

Abstract
Background:
Throughout the world, bloodstream infections (BSIs) are associated with high rates of morbidity and mortality. Rapid pathogens identification is central significance for the outcome of the patient than culture techniques for microbial identification. To develop an end point multiplex PCR to identify a group of bacteria including Enterococcus spp., Pseudomons aeruginosa, Staphylococcus spp., Acinetobacter baumannii, 16S rDNA, and Drosophila Melanogaster were used as internal control (IC).
Materials and Methods:
Design of primers was done using Mega4, Allel ID6, Oligo6 and Oligo analyzer softwares. Genetic targets for primer designing and identification of genus Enterococcus spp., Staphylococcus spp., and species of Acinetobacter baumannii, Pseudomons aeruginosa, included the rpoB, rpoB and gyrA, sss respectively. Then PCR and multiplex PCR were performed
Results:
The intended specificity was obtained for the bacteria, which used in this study and there wasn't seen any unspecific amplification by the multiplex PCR. The test showed a sensitivity ranging from 1 to 100 target copies per reaction depending on the bacterial species.
Conclusions:
The presented multiplex PCR offers a rapid and accurate molecular diagnostic tool for simultaneous detection of some pathogenic microorganisms. The IC exists in the multiplex PCR accompanied by other primers in the system, can serve as a simple, cost- effective internal control for the multiplex PCR assay.
Keywords: Conventional multiplex polymerase chain reaction, primers design, bacterimia
INTRODUCTION
Bloodstream infections (BSIs) are a life-threatening condition with a mortality rate, especially in intensive care Unit (ICU) and neutropenic patients.[1] Furthermore, bacteremias most commonly appear along with other serious infections such as urinary tract infections, endocarditits, kidney and bowel infections. Pathogenic bacteria are the most frequent causes of bloodstream infection, although fungi can also be isolated in a minority of patients. Thus, the rapid and accurate identification of bacteremia and its causative organisms are prerequisite for successful therapy. It is estimated that about 750,000 patients develop bacteremia and fungemia with associated mortality of 18% (community onset BSI) to 35% (nosocomial BSI) each year in the United State.[1,2] The current gold standard of bloodstream microbial detection and identification is automatic, continuous monitoring liquid culture, followed by gram stain, sub culturing and use of phenotypic methods.[3] Thus, in most cases, antibiotic therapy is initiated on the basis of clinical criteria. Inadequate antimicrobial treatment regimens have been associated with the emergence of drug-resistant strains, increasing treatment costs and mortality rates, especially among critically ill patients. A few methods for rapid identification of pathogenic bacteria in blood have been described in earlier studies. These were based on a fluorescence in situ hybridization assay[4,5] using fluorescent-labeled oligonucleotide probes,[6] analysis of the 16S rDNA gene,[7,8] and direct identification of bacteria using the Vitek system[9] or the polymerase chain reaction.[10,11,12] Most of the methods described to date are unspecific and use 16S “universal” primers.[13] Prevalent bacteria that might cause bacteremia have been reported. These include: Coagulase-negative staphylococci (CoNS), mainly S. epidermidis, which can cause bacteremia despite its existence in the normal human microflora.[14] Bacteremia caused by P. aeruginosa[15] and A. baumannii[16] is related to high mortality rates, especially in hospitalized patients. S. aureus and Klebsiella pneumoniae have long been known as agents that can cause nosocomial bacteremia.[17] In this study, we developed a multiplex PCR method for the molecular identification and definition of the frequent pathogens in bacteremia. A unique sequence for each bacterium was chosen in order to avoid the binding of unspecific primers.
This technique is based on synchronized amplification of distinct segments of target DNA by employment of two or more primer pairs in a single reaction tube, which, in turn, usually means reduced costs and time necessity. The aims of the present study was to develop a panel of primers for PCR amplification of specific DNA fragments of microorganisms associated with sepsis according to a unique protocol for a faster confirmatory diagnosis.
MATERIALS AND METHODS
Primer design
Firstly, specific gene included rpoB (E. faecalis, S.aureus), gyrA (A. baumannii), SSS (P. aeruginosa) 16S rDNA (Eubacteria) and chromosome X (Drosophila Melanogaster) was selected, and then the specificity of each gene was compared to all microorganisms by NCBI blast. After that, a panel of primers was carried out using Mega 4, Allel ID6 software and Oligo 6 and Oligo analyzer for checking out annealing temperature and multiplex condition respectively. The primer set chosen amplified 684, 1505, 370, 118, 190, and 246 base-pair fragment in the Drosophila Melanogaster chromosome, 16S rDNA, E. faecalis, S. aureus, P. aeruginosa, and A. baumannii and primers were obtained from Faza Biotech, Iran [Table 1]. To allow distinction between the signal of specific bacteria and internal control sequence, specific primers with different size were used. The size of internal control was 684 bp and due to different size between bacteria and internal control, they separately exactly on the agarose gel.
Table 1.
List of oligonucleotide primers used for conventional multiplex PCR amplification

Determination of minimal analytical sensitivity and specificity of bacteria and IC
The PCR products of 16S rDNA, rpoB, sss, gyrA and IC were cut out from the agarose gel, purified by the QIAquick Gel Extraction kit (Qiagen, Germany) and ligated with the pTZ57R/T (as a TA vector). The ligation mixture was transformed into E. coli DH5α strain and the recombinants were selected on LB agar containing ampicilin (100 μg/ml). Recombinant plasmid DNA was purified by standard method and subjected for further analyses. The colony PCR was done and the correct sequence of 684 bp insert was verified by sequence analysis (by Alpha sequence, Iran). A series of 10 fold dilutions of the PCR products containing plasmid, ranging from 1 to 108 copies/reaction were prepared and stored at-80 until used.
Microorganisms
Reference strains of organisms such as, A. baumannii (ATCC 19606), P. aeruginosa (ATCC 27853), E. faecalis (ATCC 29212) and S. aureus (ATCC 29213), E. coli (ATCC 25922), Enterobacter aeroginosa (ATCC 13044), Proteus mirabilis (ATCC 7002), K. pneumonia (ATCC 13882), Salmonella enterica (ATCC 35640), Shigella boydii (ATCC 9207) were purchased of reference laboratory of Iran and Pasture Institute of Iran.
DNA isolation
Bacterium genome was isolated from the standard strain maintained in solid media using the QIAamp DNA mini blood kit (Qiagen, Germany). Briefly, remove bacteria from culture plate with an inoculation loop and suspend in 180 μl of buffer ATL by vigorous stirring. Add 20 μl proteinase k, mix by vortexing and incubate at 56°C for 1-3 h, binding of the DNA to the silica membrane and washing was carried out as described in the QIAamp protocol. Pure genome was eluted from the membrane in 100 μl of TE buffer. Qualitative and quantitative assessment of DNA preparation was performed through agarose gel and spectrophotometric measurements.
Polymerase chain reaction and multiplex polymerase chain reaction
Polymerase chain reaction (PCR) conditions were optimized according to the manual of the emerald Amp MATHSP CR Master Mix 2X premix Bio Inc, (Takara, Japan). Optimal condition for annealing temperature was 60°C. As a template, 5 μl extracted DNA was added resulting in a total volume of 25 μl. Amplification started with a cycle of 4 min at 94°C, followed by 35 cycles of denaturation at 94°C for 30 second, annealing at 60°C for 30 second and extension at 72°C for 1 min, subsequently final extension at 72°C for 10 min. Amplification and detection were carried out using Mastercycler personal (eppendorf, Germany). The amplicons obtained were submitted to electrophoresis in 1% agarose gel and stained by ethidiume bromide (0.5 μg/ml) for UV light analysis and digitized (UVIDOC-CF08.XD. Multiplex PCR conditions were the same as PCR system but, two parallel reactions (Gram Positive, Gram Negative) was done that gram positive included IC, 16S r-DNA, E. faecalis, S. aureus, and gram negative included IC, 16S r-DNA, P. aeruginosa, and A. baumannii that contain the identical annealing temperature for detection of bacteremia respectively.
RESULTS
DNA extraction and assay performance
The DNA of all 10 bacterial species (for detail see Microorganisms section) was successfully extracted directly from bacterial culture, and then further analyzed by PCR using DNA free reagents. The conventional PCR was first carried out as a singleplex to show the difference gene amplicon sizes of the seven organisms. The banding patterns in the gel analysis are shown in Figure 1. Then the conventional multiplex PCR was carried out in groups of 3 targets in 4 sets which showed distinct banding patterns in the gel analysis as it was not possible to carry out all targets in a single reaction tube. This is shown in Figure 2. The 16S rDNA, rpoB, sss, gyrA and IC genes were amplified using their respective primers and they showed distinct bands in the regions corresponding to their molecular weight, which is compared using the 100 bp DNA ladder as a marker.
Figure 1.
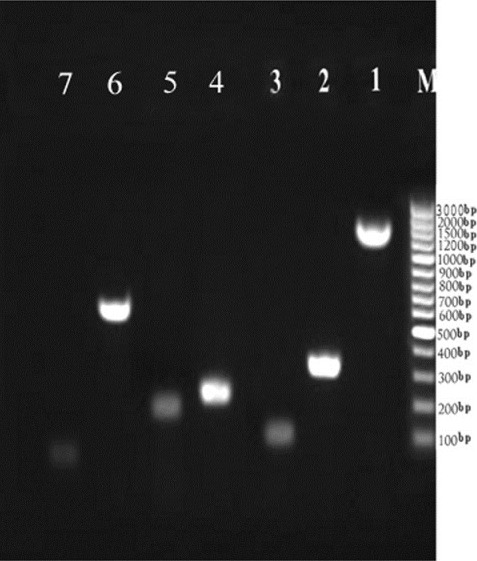
Simultaneous amplification in separate vessel (1) 16S rDNA (1505 bp), (2) E.feacalis (370 bp), (3) S.aureus (118 bp), (4) A.baumani (246 bp), (5) P.aeruginosa (190 bp), (6) Internal control (spiked IC), (7) Human DNA with optimized common annealing temperature (60°C). The bacterial concentration is (0.5 McFarland) of each strain. (M) Molecular Standard is indicated on the right (GenRuler 100 bp, Fermentas)
Figure 2.
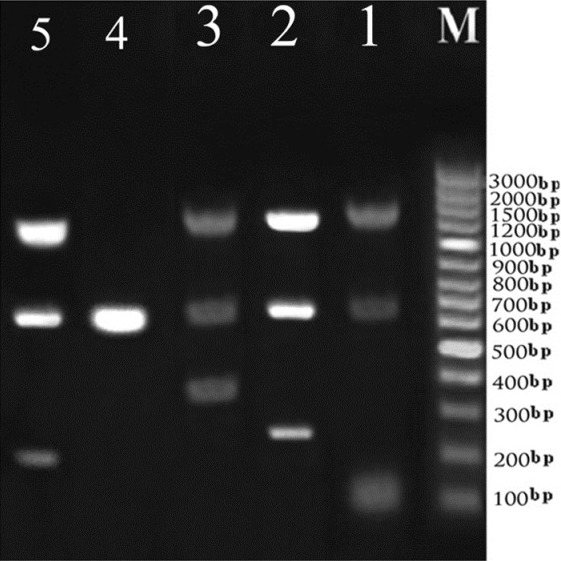
Amplification pattern of: (Lane 1) DNA derived from16S rDNA (1505 bp), spiked IC (684 bp) and S.aureus (118 bp), (lane 2) DNA derived from16S rDNA (1505 bp), spiked IC (684 bp) and A.baumanii (246 bp), (lane 3) DNA dfrived from16S rDNA (1505 bp), spiked IC (684 bp) and E.faecalis (370 bp), (lane 4) DNA derived from human DNA and spiked IC (684 bp), used as negative control, (lane 5)) DNA derived from16S rDNA(1505 bp), spiked IC (684 bp) and P.aeruginosa (189 bp) (M) Molecular Standard is indicated on the right (GenRuler 100 bp, Fermentas)
Analytical sensitivity and specificity of the test
To determine the sensitivity of the assay, 8 different dilution of IC and all of the bacteria, which mentioned above were carried out. The analytical sensitivity was 1, 100, 10, 10, 100, and 100 copies/reaction for IC, 16S rDNA, P. aeruginosa, A. baumannii, E. faecalis and S. aureus respectively. To determine the specificity of the assay, human DNA genome, A. baumannii (ATCC19606), P. aeruginosa (ATCC27853), E. faecalis (ATCC29212), S. aureus (ATCC29213) were used for control with internal control in concomitant and there was not seen any amplification [Figure 1].
DISCUSSION
The aim of the present study was to develop a rapid and simple technique for the specific detection and identification of the most common pathogenic bacteria in bacteremia. Multiplex PCR has been used successfully for rapid detection, with high specificity and sensitivity of various pathogenic bacteria from environmental water[18] and food products.[19]
After more than a century of use, the accepted widespread use of conventional blood cultures is under pressure. Improvement of culture techniques to increase sensitivity and speed of detection seems to have reached its maximum, and progress is negligible. Although the classical blood culture currently in use as imperfect gold standard for bacteremia, in many cases, blood culture is too slow or insufficiently sensitive (for fastidious organisms and in patients receiving antimicrobial treatment); therefore it has low importance as a diagnostic tool. Development of other options is essential to improve the clinical benefit of detection of pathogens in blood.[20,21,22] Conventional PCR and multiplex PCR has been employed to detect the most common pathogenic bacteria included P. aeruginosa, and A. baumannii Enterococcus spp., S. aureus and coagulase negative staphylococcus microorganisms related to sepsis show higher sensitivity and faster method than blood culture. In this study we demonstrated that the PCR method enables detection and identification of the common pathogens involved in bacteremia within a few hours of the reception samples. In our study we used IC that is introduced into each sample and coamplified with target nucleotide acid for detecting all of DNA extraction and PCR reaction process. Furthermore, direct detection and identification of above mentioned agents cause sepsis in blood or other specimens for rapid diagnosis of BSI by molecular approach is a promising idea since it will facilitate early appropriate pathogen-driven therapy.[22] The sensitivity of the assay is defined as a copy number, which will yield positive results in 1-100 copies/reaction and the specificity of the assay is yielded a detectable DNA fragment of expected molecular weight only in the presence of their respective DNA template gave negative result with other bacteria
The time required for specific bacterial pathogen diagnosis using the multiplex PCR developed in the present study is around 5 hours. The multiplex feature of this assay is optional, if so preferred it can be used as triplex assay. This makes the assay adaptable to circumstances that may not require the simultaneous detection of all the targets for a diagnostic decision.
CONCLUSION
In summary, we have developed a multiplex PCR which associated with internal control that is introduced into each test sample and is co amplified with target nucleic acid from the clinical specimens. We conclude that the molecular method developed is a reliable and fast method for detecting sepsis related to Eubacteria, A. baumannii, P. aeruginosa, E. faecalis, S. aureus. However, this method has to assess its role in clinical samples and measured its sensitivity and specificity in analytical step, but further studies must be undertaken to confirm its application.
ACKNOWLEDGMENTS
The authors are grateful to vice chancellor for research Isfahan University of medical sciences for financial support of the present study. This study is a cross-sectional model that performed in Isfahan University of medical science, Isfahan, Iran, 2011-2012 part of research project number 390011.
Footnotes
Source of Support: Isfahan University of medical sciences
Conflict of Interest: None declared
REFERENCES
- 1.Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: Analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29:1303–10. doi: 10.1097/00003246-200107000-00002. [DOI] [PubMed] [Google Scholar]
- 2.Martin GS, Mannino DM, Eaton S, Moss M. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med. 2003;348:1546–54. doi: 10.1056/NEJMoa022139. [DOI] [PubMed] [Google Scholar]
- 3.Magadia RR, Weinstein MP. Laboratory diagnosis of bacteremia and fungemia. Infect Dis Clin North Am. 2001;15:1009–24. doi: 10.1016/s0891-5520(05)70184-7. [DOI] [PubMed] [Google Scholar]
- 4.Søgaard M, Stender H, Schønheyder HC. Direct identification of major blood culture pathogens, including Pseudomonas aeruginosa and Escherichia coli, by a panel of fluorescence in situ hybridization assays using peptide nucleic acid probes. J Clin Microbiol. 2005;43:1947. doi: 10.1128/JCM.43.4.1947-1949.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jansen GJ, Mooibroek M, Idema J, Harmsen HJ, Welling GW, Degener JE. Rapid identification of bacteria in blood cultures by using fluorescently labeled oligonucleotide probes. J Clin Microbiol. 2000;38:814–7. doi: 10.1128/jcm.38.2.814-817.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Christensen JE, Stencil JA, Reed KD. Rapid identification of bacteria from positive blood cultures by terminal restriction fragment length polymorphism profile analysis of the 16S rRNA gene. J Clin Microbiol. 2003;41:3790–800. doi: 10.1128/JCM.41.8.3790-3800.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Turenne CY, Witwicki E, Hoban DJ, Karlowsky JA, Kabani AM. Rapid identification of bacteria from positive blood cultures by fluorescence-based PCR-single-strand conformation polymorphism analysis of the 16S rRNA gene. J Clin Microbiol. 2000;38:513–20. doi: 10.1128/jcm.38.2.513-520.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Cueto M, Ceballos E, Martinez-Martinez L, Perea EJ, Pascual A. Use of positive blood cultures for direct identification and susceptibility testing with the vitek 2 system. J Clin Microbiol. 2004;42:3734–8. doi: 10.1128/JCM.42.8.3734-3738.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kurupati P, Chow C, Kumarasinghe G, Poh CL. Rapid detection of Klebsiella pneumoniae from blood culture bottles by real-time PCR. J Clin Microbiol. 2004;42:1337–40. doi: 10.1128/JCM.42.3.1337-1340.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Laforgia N, Coppola B, Carbone R, Grassi A, Mautone A, Iolascon A. Rapid detection of neonatal sepsis using polymerase chain reaction. Acta Paediatr. 1997;86:1097–9. doi: 10.1111/j.1651-2227.1997.tb14815.x. [DOI] [PubMed] [Google Scholar]
- 11.Louie L, Goodfellow J, Mathieu P, Glatt A, Louie M, Simor AE. Rapid detection of methicillin-resistant staphylococci from blood culture bottles by using a multiplex PCR assay. J Clin Microbiol. 2002;40:2786–90. doi: 10.1128/JCM.40.8.2786-2790.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rothman RE, Majmudar MD, Kelen GD, Madico G, Gaydos CA, Walker T, et al. Detection of bacteremia in emergency department patients at risk for infective endocarditis using universal 16S rRNA primers in a decontaminated polymerase chain reaction assay. J Infect Dis. 2002;186:1677–81. doi: 10.1086/345367. [DOI] [PubMed] [Google Scholar]
- 13.Fujita S, Senda Y, Iwagami T, Hashimoto T. Rapid identification of staphylococcal strains from positive-testing blood culture bottles by internal transcribed spacer PCR followed by microchip gel electrophoresis. J Clin Microbiol. 2005;43:1149–57. doi: 10.1128/JCM.43.3.1149-1157.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kang CI, Kim SH, Kim HB, Park SW, Choe YJ, Oh MD, et al. Pseudomonas aeruginosa bacteremia: Risk factors for mortality and influence of delayed receipt of effective antimicrobial therapy on clinical outcome. Clin Infect Dis. 2003;37:745–51. doi: 10.1086/377200. [DOI] [PubMed] [Google Scholar]
- 15.Bergogne-Bérézin E, Towner KJ. Acinetobacter spp. as nosocomial pathogens: Microbiological, clinical, and epidemiological features. Clin Microbiol Rev. 1996;9:148–65. doi: 10.1128/cmr.9.2.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ca atay AA, Ozcan PE, Gulec L, Ince N, Tugrul S, Ozsut H, et al. Risk factors for mortality of nosocomial bacteraemia in intensive care units. Med Princ Pract. 2007;16:187–92. doi: 10.1159/000100388. [DOI] [PubMed] [Google Scholar]
- 17.Cheng AC, West TE, Peacock SJ. Surviving sepsis in developing countries. Crit Care Med. 2008;36:2487–8. doi: 10.1097/CCM.0b013e318177762d. [DOI] [PubMed] [Google Scholar]
- 18.Bej AK, DiCesare JL, Haff L, Atlas RM. Detection of Escherichia coli and Shigella spp. in water by using the polymerase chain reaction and gene probes for uid. Appl Environ Microbiol. 1991;57:1013–7. doi: 10.1128/aem.57.4.1013-1017.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sabet NS, Subramaniam G, Navaratnam P, Sekaran SD. Simultaneous species identification and detection of methicillin resistance in staphylococci using triplex real-time PCR assay. Diagn Microbiol Infect Dis. 2006;56:13–8. doi: 10.1016/j.diagmicrobio.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 20.Xu J, Millar BC, Moore JE, Murphy K, Webb H, Fox AJ, et al. Employment of broad-range 16S rRNA PCR to detect aetiological agents of infection from clinical specimens in patients with acute meningitis-rapid separation of 16S rRNA PCR amplicons without the need for cloning. J Appl Microbiol. 2003;94:197–206. doi: 10.1046/j.1365-2672.2003.01839.x. [DOI] [PubMed] [Google Scholar]
- 21.Mancini N, Carletti S, Ghidoli N, Cichero P, Burioni R, Clementi M. The era of molecular and other non-culture-based methods in diagnosis of sepsis. Clin Microbiol Rev. 2010;23:235–51. doi: 10.1128/CMR.00043-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reier-Nilsen T, Farstad T, Nakstad B, Lauvrak V, Steinbakk M. Comparison of broad range 16S rDNA PCR and conventional blood culture for diagnosis of sepsis in the newborn: A case control study. BMC Pediatr. 2009;9:5. doi: 10.1186/1471-2431-9-5. [DOI] [PMC free article] [PubMed] [Google Scholar]