pol LS*. Both forms of LS*wtEGFP migrate slightly slower due to their greater size. However, the expression pattern of LS*49+50 can be seen to differ from that of the other mutants by the presence of a new band that migrates at a speed of ∼180 kDa. This corresponds to a previously described form of Pol II LS (Pol IIb) originally isolated from calf thymus (1,51), and later suggested to be an artefact of sample preparation that does not occur in vivo (52). Since all our protein samples were prepared in the presence of a protease inhibitor cocktail and EDTA, it is unlikely that the degradation we see is due to poor handling. Furthermore, for each experiment, all samples were produced using the same buffer; the IIb form was only reproducibly detected in samples from mutants lacking CTD52. We are, therefore, overwhelmingly convinced that the appearance of the IIb form is not artefactual. From these data, we can conclude that since the HA tag in our mutants is N-terminal, a fragment of ∼40 kDa, similar in size to the entire CTD, is removed from the large subunit C-terminus, a property of Pol IIb. To confirm these data, mutants LS*49+50 and LS*49+52 were stably transfected into the BL cell lines BL29 and Elijah, and the LCL Rosi, where the same result was seen (Fig. 4B). In conclusion, the reduced transcription observed in Raji LS*49+50 cells, arises not from its reduced expression, but rather its destruction to the IIb form.
Figure 4.
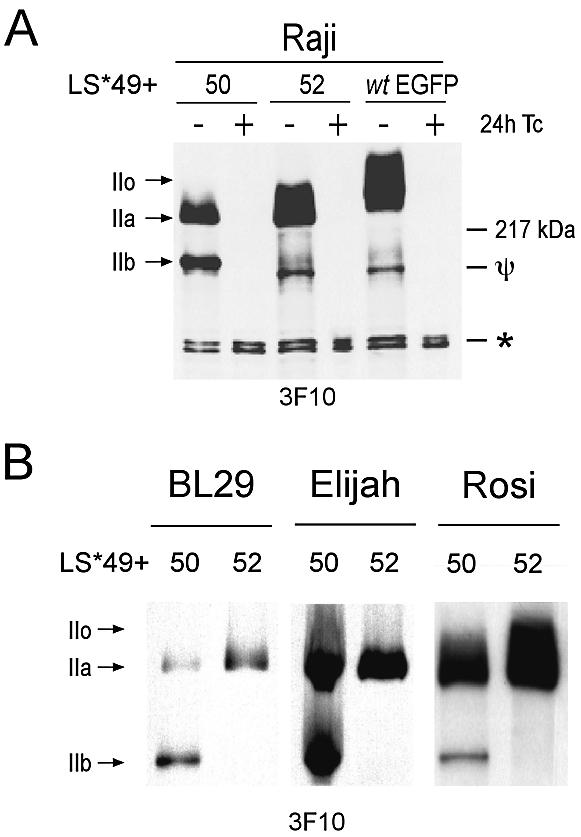
Appearance of the Pol IIb in cell lines expressing a mutant lacking CTD52. (A) RIPA extracts of stably transfected cell lines grown in the presence or absence of Tc for 24 h were examined following western blot using anti-HA antibody (3F10). The asterisk denotes a non-specific background band, which also demonstrates equal loading. Ψ signifies a degradation product of Pol II LS* other than the IIb form. (B) Expression profiling of mutants containing or lacking the last CTD repeat in the BL cell lines BL29 and Elijah, and the LCL Rosi.
CTD52 is permanently phosphorylated by CKII in vivo
Dahmus and colleagues first investigated a role for CKII in the regulation of Pol II >20 years ago (40,53,54). It was eventually established that CKII phosphorylates the CTD of Pol II LS once in vitro, probably in CTD52 (15). To investigate this interaction in vivo, polyclonal antibodies were raised against synthetic peptides encoding a portion of CTD52 (Fig. 5A). A GST–CTD fusion protein was phosphorylated in vitro by CKII before western blot analysis. Both CKII-phosphorylated, and non-CKII-phosphorylated GST–CTD were recognized by 8WG16, an antibody that recognizes consensus CTD repeats. The polyclonal antibodies DEEP and DEEN specifically recognized CKII-phosphorylated, or non-phosphorylated CTD, respectively (Fig. 5B). The reactivity of these antibodies against a panel of HeLa cell extracts was tested (Fig. 5C). The DEEP antibodies recognized all forms of Pol II LS, under all conditions, whereas no reactivity was seen using DEEN. The antibody POL 3/3 recognizes a non-CTD epitope in Pol II LS, allowing all forms to be identified. DRB treatment inhibits cyclin-dependent kinases, thus preventing CTD phosphorylation and the appearance of the IIo form. CKII is also inhibited by DRB, but no reactivity with DEEN antibodies was observed following treatment, nor was a change in the reactivity of the IIa form with DEEP observed compared with the control. Treatment with actinomycin D, or heat shock, affected the levels of the IIa and IIo forms. In addition, actinomycin D also affected the IIo/IIa ratio. Reactivity of HeLa extracts with DEEN could, however, be induced by pre-incubation of extracts with alkaline phosphatase (Fig. 5D). These data suggest that CKII phosphorylation of CTD52 is an event that takes place soon after translation of the Pol II LS, since a non-CKII-phosphorylated form was not detectable in vivo.
Disruption or removal of CTD52, but not mutation of its CKII sites, induces the Pol IIb form
The mutants tested in the previous experiments indicate that elements contained in the last repeat of Pol II CTD are important for its stability and cell viability. This element is apparently undisturbed by the addition of a small, random amino acid sequence (LS*wtMCS; Fig. 6B), or a much larger domain (LS*wtEGFP; Fig. 4A).
Figure 6.

Expression profiling of Pol II LS mutants. (A) The amino acid sequence of the last CTD repeat is shown for the mutants produced in this study. LS*wtMCS consists of the wild-type 52 repeats plus an additional seven amino acids resulting from the introduction of a multiple cloning site in the DNA sequence (MCS). Green boxes signify regions containing the CKII consensus recognition sequence S/TxxD/E. LS*49+52 consists of a total of 50 repeats, whereby the last repeat corresponds to the sequence of CTD52. Variations of this mutant were produced where one or both potential sites for phosphorylation by CKII are mutated (S→A; labelled red). Similarly, mutants LS*49+NS and LS*49+ATM contain a total of 50 repeats, where the last repeat is either a scrambled CTD52 (NS) or consists of repeat 50 of the wild-type sequence plus the c-Abl interaction domain of ATM. In addition, a mutant truncated to the 50th repeat (LS*49+50) was also produced. (B) Cell lines were cultivated in the absence of Tc for 24 h before the addition of α-amanitin and harvesting a further 24 h later. The induction of the recombinant large subunits was analysed by western blot using HA-specific antibodies (3F10). Samples were prepared using Laemmli buffer. The same blot was stripped and re-probed using CTD-specific antibodies (8WG16). The same samples were also screened using antibodies directed against CKII-phosphorylated CTD (DEEP).
In addition to its interaction with CKII, CTD52 has been identified as the essential recognition site for the tyrosine kinases Abl1 and Abl2 (6,7). In contrast to Pol II LSwt, a mutant lacking CTD52 shows no increase in tyrosine phosphorylation following exposure to IR (6). A further series of mutants were produced to better identify the important elements within CTD52 (Fig. 6A). Two mutants were produced with one point mutation, resulting in the change of a serine to an alanine in either one of the two CKII sites (LS*49+52 S9A and LS*49+52 S13A), and also a double point mutant where both sites were mutated (LS*49+52 S9/13A). To disturb Abl1 interaction, a nonsense mutant was produced where the amino acid sequence of CTD52 was randomly reconstituted (LS*49+NS52) (6). Additionally, a mutant was produced, containing repeat 50, plus a 10 amino acid sequence known to be the Abl1 recognition motif in ATM (LS*49+50ATM) (55). Whether this short motif can indeed interact with Abl1 in this context is not known, but nevertheless it provides another useful control: like CTD52, a non-consensus repeat (YSPTSPG) is flanked by an additional 10 amino acids (DPAPNPPHFP).
Stable Raji cell lines were produced for all mutants. Expression of the mutants was induced, and the cells were grown a further 24 h in α-amanitin to remove the background of the endogenous Pol II LS. Figure 6B shows the reactivity of the different mutants to a panel of antibodies, following western blotting. A cell line expressing a mutant with just five CTD repeats (including the wild-type last CTD repeat), LS*Δ5, was also included for size comparison (10). The anti-HA antibody (3F10) recognizes all mutants and forms thereof. With the exception of LS*Δ5, which cannot be so extensively phosphorylated, all mutants were present in both Pol IIa and IIo forms. Again, the Pol IIb form is present in some cell lines, migrating slightly faster than LS*Δ5. The Pol IIb form can be seen in mutants LS*49+50, LS*49+50ATM and LS*49+52NS, where CTD52 is absent or severely disrupted. The Pol IIb form was not seen in any of the CKII site mutants. The anti-CTD antibody (8WG16) shows reactivity with both the Pol IIa and IIo forms, but not with the IIb form, or the LS*Δ5 mutant. The DEEP antibody shows reactivity towards mutants containing CTD52 (LS*Δ5 and LS*49+52). DEEP does not, however, recognize a mutant containing an extended CTD52 (LS*wtMCS), indicating either that this mutant is not CKII phosphorylated, or that the extension impedes antibody recognition. Importantly, DEEP recognizes LS*49+52 S9A, but not LS*49+52 S13A, confirming that Ser13 is the target of CKII phosphorylation. These data suggest that interaction motifs within CTD52, other than those for CKII, are important for regulating the stability of Pol II LS.
DISCUSSION
Transcription and the processing of pre-mRNA are intrinsically linked events, that both require the phosphorylated CTD of the Pol II LS (36). The number of repeats that comprise the Pol II LS CTD in different organisms appears to correlate with their requirement for complex pre-mRNA processing events and transcriptional control. By both increasing the number of repeats and diverging their sequence, a greater number and diversity of factors may bind the CTD. CTD52 of the mammalian Pol II provides a potentially unique site for the binding of other factors, including the only known CTD tyrosine kinases, Abl1 and Abl2. An advanced mutational analysis of CTD52 revealed that a Pol II LS* lacking the last CTD repeat was unable to support the viability of Raji cells (Fig. 2). These data are surprising, since a mouse with 39 repeats is viable (8). However, the CTD mutant used in that study, like many others, was produced through internal deletion and contained CTD52. Abl1 has a DNA-binding domain (56), binds several transcription factors (55,57–59) and has been implicated in the regulation of c-myc (60). The activity of nuclear Abl1 can be stimulated by IR (38). We compared the density of two mutant polymerases on 1176 genes in vivo (Fig. 3). No reproducible differences could be observed between Pol II LS* mutants containing or lacking CTD52, with or without IR. This suggests that tyrosine phosphorylation of the CTD by Abl1 and Abl2 does not lead to a specific change in the density of Pol II complexes on any of the genes in our screen. However, a significant reduction in the density of polymerases on class II genes was consistently observed in all experiments when CTD52 was absent. This same effect was observed recently for the β-globin gene, also using a Pol II LS* lacking the last CTD repeat (61).
The removal of CTD52 induced the truncation of Pol II LS* to what appears to be the CTD-less, Pol IIb form (Fig. 4). A panel of mutants was produced in order to identify the element responsible for this event. A detailed examination of the CKII sites within CTD52 revealed that Ser13 is constitutively phosphorylated in vivo. However, the Pol IIb form was not observed for mutants with CTD52-CKII site mutations (Fig. 6B), indicating that CKII does not govern this event. Moreover, no truncation of Pol II LS* mutants containing a C-terminal extension (LS*wtMCS and LS*wtEGFP) was observed, indicating that the precise position of CTD52 in relation to the C-terminus is not important. Additionally, the IIb form was not observed in a truncation mutant of 31 repeats, where CTD52 was conserved, further suggesting that the presence of CTD52, and not its position, is the critical factor controlling degradation (10). An inhibitor (STI571) of Abl1 and Abl2 fails to induce the IIb form in vivo, suggesting that their kinase activities are not responsible for this effect (data not shown).
The Pol IIb form was originally identified as the largest subunit of Pol IIB (62), and was studied for more than a decade before being dismissed as an artefact of sample preparation (52). A comparison of the abilities of all three, purified Pol II subspecies (IIA, IIO and IIB) to transcribe the major late promoter of adenovirus-2 revealed that only the IIA and IIO forms are capable of promoter-specific initiation and transcription in vitro. However, all three forms were able to transcribe calf thymus DNA, which is known to contain breaks and nicks, and thus permits non-specific initiation and transcription (63). A mutant of similar size to Pol IIb, containing just five CTD repeats, is not able to initiate and transcribe a chromatin template (9,10). It is thus unlikely that the IIB form participates in transcription in vivo. Due to the precautions taken during these experiments to prevent sample degradation, and given the reproducible appearance of the IIb form only following the expression of certain mutants, we are confident now that the IIb form is not an artefact of preparation, rather a form produced in vivo by an, as yet, unknown mechanism involving CTD52.
Our data suggest that the IIb form is produced by cleavage of the CTD from the main body of the Pol II LS in the region where the linker and CTD join [for Pol II architecture see Cramer et al. (64)]. We were not able to detect the cleaved CTD fragment: it is not clear whether it is removed by a single cut, or by a protease that cuts each repeat. CTD fragments may have a negative effect on transcription elongation through titration of cyclin T (65). This observation, along with the destruction of Pol II to the inactive IIB form, may account for the observed global decrease in transcription.
Why remove the Pol II CTD, and what could be the consequences? The proteins known to interact with CTD52 are implicated in both stress responses and cell fate decisions (41,66–68). Removal of the CTD from actively transcribing polymerases may affect transcription, but would have the much greater effect of freeing Pol II from the pre-mRNA processing machinery (69). Inhibition of mRNA 3′ processing has been shown to be a consequence of DNA repair (70). It is suggested that the destruction of stalled Pol II complexes from sites of DNA damage is required to allow access of the repair machinery (71). This is not an unreasonable waste of polymerase: UV radiation induces the degradation of the whole Pol II LS via the ubiquitin–proteasome pathway (72,73).
Our findings suggest that the removal of CTD52 induces CTD degradation, whereas the remaining portion of the LS (IIb) remains relatively stable. This could be explained in that the CTD may be required for the efficient degradation of Pol II LS by the ubiquitin–proteasome pathway (74,75). The putative Ub-ligase for Pol II, Rsp5, binds to the CTD via its WW domain (76). Inefficient ubiquitylation of the CTD-less Pol IIb form could result in its increased stability. The cleavage and destruction of the CTD during or after transcription could result in the inhibition or partial completion of pre-mRNA processing. Indeed, Pol II LS* mutants lacking the last CTD repeat are deficient in the splicing and 3′ processing of mRNA precursors (61). Since these processes occur successfully in lower organisms, where a similar motif is not present (exception: Drosophila melanogaster also has an acidic C-terminal motif, FEESED), it is unlikely that these processes have become uniquely dependent on the last CTD repeat through evolution. We suggest that this motif may bind factors which, when displaced, target Pol II for degradation.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Marie Françoise Dubois for her interest in this study, and Elisabeth Kremmer for antibody production. This work was supported by grants from the Association pour la Recherche sur le Cancer to O.B. and from the Deutsche Forschungsgemeinschaft, SFB/Transregio 5 and Fonds der Chemischen Industrie to D.E.
REFERENCES
- 1.Corden J.L., Cadena,D.L., Ahearn,J.M.,Jr and Dahmus,M.E. (1985) A unique structure at the carboxyl terminus of the largest subunit of eukaryotic RNA polymerase II. Proc. Natl Acad. Sci. USA, 82, 7934–7938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stiller J.W. and Hall,B.D. (2002) Evolution of the RNA polymerase II C-terminal domain. Proc. Natl Acad. Sci. USA, 99, 6091–6096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fong N. and Bentley,D.L. (2001) Capping, splicing and 3′ processing are independently stimulated by RNA polymerase II: different functions for different segments of the CTD. Genes Dev., 15, 1783–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Allison L.A., Wong,J.K., Fitzpatrick,V.D., Moyle,M. and Ingles,C.J. (1988) The C-terminal domain of the largest subunit of RNA polymerase II of Saccharomyces cerevisiae, Drosophila melanogaster and mammals: a conserved structure with an essential function. Mol. Cell. Biol., 8, 321–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pinna L.A. (1990) Casein kinase 2: an ‘eminence grise’ in cellular regulation? Biochim. Biophys. Acta, 1054, 267–284. [DOI] [PubMed] [Google Scholar]
- 6.Baskaran R., Escobar,S.R. and Wang,J.Y. (1999) Nuclear c-Abl is a COOH-terminal repeated domain (CTD)-tyrosine (CTD)-tyrosine kinase-specific for the mammalian RNA polymerase II: possible role in transcription elongation. Cell Growth Differ., 10, 387–396. [PubMed] [Google Scholar]
- 7.Baskaran R., Chiang,G.G., Mysliwiec,T., Kruh,G.D. and Wang,J.Y. (1997) Tyrosine phosphorylation of RNA polymerase II carboxyl-terminal domain by the Abl-related gene product. J. Biol. Chem., 272, 18905–18909. [DOI] [PubMed] [Google Scholar]
- 8.Litingtung Y., Lawler,A.M., Sebald,S.M., Lee,E., Gearhart,J.D., Westphal,H. and Corden,J.L. (1999) Growth retardation and neonatal lethality in mice with a homozygous deletion in the C-terminal domain of RNA polymerase II. Mol. Gen. Genet., 261, 100–105. [DOI] [PubMed] [Google Scholar]
- 9.Meininghaus M. and Eick,D. (1999) Requirement of the carboxy-terminal domain of RNA polymerase II for the transcriptional activation of chromosomal c-fos and hsp70A genes. FEBS Lett., 446, 173–176. [DOI] [PubMed] [Google Scholar]
- 10.Meininghaus M., Chapman,R.D., Horndasch,M. and Eick,D. (2000) Conditional expression of RNA polymerase II in mammalian cells. Deletion of the carboxyl-terminal domain of the large subunit affects early steps in transcription. J. Biol. Chem., 275, 24375–24382. [DOI] [PubMed] [Google Scholar]
- 11.McCracken S., Fong,N., Yankulov,K., Ballantyne,S., Pan,G., Greenblatt,J., Patterson,S.D., Wickens,M. and Bentley,D.L. (1997) The C-terminal domain of RNA polymerase II couples mRNA processing to transcription. Nature, 385, 357–361. [DOI] [PubMed] [Google Scholar]
- 12.Dahmus M.E. (1996) Reversible phosphorylation of the C-terminal domain of RNA polymerase II. J. Biol. Chem., 271, 19009–19012. [DOI] [PubMed] [Google Scholar]
- 13.Palancade B. and Bensaude,O. (2003) Investigating RNA polymerase II carboxyl-terminal domain (CTD) phosphorylation. Eur. J. Biochem., 270, 1–12. [DOI] [PubMed] [Google Scholar]
- 14.Cadena D.L. and Dahmus,M.E. (1987) Messenger RNA synthesis in mammalian cells is catalyzed by the phosphorylated form of RNA polymerase II. J. Biol. Chem., 262, 12468–12474. [PubMed] [Google Scholar]
- 15.Payne J.M., Laybourn,P.J. and Dahmus,M.E. (1989) The transition of RNA polymerase II from initiation to elongation is associated with phosphorylation of the carboxyl-terminal domain of subunit IIa. J. Biol. Chem., 264, 19621–19629. [PubMed] [Google Scholar]
- 16.Majello B. and Napolitano,G. (2001) Control of RNA polymerase II activity by dedicated CTD kinases and phosphatases. Front. Biosci., 6, D1358–D1368. [DOI] [PubMed] [Google Scholar]
- 17.Bregman D.B., Pestell,R.G. and Kidd,V.J. (2000) Cell cycle regulation and RNA polymerase II. Front. Biosci., 5, D244–D257. [DOI] [PubMed] [Google Scholar]
- 18.Kobor M. and Greenblatt,J. (2002) Regulation of transcription elongation by phosphorylation. Biochim. Biophys. Acta, 1577, 261. [DOI] [PubMed] [Google Scholar]
- 19.Oelgeschlager T. (2002) Regulation of RNA polymerase II activity by CTD phosphorylation and cell cycle control. J. Cell. Physiol., 190, 160–169. [DOI] [PubMed] [Google Scholar]
- 20.Bonnet F., Vigneron,M., Bensaude,O. and Dubois,M.F. (1999) Transcription-independent phosphorylation of the RNA polymerase II C-terminal domain (CTD) involves ERK kinases (MEK1/2). Nucleic Acids Res., 27, 4399–4404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dubois M.F., Nguyen,V.T., Dahmus,M.E., Pages,G., Pouyssegur,J. and Bensaude,O. (1994) Enhanced phosphorylation of the C-terminal domain of RNA polymerase II upon serum stimulation of quiescent cells: possible involvement of MAP kinases. EMBO J., 13, 4787–4797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hengartner C.J., Myer,V.E., Liao,S.M., Wilson,C.J., Koh,S.S. and Young,R.A. (1998) Temporal regulation of RNA polymerase II by Srb10 and Kin28 cyclin-dependent kinases. Mol. Cell, 2, 43–53. [DOI] [PubMed] [Google Scholar]
- 23.Baskaran R., Dahmus,M.E. and Wang,J.Y. (1993) Tyrosine phosphorylation of mammalian RNA polymerase II carboxyl-terminal domain. Proc. Natl Acad. Sci. USA, 90, 11167–11171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang J. and Corden,J.L. (1991) Identification of phosphorylation sites in the repetitive carboxyl-terminal domain of the mouse RNA polymerase II largest subunit. J. Biol. Chem., 266, 2290–2296. [PubMed] [Google Scholar]
- 25.Komarnitsky P., Cho,E.J. and Buratowski,S. (2000) Different phosphorylated forms of RNA polymerase II and associated mRNA processing factors during transcription. Genes Dev., 14, 2452–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sudol M., Sliwa,K. and Russo,T. (2001) Functions of WW domains in the nucleus. FEBS Lett., 490, 190–195. [DOI] [PubMed] [Google Scholar]
- 27.Lu P.J., Zhou,X.Z., Shen,M. and Lu,K.P. (1999) Function of WW domains as phosphoserine- or phosphothreonine-binding modules. Science, 283, 1325–1328. [DOI] [PubMed] [Google Scholar]
- 28.Duyster J., Baskaran,R. and Wang,J.Y. (1995) Src homology 2 domain as a specificity determinant in the c-Abl-mediated tyrosine phosphorylation of the RNA polymerase II carboxyl-terminal repeated domain. Proc. Natl Acad. Sci. USA, 92, 1555–1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cho E.J., Takagi,T., Moore,C.R. and Buratowski,S. (1997) mRNA capping enzyme is recruited to the transcription complex by phosphorylation of the RNA polymerase II carboxy-terminal domain. Genes Dev., 11, 3319–3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pei Y., Hausmann,S., Ho,C.K., Schwer,B. and Shuman,S. (2001) The length, phosphorylation state and primary structure of the RNA polymerase II carboxyl-terminal domain dictate interactions with mRNA capping enzymes. J. Biol. Chem., 276, 28075–28082. [DOI] [PubMed] [Google Scholar]
- 31.Morris D.P. and Greenleaf,A.L. (2000) The splicing factor, Prp40, binds the phosphorylated carboxyl-terminal domain of RNA polymerase II. J. Biol. Chem., 275, 39935–39943. [DOI] [PubMed] [Google Scholar]
- 32.Hirose Y., Tacke,R. and Manley,J.L. (1999) Phosphorylated RNA polymerase II stimulates pre-mRNA splicing. Genes Dev., 13, 1234–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goldstrohm A.C., Albrecht,T.R., Sune,C., Bedford,M.T. and Garcia-Blanco,M.A. (2001) The transcription elongation factor CA150 interacts with RNA polymerase II and the pre-mRNA splicing factor SF1. Mol. Cell. Biol., 21, 7617–7628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dichtl B., Blank,D., Sadowski,M., Hubner,W., Weiser,S. and Keller,W. (2002) Yhh1p/Cft1p directly links poly(A) site recognition and RNA polymerase II transcription termination. EMBO J., 21, 4125–4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Licatalosi D.D., Geiger,G., Minet,M., Schroeder,S., Cilli,K., McNeil,J.B. and Bentley,D.L. (2002) Functional interaction of yeast pre-mRNA 3′ end processing factors with RNA polymerase II. Mol. Cell, 9, 1101–1111. [DOI] [PubMed] [Google Scholar]
- 36.Proudfoot N.J., Furger,A. and Dye,M.J. (2002) Integrating mRNA processing with transcription. Cell, 108, 501–512. [DOI] [PubMed] [Google Scholar]
- 37.Howe K. (2002) RNA polymerase II conducts a symphony of pre-mRNA processing activities. Biochim. Biophys. Acta, 1577, 308. [DOI] [PubMed] [Google Scholar]
- 38.Baskaran R., Wood,L.D., Whitaker,L.L., Canman,C.E., Morgan,S.E., Xu,Y., Barlow,C., Baltimore,D., Wynshaw-Boris,A., Kastan,M.B. et al. (1997) Ataxia telangiectasia mutant protein activates c-Abl tyrosine kinase in response to ionizing radiation. Nature, 387, 516–519. [DOI] [PubMed] [Google Scholar]
- 39.Perego R., Ron,D. and Kruh,G.D. (1991) Arg encodes a widely expressed 145 kDa protein-tyrosine kinase. Oncogene, 6, 1899–1902. [PubMed] [Google Scholar]
- 40.Dahmus M.E. (1981) Phosphorylation of eukaryotic DNA-dependent RNA polymerase. Identification of calf thymus RNA polymerase subunits phosphorylated by two purified protein kinases, correlation with in vivo sites of phosphorylation in HeLa cell RNA polymerase II. J. Biol. Chem., 256, 3332–3339. [PubMed] [Google Scholar]
- 41.Allende J.E. and Allende,C.C. (1995) Protein kinases. 4. Protein kinase CK2: an enzyme with multiple substrates and a puzzling regulation. FASEB J., 9, 313–323. [DOI] [PubMed] [Google Scholar]
- 42.Egyhazi E., Ossoinak,A., Filhol-Cochet,O., Cochet,C. and Pigon,A. (1999) The binding of the alpha subunit of protein kinase CK2 and RAP74 subunit of TFIIF to protein-coding genes in living cells is DRB sensitive. Mol. Cell. Biochem., 191, 149–159. [PubMed] [Google Scholar]
- 43.Trembley J.H., Hu,D., Slaughter,C.A., Lahti,J.M. and Kidd,V.J. (2003) Casein kinase 2 interacts with cyclin-dependent kinase II (CDKII) in vivo and phosphorylates both the RNA polymerase II carboxyl-terminal domain and CDKII in vitro. J. Biol. Chem., 278, 2265–2270. [DOI] [PubMed] [Google Scholar]
- 44.Pulvertaft R.J.V. (1964) Cytology of Burkitt’s tumour (African lymphoma). Lancet, 1, 238–240. [DOI] [PubMed] [Google Scholar]
- 45.Dignam J.D., Lebovitz,R.M. and Roeder,R.G. (1983) Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res., 11, 147–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Palancade B., Dubois,M.F. and Bensaude,O. (2002) FCP1 phosphorylation by casein kinase 2 enhances binding to TFIIF and RNA polymerase II carboxyl-terminal domain phosphatase activity. J. Biol. Chem., 277, 36061–36067. [DOI] [PubMed] [Google Scholar]
- 47.Thompson N.E., Steinberg,T.H., Aronson,D.B. and Burgess,R.R. (1989) Inhibition of in vivo and in vitro transcription by monoclonal antibodies prepared against wheat germ RNA polymerase II that react with the heptapeptide repeat of eukaryotic RNA polymerase II. J. Biol. Chem., 264, 11511–11520. [PubMed] [Google Scholar]
- 48.Kontermann R.E., Liu,Z., Schulze,R.A., Sommer,K.A., Queitsch,I., Dubel,S., Kipriyanov,S.M., Breitling,F. and Bautz,E.K. (1995) Characterization of the epitope recognized by a monoclonal antibody directed against the largest subunit of Drosophila RNA polymerase II. Biol. Chem. Hoppe Seyler, 376, 473–481. [DOI] [PubMed] [Google Scholar]
- 49.Peterson S.R., Dvir,A., Anderson,C.W. and Dynan,W.S. (1992) DNA binding provides a signal for phosphorylation of the RNA polymerase II heptapeptide repeats. Genes Dev., 6, 426–438. [DOI] [PubMed] [Google Scholar]
- 50.Gerber H.P., Hagmann,M., Seipel,K., Georgiev,O., West,M.A., Litingtung,Y., Schaffner,W. and Corden,J.L. (1995) RNA polymerase II C-terminal domain required for enhancer-driven transcription. Nature, 374, 660–662. [DOI] [PubMed] [Google Scholar]
- 51.Dahmus M.E. (1983) Structural relationship between the large subunits of calf thymus RNA polymerase II. J. Biol. Chem., 258, 3956–3960. [PubMed] [Google Scholar]
- 52.Kim W.Y. and Dahmus,M.E. (1986) Immunochemical analysis of mammalian RNA polymerase II subspecies. Stability and relative in vivo concentration. J. Biol. Chem., 261, 14219–14225. [PubMed] [Google Scholar]
- 53.Dahmus M.E. (1981) Purification and properties of calf thymus casein kinases I and II. J. Biol. Chem., 256, 3319–3325. [PubMed] [Google Scholar]
- 54.Dahmus G.K., Glover,C.V., Brutlag,D.L. and Dahmus,M.E. (1984) Similarities in structure and function of calf thymus and Drosophila casein kinase II. J. Biol. Chem., 259, 9001–9006. [PubMed] [Google Scholar]
- 55.Shafman T., Khanna,K.K., Kedar,P., Spring,K., Kozlov,S., Yen,T., Hobson,K., Gatei,M., Zhang,N., Watters,D. et al. (1997) Interaction between ATM protein and c-Abl in response to DNA damage. Nature, 387, 520–523. [DOI] [PubMed] [Google Scholar]
- 56.Kipreos E.T. and Wang,J.Y. (1992) Cell cycle-regulated binding of c-Abl tyrosine kinase to DNA. Science, 256, 382–385. [DOI] [PubMed] [Google Scholar]
- 57.Welch P.J. and Wang,J.Y. (1993) A C-terminal protein-binding domain in the retinoblastoma protein regulates nuclear c-Abl tyrosine kinase in the cell cycle. Cell, 75, 779–790. [DOI] [PubMed] [Google Scholar]
- 58.Yuan Z.M., Shioya,H., Ishiko,T., Sun,X., Gu,J., Huang,Y.Y., Lu,H., Kharbanda,S., Weichselbaum,R. and Kufe,D. (1999) p73 is regulated by tyrosine kinase c-Abl in the apoptotic response to DNA damage. Nature, 399, 814–817. [DOI] [PubMed] [Google Scholar]
- 59.Kharbanda S., Pandey,P., Jin,S., Inoue,S., Bharti,A., Yuan,Z.M., Weichselbaum,R., Weaver,D. and Kufe,D. (1997) Functional interaction between DNA-PK and c-Abl in response to DNA damage. Nature, 386, 732–735. [DOI] [PubMed] [Google Scholar]
- 60.Birchenall-Roberts M.C., Yoo,Y.D., Bertolette,D.C., 3rd, Lee,K.H., Turley,J.M., Bang,O.S., Ruscetti,F.W. and Kim,S.J. (1997) The p120-v-Abl protein interacts with E2F-1 and regulates E2F-1 transcriptional activity. J. Biol. Chem., 272, 8905–8911. [DOI] [PubMed] [Google Scholar]
- 61.Fong N., Bird,G., Vigneron,M. and Bentley,D.L. (2003) A 10 residue motif at the C-terminus of the RNA pol II CTD is required for transcription, splicing and 3′ end processing. EMBO J., 22, 4274–4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schwartz L.B. and Roeder,R.G. (1975) Purification and subunit structure of deoxyribonucleic acid-dependent ribonucleic acid polymerase II from the mouse plasmacytoma, MOPC 315. J. Biol. Chem., 250, 3221–3228. [PubMed] [Google Scholar]
- 63.Dahmus M.E. and Kedinger,C. (1983) Transcription of adenovirus-2 major late promoter inhibited by monoclonal antibody directed against RNA polymerases IIO and IIA. J. Biol. Chem., 258, 2303–2307. [PubMed] [Google Scholar]
- 64.Cramer P., Bushnell,D.A., Fu,J., Gnatt,A.L., Maier-Davis,B., Thompson,N.E., Burgess,R.R., Edwards,A.M., David,P.R. and Kornberg,R.D. (2000) Architecture of RNA polymerase II and implications for the transcription mechanism. Science, 288, 640–649. [DOI] [PubMed] [Google Scholar]
- 65.Zhang F., Barboric,M., Blackwell,T.K. and Peterlin,B.M. (2003) A model of repression: CTD analogs and PIE-1 inhibit transcriptional elongation by P-TEFb. Genes Dev., 17,748–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang J.Y. (2000) Regulation of cell death by the Abl tyrosine kinase. Oncogene, 19, 5643–5650. [DOI] [PubMed] [Google Scholar]
- 67.Wang J.Y. and Ki,S.W. (2001) Choosing between growth arrest and apoptosis through the retinoblastoma tumour suppressor protein, Abl and p73. Biochem. Soc. Trans, 29, 666–673. [DOI] [PubMed] [Google Scholar]
- 68.Cao C., Ren,X., Kharbanda,S., Koleske,A., Prasad,K.V. and Kufe,D. (2001) The ARG tyrosine kinase interacts with Siva-1 in the apoptotic response to oxidative stress. J. Biol. Chem., 276, 11465–11468. [DOI] [PubMed] [Google Scholar]
- 69.Misteli T. and Spector,D.L. (1999) RNA polymerase II targets pre-mRNA splicing factors to transcription sites in vivo. Mol. Cell, 3, 697–705. [DOI] [PubMed] [Google Scholar]
- 70.Kleiman F.E. and Manley,J.L. (2001) The BARD1–CstF-50 interaction links mRNA 3′ end formation to DNA damage and tumor suppression. Cell, 104, 743–753. [DOI] [PubMed] [Google Scholar]
- 71.Citterio E., Vermeulen,W. and Hoeijmakers,J.H. (2000) Transcriptional healing. Cell, 101, 447–450. [DOI] [PubMed] [Google Scholar]
- 72.Ratner J.N., Balasubramanian,B., Corden,J., Warren,S.L. and Bregman,D.B. (1998) Ultraviolet radiation-induced ubiquitination and proteasomal degradation of the large subunit of RNA polymerase II. Implications for transcription-coupled DNA repair. J. Biol. Chem., 273, 5184–5189. [DOI] [PubMed] [Google Scholar]
- 73.Luo Z., Zheng,J., Lu,Y. and Bregman,D.B. (2001) Ultraviolet radiation alters the phosphorylation of RNA polymerase II large subunit and accelerates its proteasome-dependent degradation. Mutat. Res., 486, 259–274. [DOI] [PubMed] [Google Scholar]
- 74.Mitsui A. and Sharp,P.A. (1999) Ubiquitination of RNA polymerase II large subunit signaled by phosphorylation of carboxyl-terminal domain. Proc. Natl Acad. Sci. USA, 96, 6054–6059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Muratani M. and Tansey,W.P. (2003) How the ubiquitin–proteasome system controls transcription. Nature Rev. Mol. Cell. Biol., 4, 192–201. [DOI] [PubMed] [Google Scholar]
- 76.Chang A., Cheang,S., Espanel,X. and Sudol,M. (2000) Rsp5 WW domains interact directly with the carboxyl-terminal domain of RNA polymerase II. J. Biol. Chem., 275, 20562–20571. [DOI] [PubMed] [Google Scholar]